Dysbacteriosis-Derived Lipopolysaccharide Causes Embryonic Osteopenia through Retinoic-Acid-Regulated DLX5 Expression

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
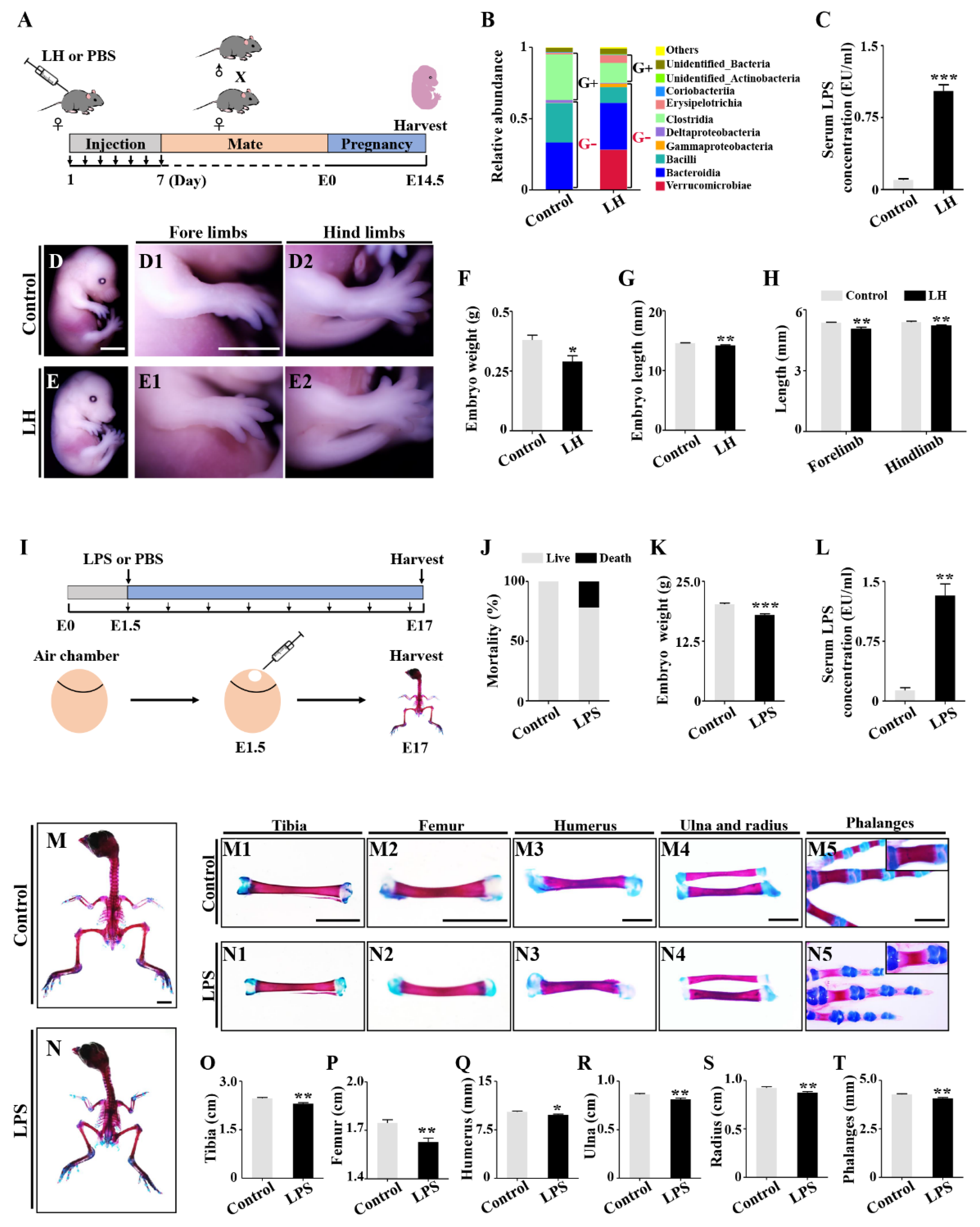
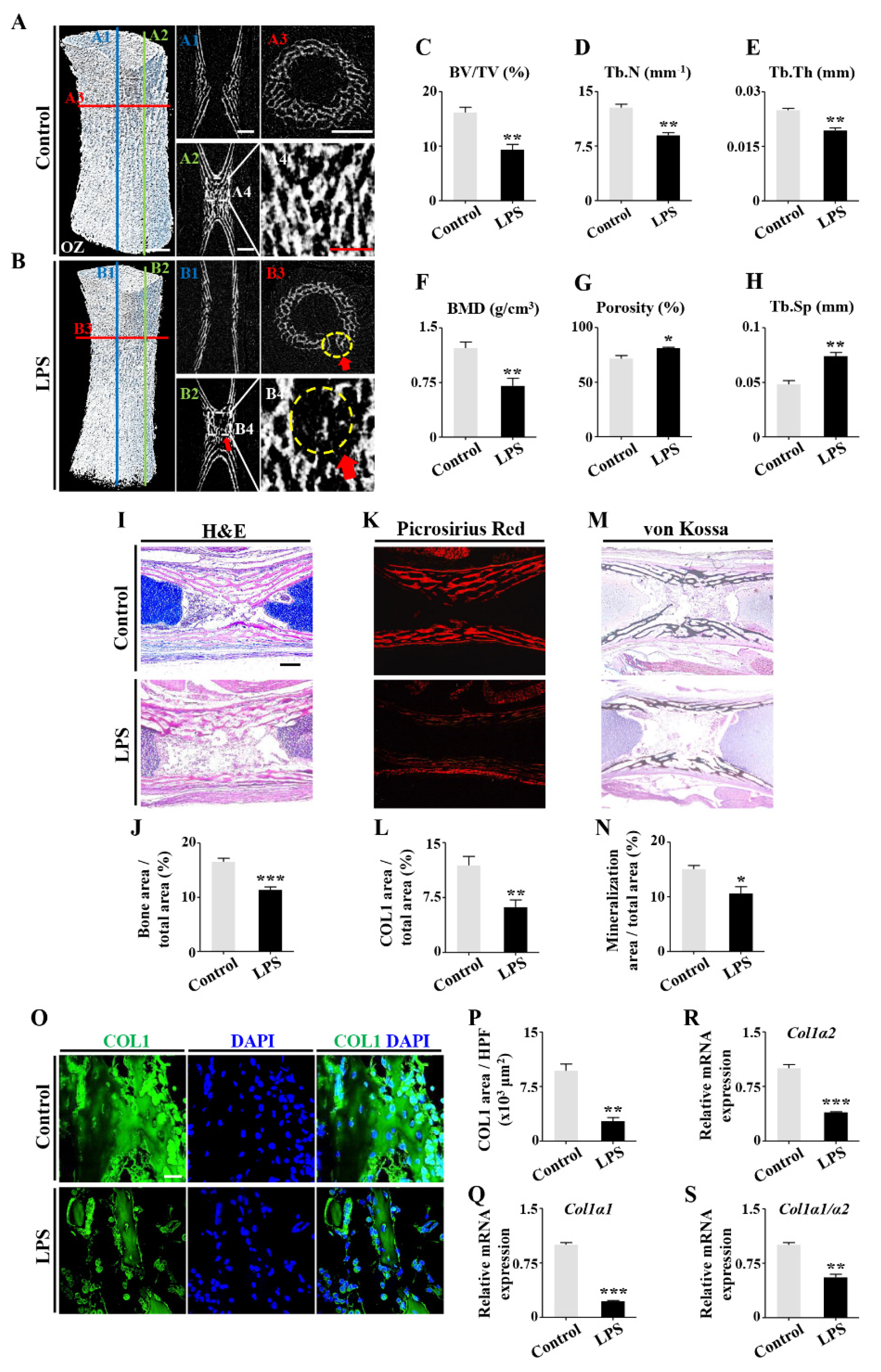
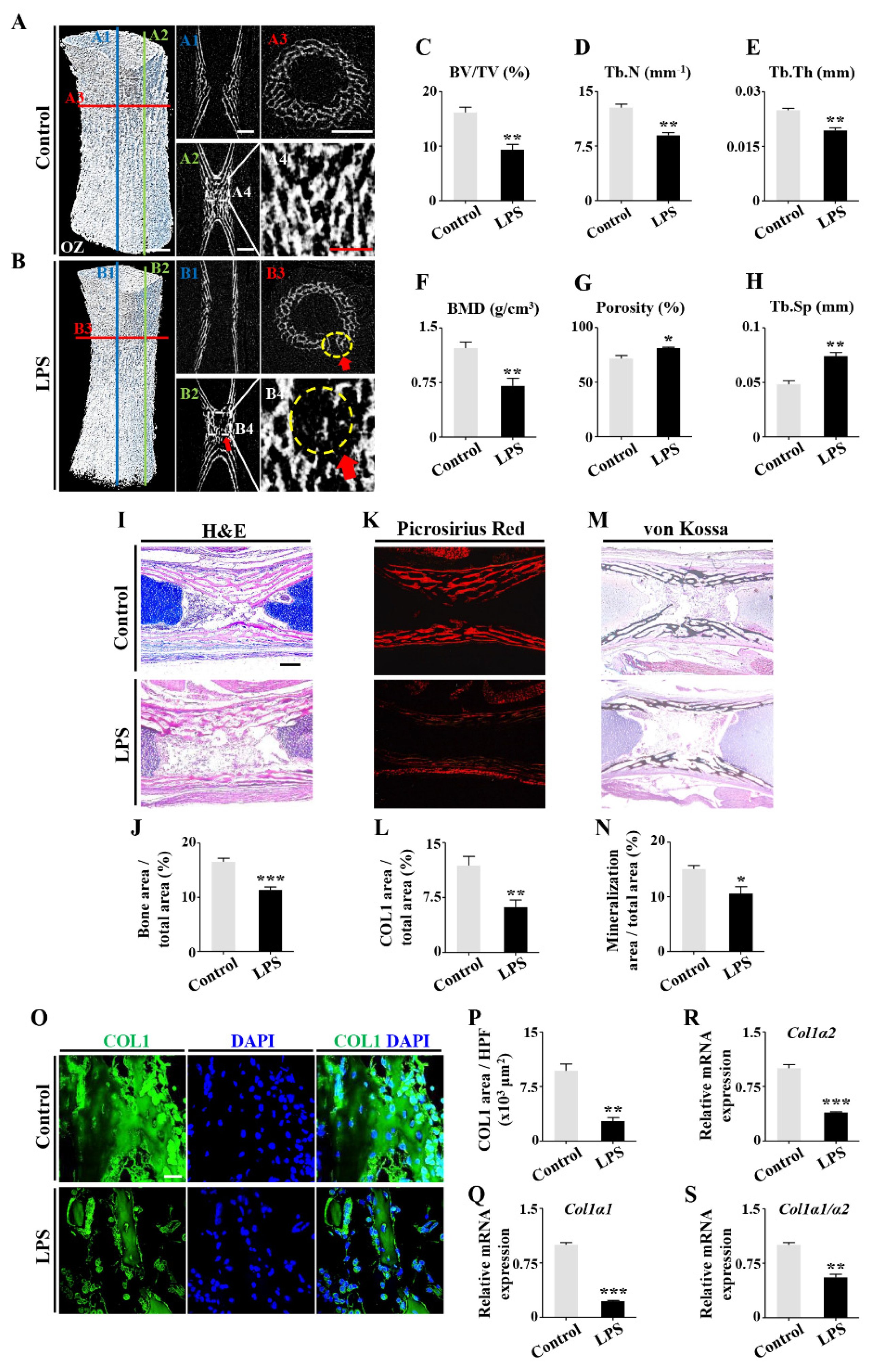
2.1. Retardation of Embryonic Skeleton Development Was Observed in Both Pregnant Mice with Lincomycin Hydrochloride (LH)-Induced Dysbacteriosis and Chick Embryos Treated by LPS
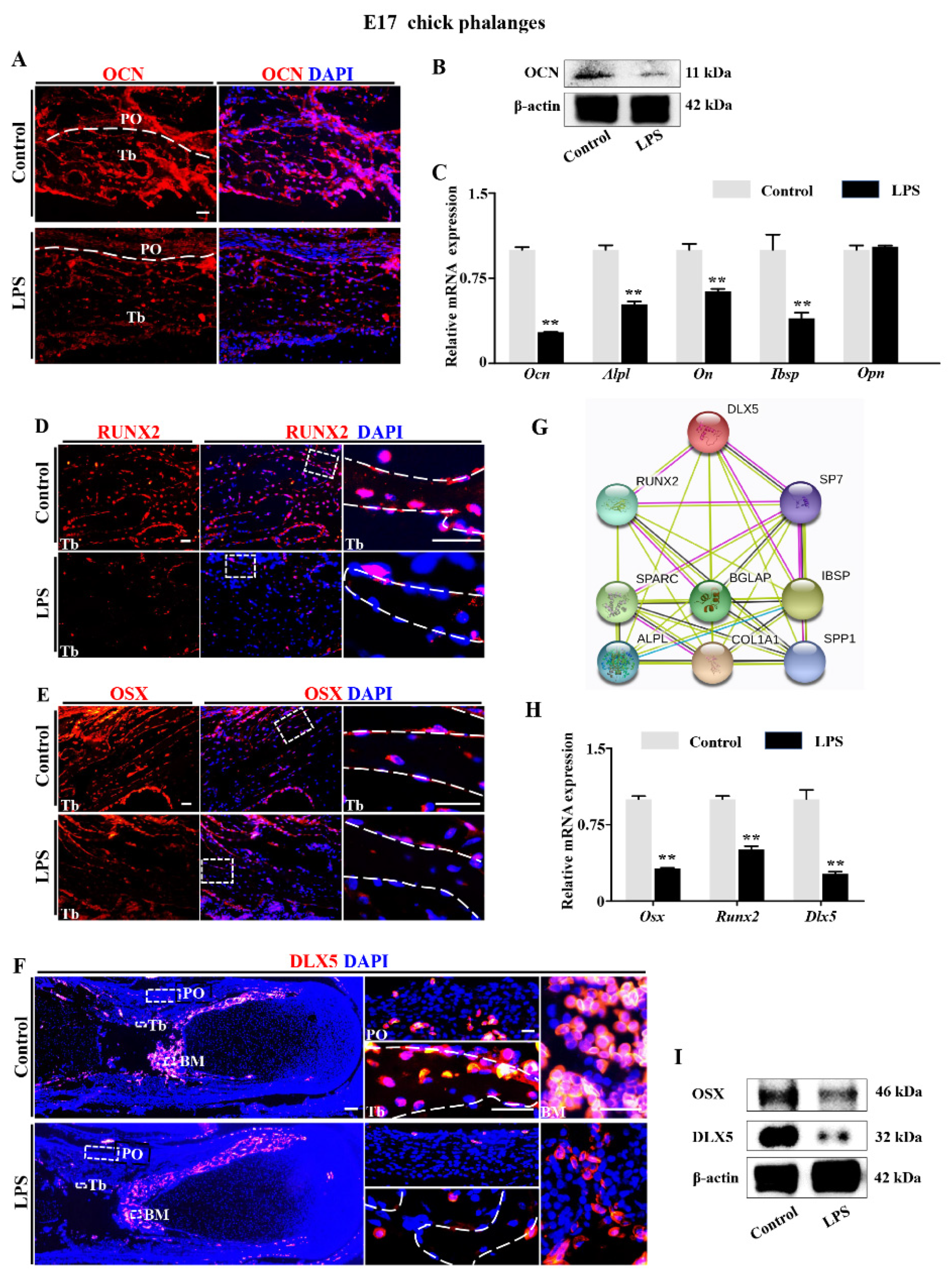
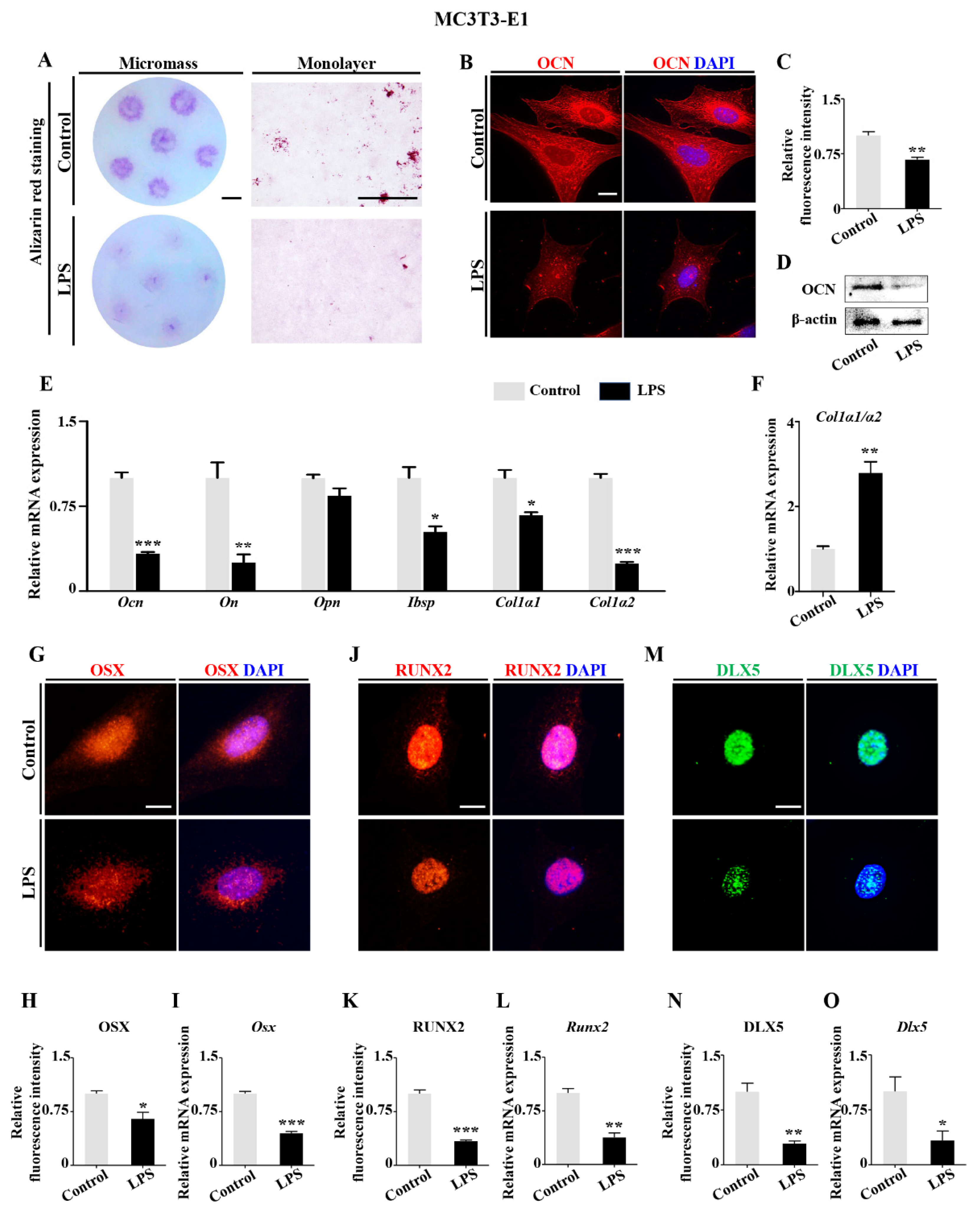
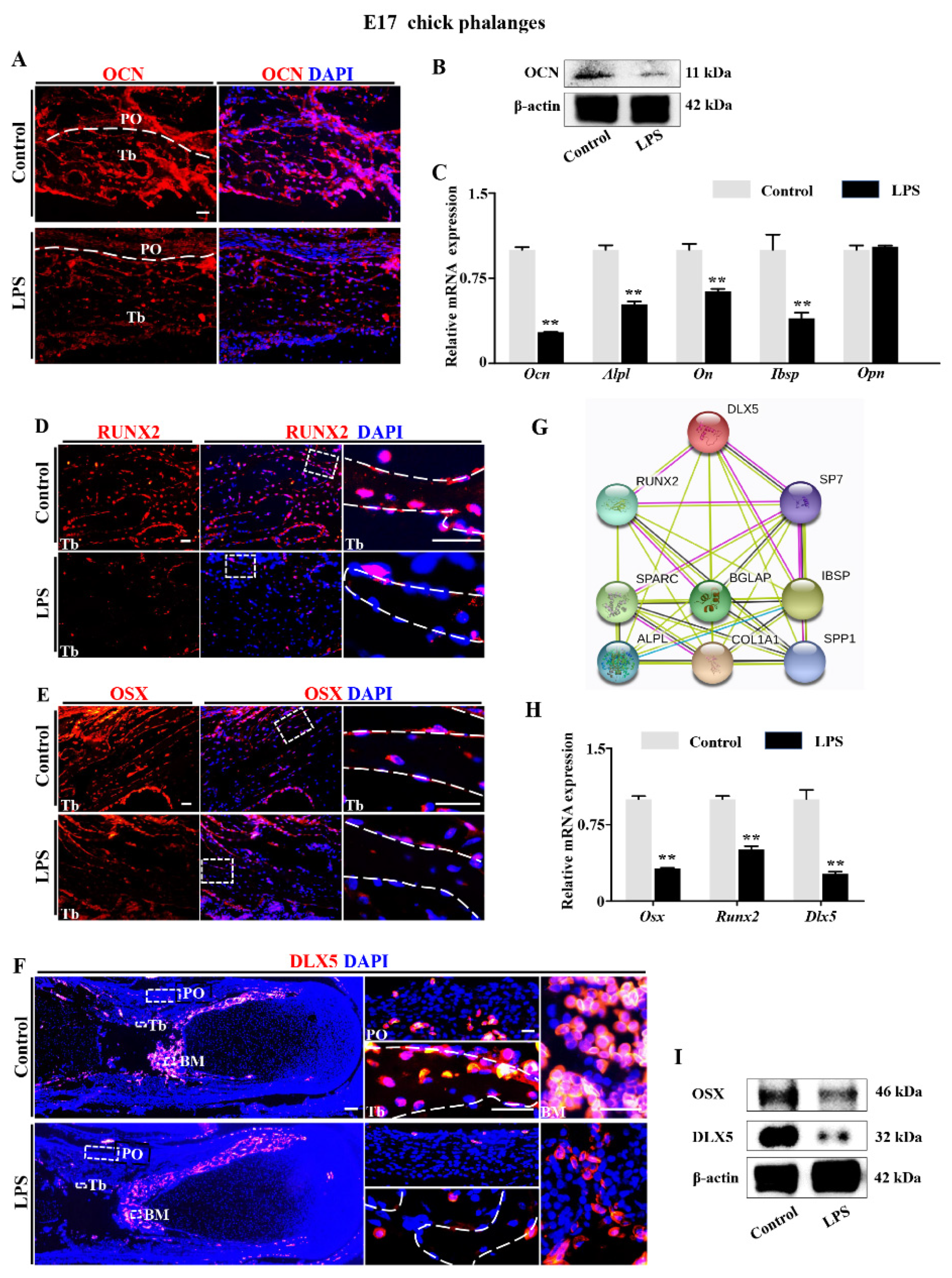
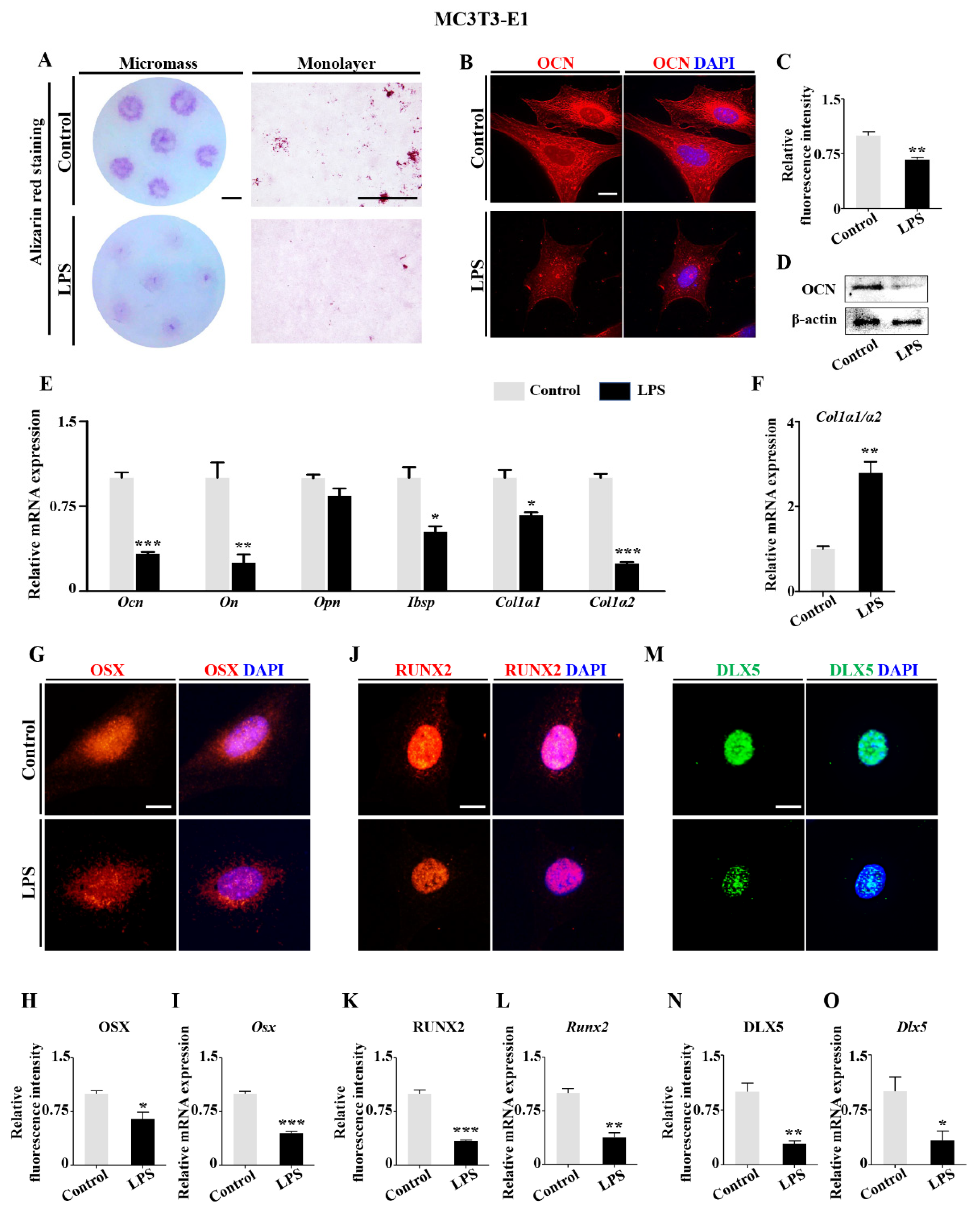
2.2. Downregulation of DLX5 Was Responsible for Aberrant Skeleton Development Induced by LPS Exposure in Chick Embryos
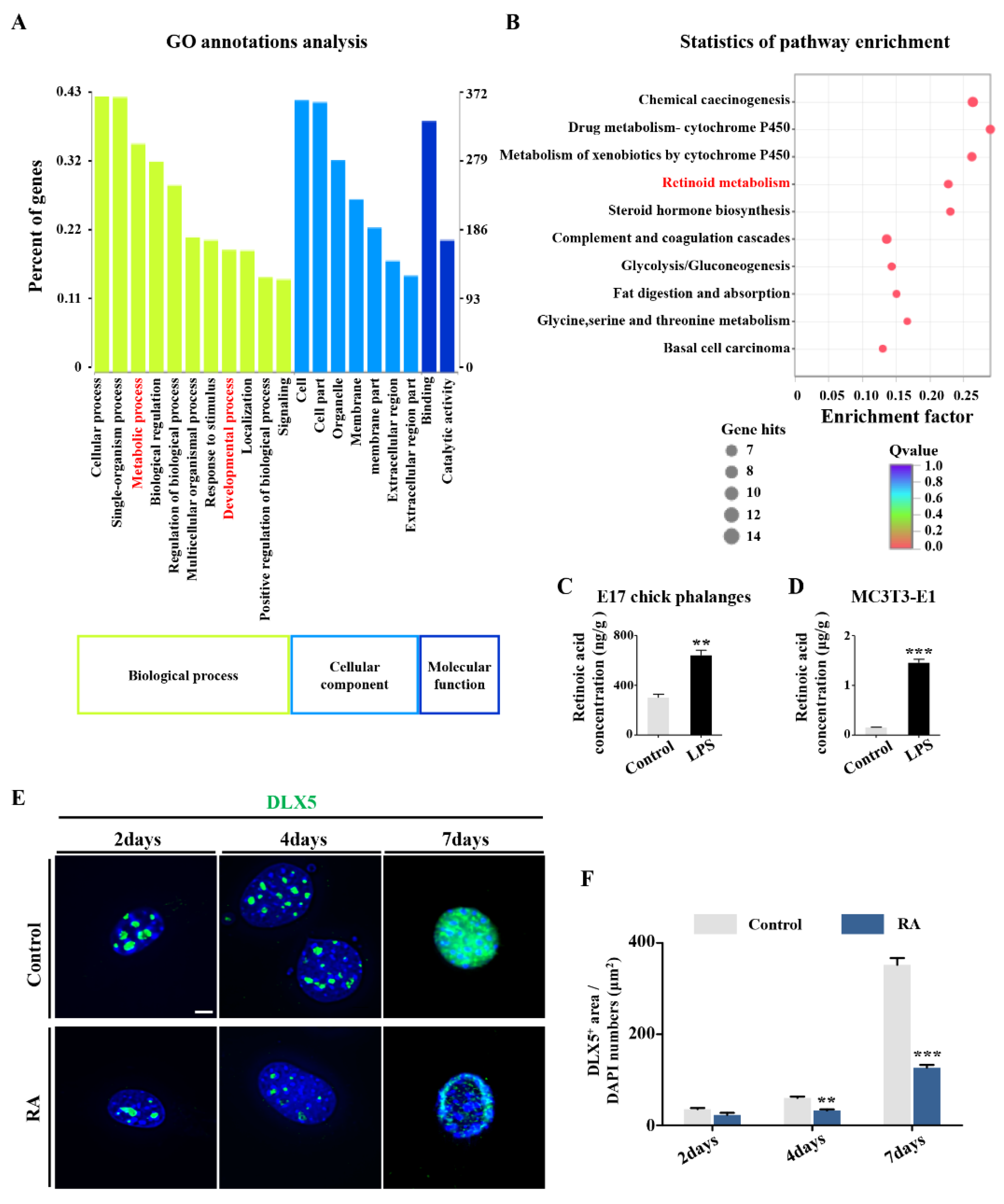
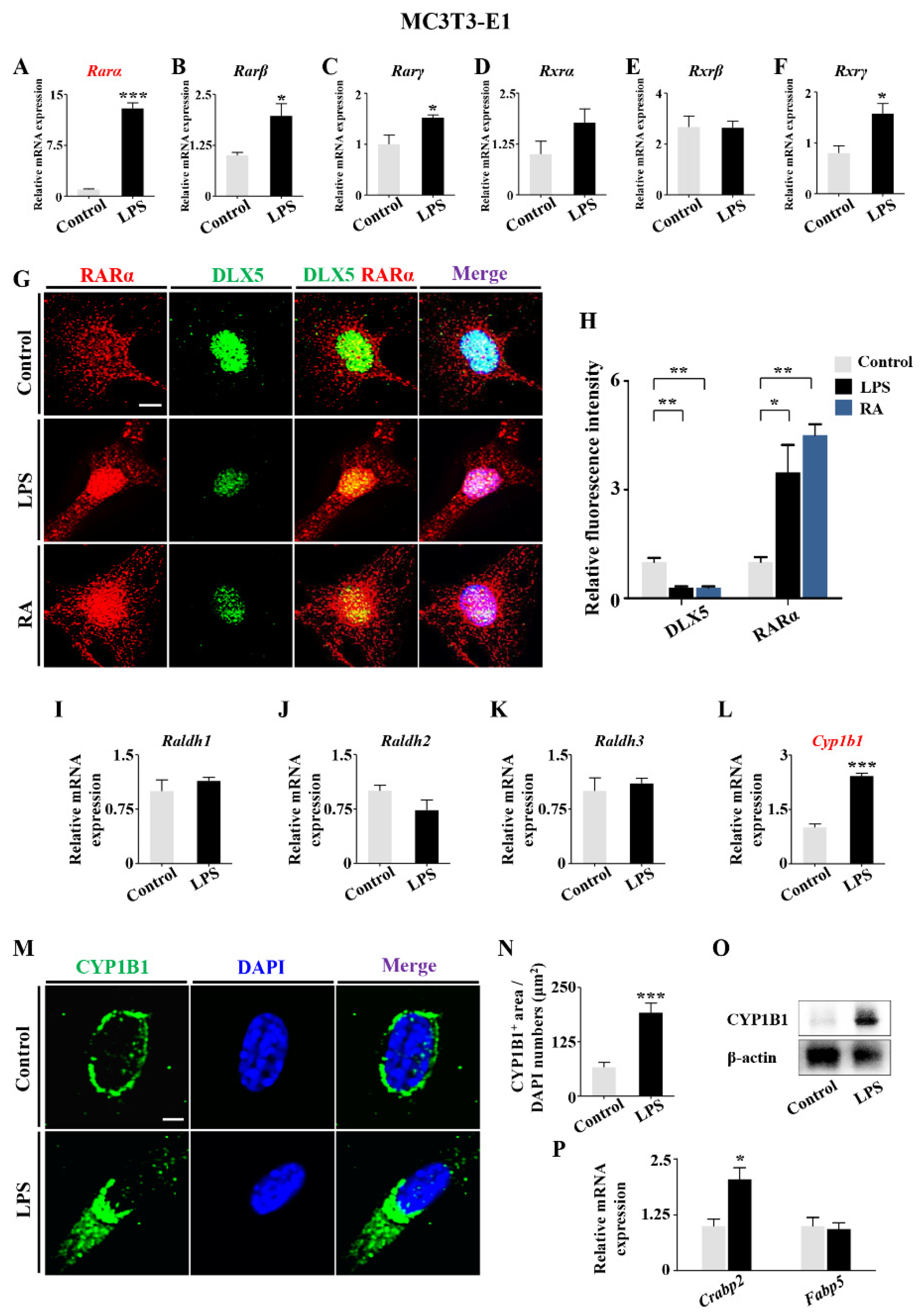
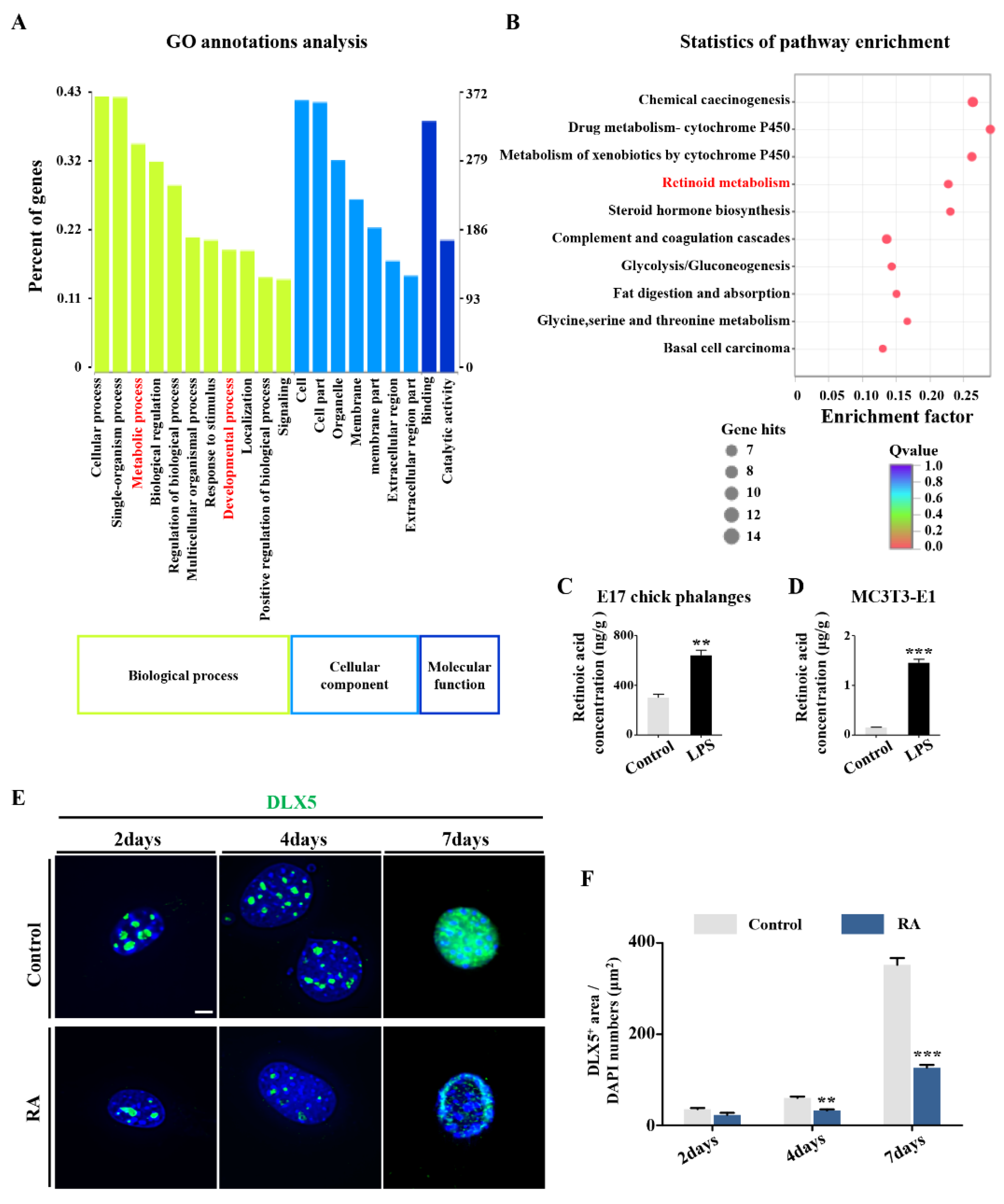
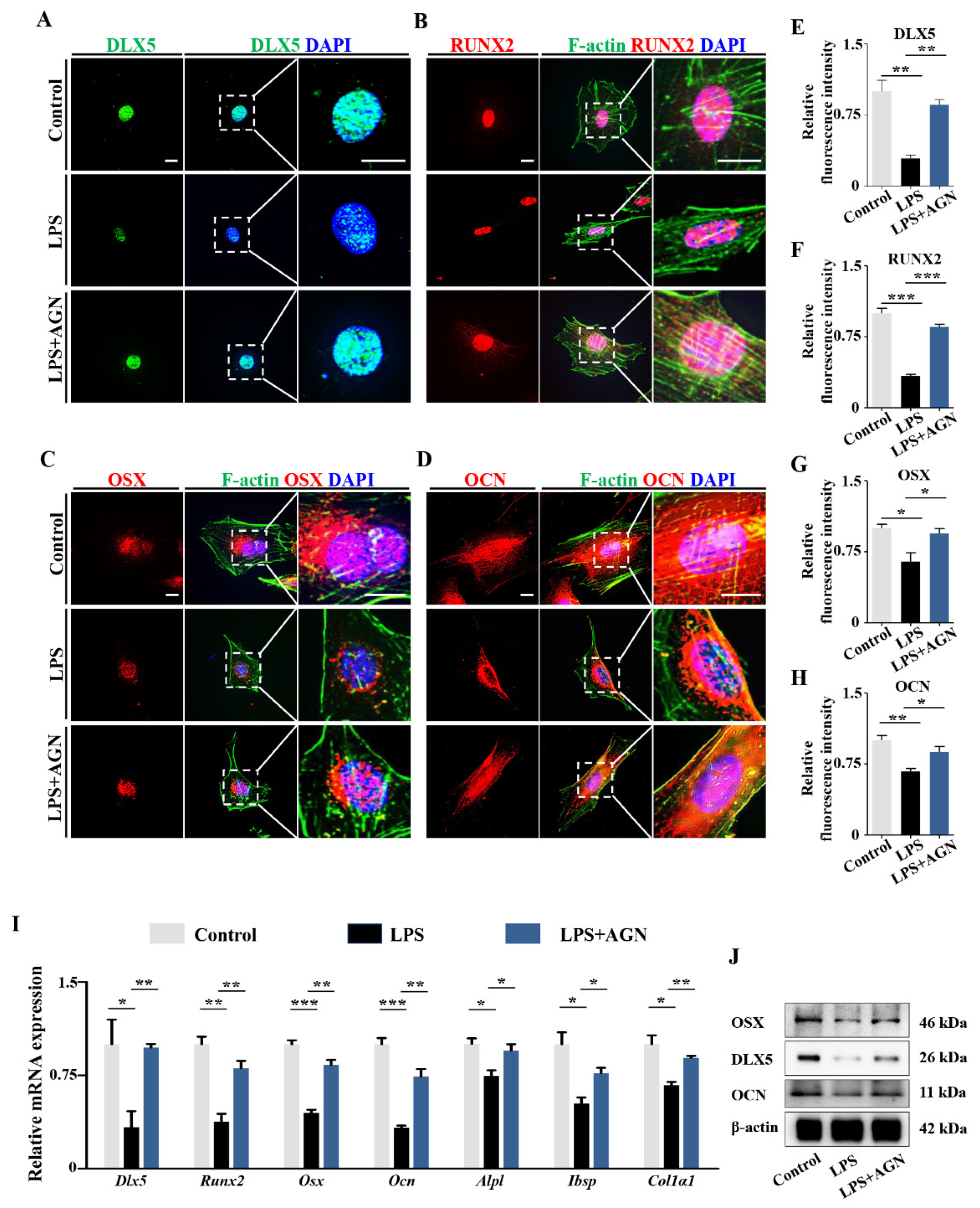
2.3. RA Metabolism Played an Important Role on LPS-Induced Abnormal DLX5 Expressions
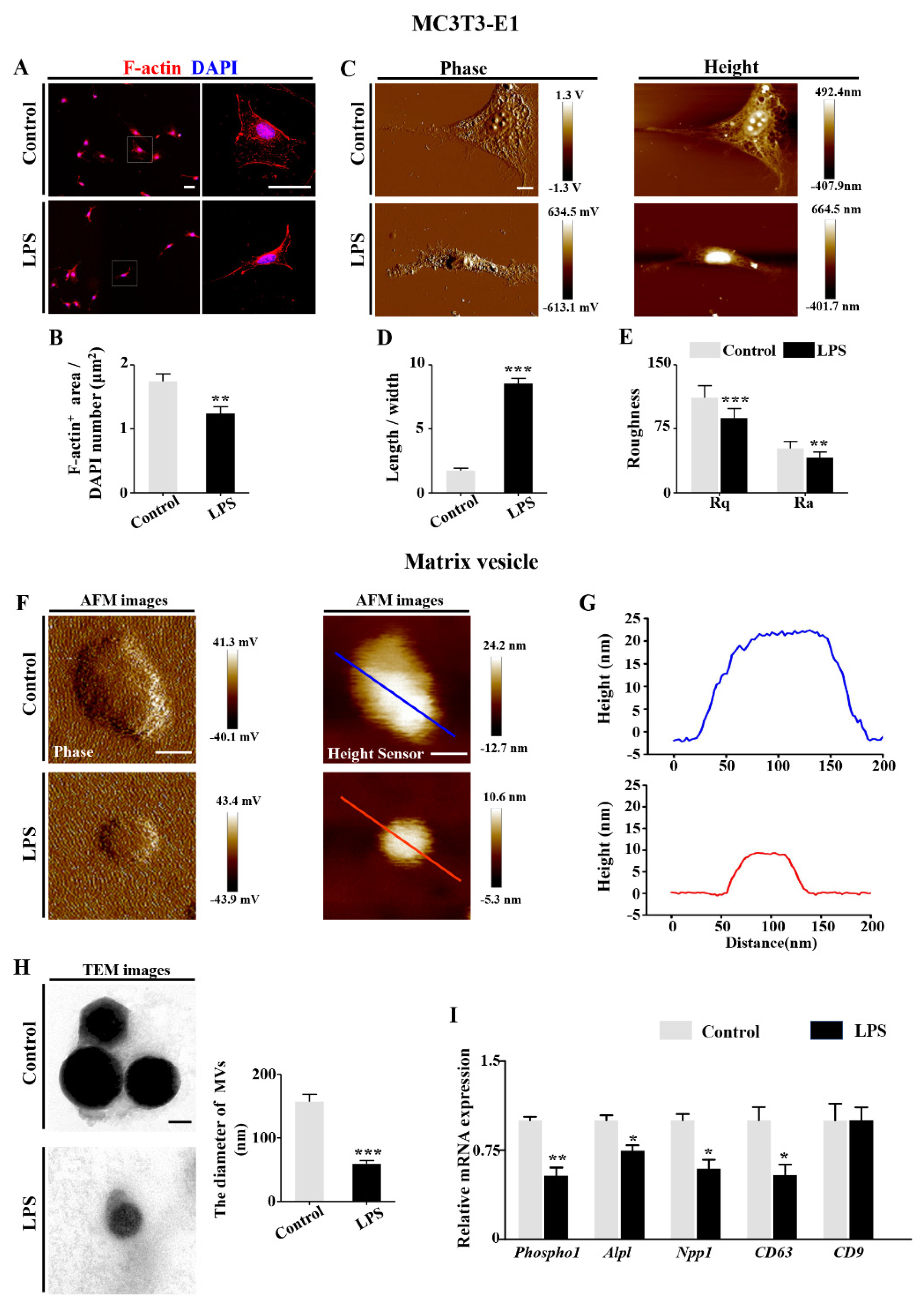
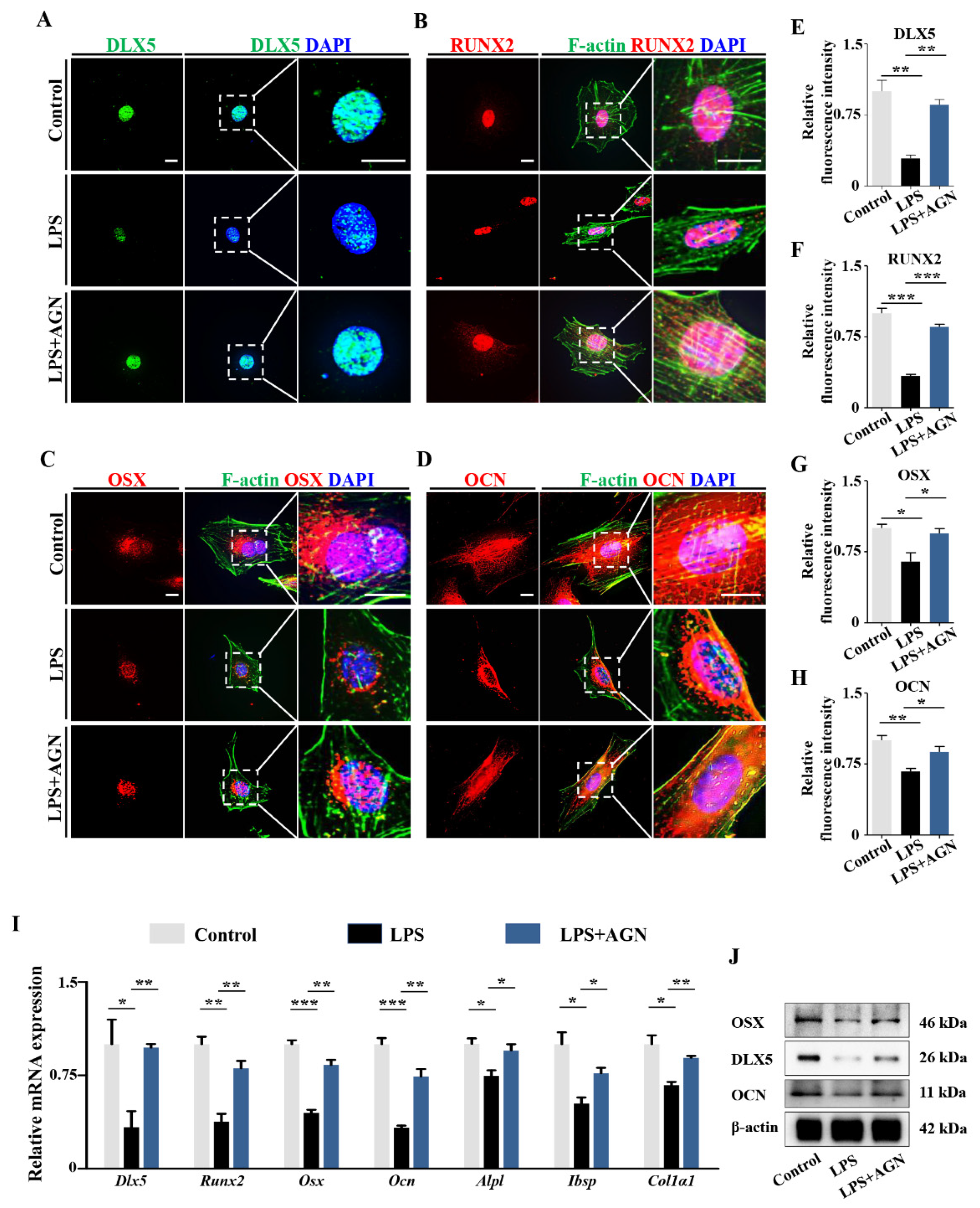
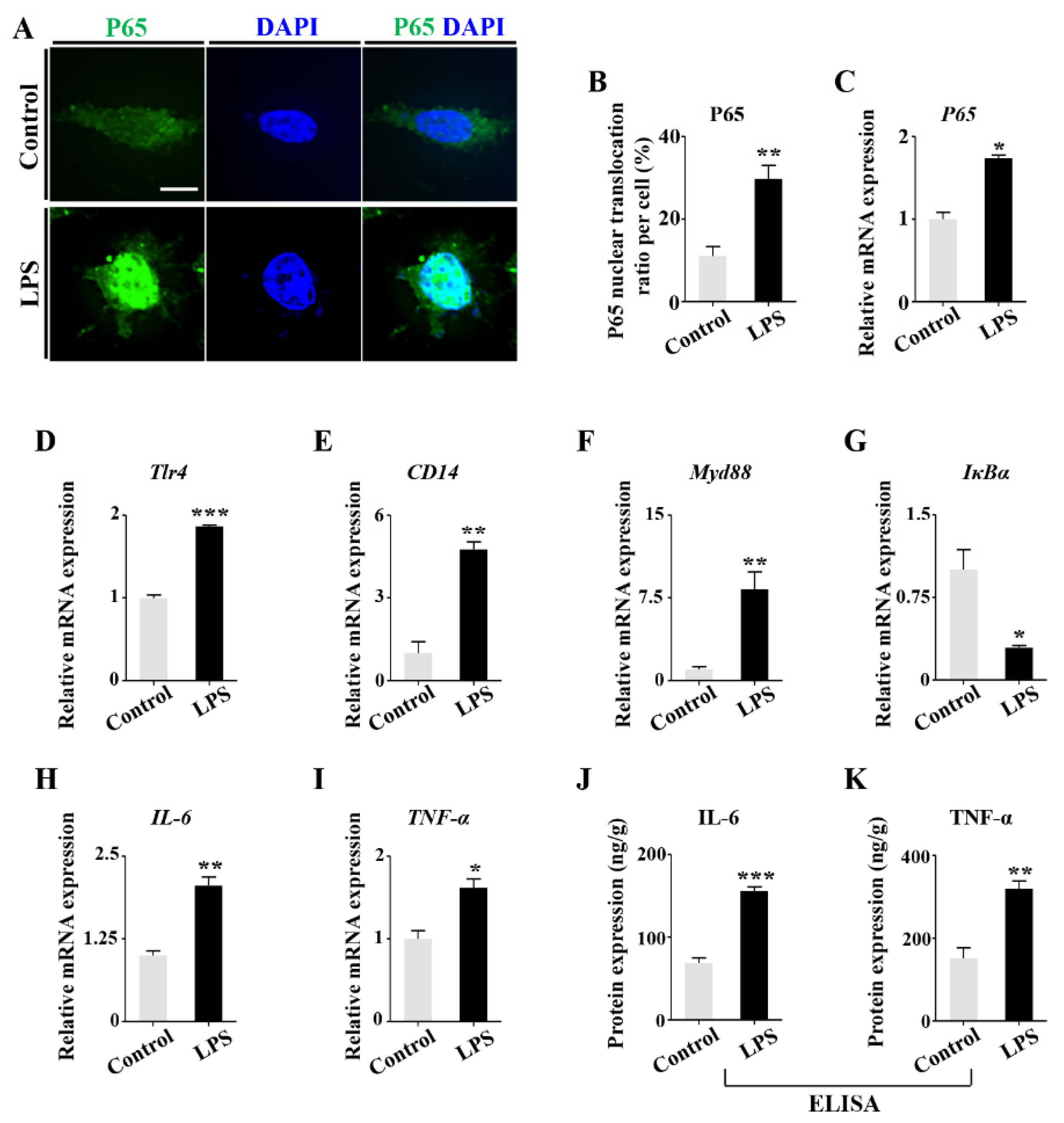
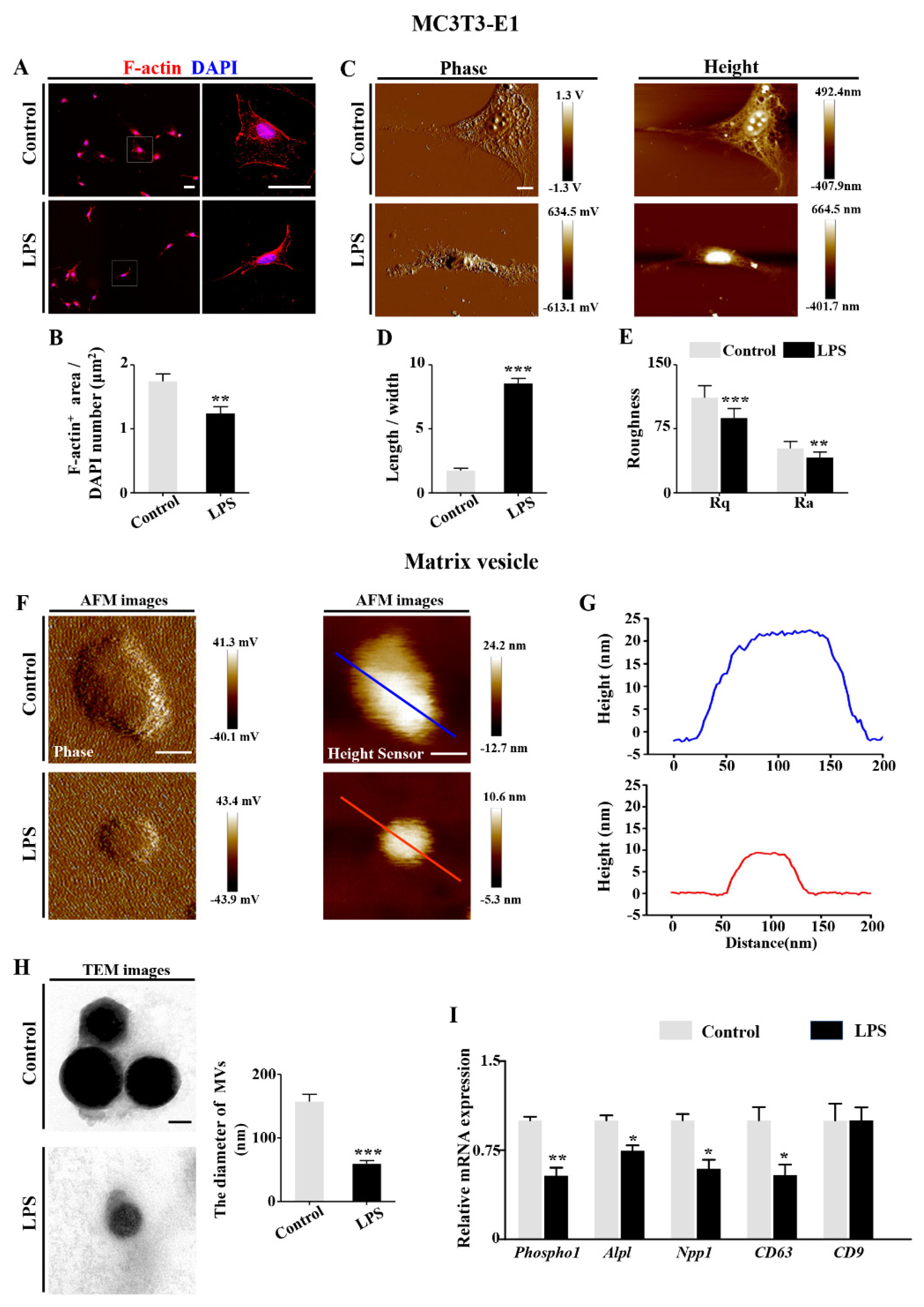
2.4. LPS-Induced Inflammatory Cytokines Lead to RA Increases in Osteoblasts
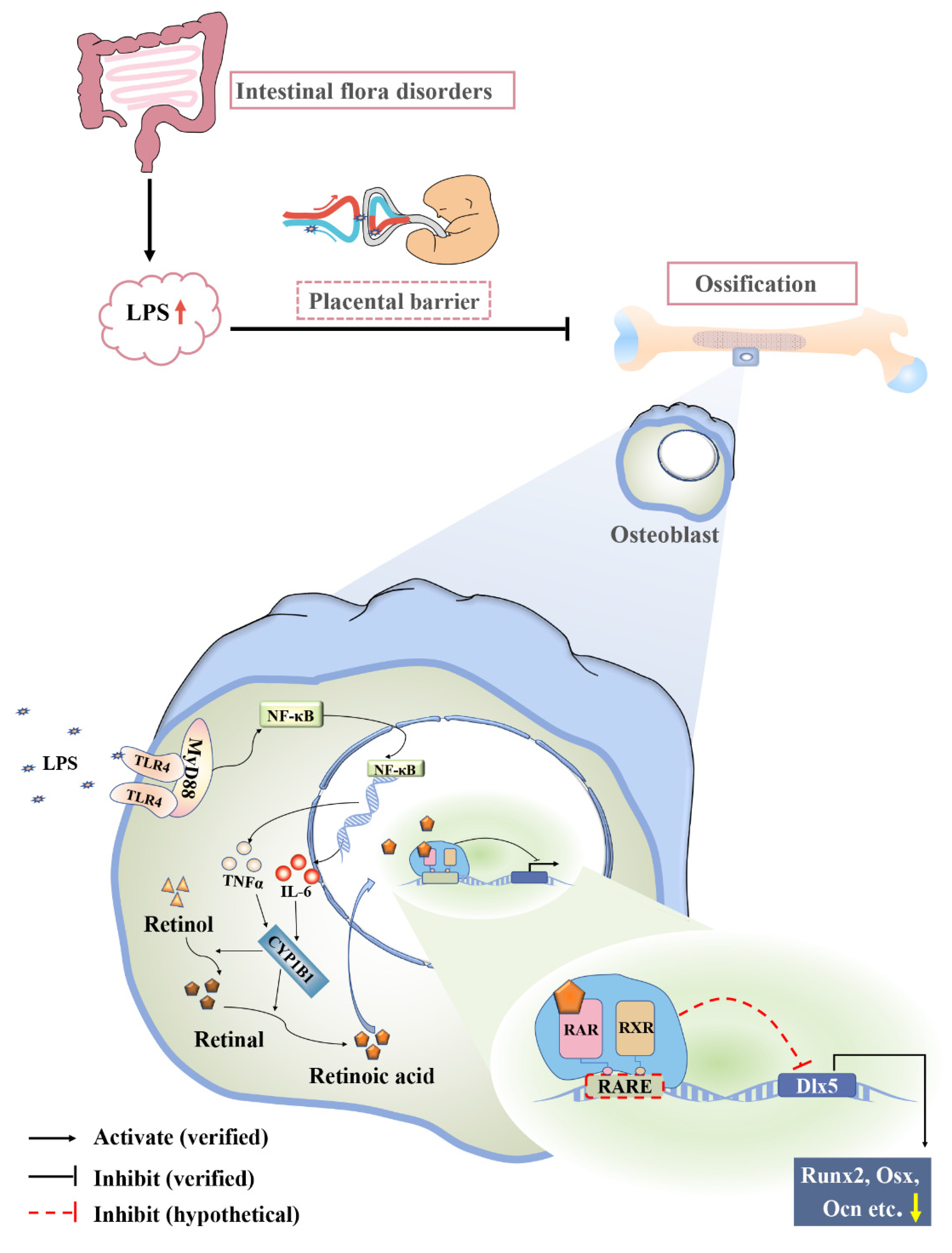
3. Discussion
4. Materials and Methods
4.1. Ethic Statement
4.2. Experimental Mouse and Chick Embryos
4.3. High-Throughput Sequencing Analysis
4.4. Serum Collection and LPS Concentration Determination
4.5. Alcian Blue and Alizarin Red Staining
4.6. Micro-Computed Tomography (Micro-CT)
4.7. Histology
4.8. Cell Culture
4.9. Immunofluorescent Staining and F-Actin Staining
4.10. RNA Isolation and qPCR
4.11. RNA Sequencing Transcript Profiling
4.12. Western Blotting
4.13. Enzyme-Linked Immunosorbent Assay (ELISA)
4.14. Isolation and Purification of Matrix Vesicles (MVs)
4.15. Atomic Force Microscopy (AFM) Morphological Imaging and Membrane Ultrastructure Analysis
4.16. Transmission Electron Microscopy (TEM)
4.17. Data Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LPS | lipopolysaccharide |
| LH Dlx5 | lincomycin hydrochloride distal-less homeobox 5 |
| RA | retinoic acid |
| RARα RARE OZ BMD BV Tb PO BM MVs TRAP | retinoic acid receptor α retinoic acid receptor element ossification zone bone mineral density bone volume trabecular bone periosteum bone marrow matrix vesicles tartrate-resistant acid phosphatase |
References
- Simon, G.L.; Gorbach, S.L. Intestinal flora in health and disease. Gastroenterology 1984, 86, 174–193. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.-R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Trajkovski, M.; Wollheim, C.B. Physiology: Microbial signals to the brain control weight. Nature 2016, 534, 185–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, F.; Backhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Roberfroid, M.; Bornet, F.; Bouley, C.e.; Cummings, J. Colonic microflora: Nutrition and health. Summary and conclusions of an International Life Sciences Institute (ILSI) [Europe] workshop held in Barcelona, Spain. Nutr. Rev. 1995, 53, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Serino, M.; Blasco-Baque, V.; Nicolas, S.; Burcelin, R. Far from the eyes, close to the heart: Dysbiosis of gut microbiota and cardiovascular consequences. Curr. Cardiol. Rep. 2014, 16, 540. [Google Scholar] [CrossRef] [Green Version]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [Green Version]
- Kurita, N.; Yamashiro, K.; Kuroki, T.; Tanaka, R.; Ueno, Y.; Urabe, T.; Nomoto, K.; Takahashi, T.; Tsuji, H.; Asahara, T. Gut dysbiosis induces lipopolysaccharide-mediated inflammation after cerebral ischemia in type 2 diabetic mice. J. Neurol. Sci. 2017, 381, 875. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.X.; Wang, H.; Zhao, L.; Ning, H.; Chen, Y.H.; Zhang, C. Effects of low-dose lipopolysaccharide (LPS) pretreatment on LPS-induced intra-uterine fetal death and preterm labor. Toxicology 2007, 234, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.B.; Keating, D.J.; Young, R.L.; Wong, M.L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Song, X.; Huang, Y.; Yao, J.; Zhou, M.; Li, Z.; You, Q.; Guo, Q.; Lu, N. Wogonin inhibits LPS-induced tumor angiogenesis via suppressing PI3K/Akt/NF-kappaB signaling. Eur. J. Pharm. 2014, 737, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Aisemberg, J.; Vercelli, C.A.; Bariani, M.V.; Billi, S.C.; Wolfson, M.L.; Franchi, A.M. Progesterone is essential for protecting against LPS-induced pregnancy loss. LIF as a potential mediator of the anti-inflammatory effect of progesterone. PLoS ONE 2013, 8, e56161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.A.; Zoellner, M.J.; Carney, E.W. In Vivo and In Vitro developmental toxicity in LPS-induced zinc-deficient rabbits. Reprod. Toxicol. 1997, 11, 771–779. [Google Scholar] [CrossRef]
- Wang, H.; Yang, L.L.; Hu, Y.F.; Wang, B.W.; Huang, Y.Y.; Zhang, C.; Chen, Y.H.; Xu, D.X. Maternal LPS exposure during pregnancy impairs testicular development, steroidogenesis and spermatogenesis in male offspring. PLoS ONE 2014, 9, e106786. [Google Scholar] [CrossRef]
- Roughley, P.J. The structure and function of cartilage proteoglycans. Eur. Cells Mater. 2006, 12, 92–101. [Google Scholar] [CrossRef]
- Jaiswal, Y.K.; Jaiswal, M.K.; Agrawal, V.; Chaturvedi, M.M. Bacterial endotoxin (LPS)-induced DNA damage in preimplanting embryonic and uterine cells inhibits implantation. Fertil. Steril. 2009, 91, 2095–2103. [Google Scholar] [CrossRef]
- Collins, J.J.; Kuypers, E.; Nitsos, I.; Jane Pillow, J.; Polglase, G.R.; Kemp, M.W.; Newnham, J.P.; Cleutjens, J.P.; Frints, S.G.; Kallapur, S.G.; et al. LPS-induced chorioamnionitis and antenatal corticosteroids modulate Shh signaling in the ovine fetal lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L778–L787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Xu, D.X.; Wang, J.P.; Wang, H.; Wei, L.Z.; Sun, M.F.; Wei, W. Melatonin protects against lipopolysaccharide-induced intra-uterine fetal death and growth retardation in mice. J. Pineal Res. 2006, 40, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Romano, T.; Wark, J.D.; Owens, J.A.; Wlodek, M.E. Prenatal growth restriction and postnatal growth restriction followed by accelerated growth independently program reduced bone growth and strength. Bone 2009, 45, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Giussani, D.A.; Forhead, A.J. Endocrine and metabolic programming during intrauterine development. Early Hum. Dev. 2005, 81, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samee, N.; Geoffroy, V.; Marty, C.; Schiltz, C.; Vieux-Rochas, M.; Levi, G.; de Vernejoul, M.C. Dlx5, a positive regulator of osteoblastogenesis, is essential for osteoblast-osteoclast coupling. Am. J. Pathol. 2008, 173, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Kim, Y.J.; Yoon, W.J.; Kim, J.I.; Kim, B.G.; Hwang, Y.S.; Wozney, J.M.; Chi, X.Z.; Bae, S.C.; Choi, K.Y.; et al. Dlx5 specifically regulates Runx2 type II expression by binding to homeodomain-response elements in the Runx2 distal promoter. J. Biol. Chem. 2005, 280, 35579–35587. [Google Scholar] [CrossRef] [Green Version]
- Newberry, E.P.; Latifi, T.; Towler, D.A. Reciprocal regulation of osteocalcin transcription by the homeodomain proteins Msx2 and Dlx5. Biochemistry 1998, 37, 16360–16368. [Google Scholar] [CrossRef]
- Ferrari, D.; Harrington, A.; Dealy, C.N.; Kosher, R.A. Dlx-5 in limb initiation in the chick embryo. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 1999, 216, 10–15. [Google Scholar] [CrossRef]
- Patel, S.A.; Bhambra, U.; Charalambous, M.P.; David, R.M.; Edwards, R.J.; Lightfoot, T.; Boobis, A.R.; Gooderham, N.J. Interleukin-6 mediated upregulation of CYP1B1 and CYP2E1 in colorectal cancer involves DNA methylation, miR27b and STAT3. Br. J. Cancer 2014, 111, 2287–2296. [Google Scholar] [CrossRef] [Green Version]
- Umannova, L.; Machala, M.; Topinka, J.; Novakova, Z.; Milcova, A.; Kozubik, A.; Vondracek, J. Tumor necrosis factor-alpha potentiates genotoxic effects of benzo[a]pyrene in rat liver epithelial cells through upregulation of cytochrome P450 1B1 expression. Mutat. Res. 2008, 640, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Aleshukina, A.V. Pathogenesis of intestinal dysbacteriosis. Zhurnal Mikrobiol. Epidemiol. I Immunobiol. 2012, 3, 74–78. [Google Scholar]
- Zhang, J.; Wang, G.; Liu, J.; Gao, L.R.; Liu, M.; Wang, C.J.; Chuai, M.; Bao, Y.; Li, G.; Li, R.M.; et al. Gut microbiota-derived endotoxin enhanced the incidence of cardia bifida during cardiogenesis. J. Cell. Physiol. 2018, 233, 9271–9283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Li, P.Z.; Wang, G.; Yan, Y.; Li, K.; Brand-Saberi, B.; Yang, X. Microbiota-derived lipopolysaccharide retards chondrocyte hypertrophy in the growth plate through elevating Sox9 expression. J. Cell. Physiol. 2019, 234, 2593–2605. [Google Scholar] [CrossRef]
- Nuriel-Ohayon, M.; Neuman, H.; Koren, O. Microbial Changes during Pregnancy, Birth, and Infancy. Front. Microbiol. 2016, 7, 1031. [Google Scholar] [CrossRef] [Green Version]
- Boskey, A.L.; Robey, P.G. The regulatory role of matrix proteins in mineralization of bone. In Osteoporosis; Elsevier: Amsterdam, The Netherlands, 2013; pp. 235–255. [Google Scholar]
- Golub, E.E.; Boesze-Battaglia, K. The role of alkaline phosphatase in mineralization. Curr. Opin. Orthop. 2007, 18, 444–448. [Google Scholar] [CrossRef]
- Komori, T. Regulation of osteoblast differentiation by transcription factors. J. Cell. Biochem. 2006, 99, 1233–1239. [Google Scholar] [CrossRef]
- Kawane, T.; Komori, H.; Liu, W.; Moriishi, T.; Miyazaki, T.; Mori, M.; Matsuo, Y.; Takada, Y.; Izumi, S.; Jiang, Q.; et al. Dlx5 and mef2 regulate a novel runx2 enhancer for osteoblast-specific expression. J. Bone Min. Res. 2014, 29, 1960–1969. [Google Scholar] [CrossRef]
- Plaut, J.S.; Strzelecka-Kiliszek, A.; Bozycki, L.; Pikula, S.; Buchet, R.; Mebarek, S.; Chadli, M.; Bolean, M.; Simao, A.M.S.; Ciancaglini, P.; et al. Quantitative atomic force microscopy provides new insight into matrix vesicle mineralization. Arch. Biochem. Biophys. 2019, 667, 14–21. [Google Scholar] [CrossRef]
- Perez-Campo, F.M.; Santurtun, A.; Garcia-Ibarbia, C.; Pascual, M.A.; Valero, C.; Garces, C.; Sanudo, C.; Zarrabeitia, M.T.; Riancho, J.A. Osterix and RUNX2 are transcriptional regulators of sclerostin in human bone. Calcif. Tissue Int. 2016, 99, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Burcelin, R. Gut microbiota and immune crosstalk in metabolic disease. Mol. Metab. 2016, 5, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Tanugi, A.; Forest, N. Retinoic acid suppresses the osteogenic differentiation capacity of murine osteoblast-like 3/A/1D-1M cell cultures. Differ. Res. Biol. Divers. 1998, 63, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Green, A.C.; Kocovski, P.; Jovic, T.; Walia, M.K.; Chandraratna, R.A.S.; Martin, T.J.; Baker, E.K.; Purton, L.E. Retinoic acid receptor signalling directly regulates osteoblast and adipocyte differentiation from mesenchymal progenitor cells. Exp. Cell Res. 2017, 350, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Promislow, J.H.; Goodman-Gruen, D.; Slymen, D.J.; Barrett-Connor, E. Retinol intake and bone mineral density in the elderly: The rancho bernardo study. J. Bone Min. Res. 2002, 17, 1349–1358. [Google Scholar] [CrossRef]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, Y.; Li, R.; Chen, G. Transcriptional factors mediating retinoic acid signals in the control of energy metabolism. Int. J. Mol. Sci. 2015, 16, 14210–14244. [Google Scholar] [CrossRef] [Green Version]
- Podvinec, M.; Kaufmann, M.R.; Handschin, C.; Meyer, U.A. NUBIScan, an in silico approach for prediction of nuclear receptor response elements. Mol. Endocrinol. 2002, 16, 1269–1279. [Google Scholar] [CrossRef]
- Guo, W.; Li, Y.; Wang, L.; Wang, J.; Xu, Q.; Yan, T.; Xue, B. Evaluation of composition and individual variability of rumen microbiota in yaks by 16S rRNA high-throughput sequencing technology. Anaerobe 2015, 34, 74–79. [Google Scholar] [CrossRef]
- Wang, L.; Jin, L.; Xue, B.; Wang, Z.; Peng, Q. Characterizing the bacterial community across the gastrointestinal tract of goats: Composition and potential function. Microbiologyopen 2019, 8, e00820. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, J.; Xu, L.; Zhang, J.; Chan, K.; Pan, X.; Li, G. MiR-503 promotes bone formation in distraction osteogenesis through suppressing smurf1 expression. Sci. Rep. 2017, 7, 409. [Google Scholar] [CrossRef] [Green Version]
- Kertesz, Z.; Gyori, D.; Kormendi, S.; Fekete, T.; Kis-Toth, K.; Jakus, Z.; Schett, G.; Rajnavolgyi, E.; Dobo-Nagy, C.; Mocsai, A. Phospholipase Cgamma2 is required for basal but not oestrogen deficiency-induced bone resorption. Eur. J. Clin. Investig. 2012, 42, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Yan, Y.; Chen, J.L.; Ma, Z.L.; Yang, R.H.; Wang, G.; Chuai, M.; Ka Ho Lee, K.; Yang, X. Dexamethasone exposure accelerates endochondral ossification of chick embryos via angiogenesis. Toxicol. Sci. Off. J. Soc. Toxicol. 2016, 149, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.S.; Guo, R.; Li, P.Z.; Li, K.; Yan, Y.; Chen, J.L.; Wang, G.; Brand-Saberi, B.; Yang, X.S.; Cheng, X. Dexamethasone interferes with osteoblasts formation during osteogenesis through altering IGF-1-mediated angiogenesis. J. Cell. Physiol. 2019, 234, 15167–15181. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.C.; Kuzynski, M.; Bottini, M.; Beniash, E.; Dokland, T.; Mobley, C.G.; Yadav, M.C.; Poliard, A.; Kellermann, O.; Millan, J.L.; et al. Phosphate induces formation of matrix vesicles during odontoblast-initiated mineralization In Vitro. Matrix Biol. J. Int. Soc. Matrix Biol. 2016, 52–54, 284–300. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, K.; Wang, Y.; Li, M.; Sun, X.; Liang, Z.; Wang, L.; Chen, L.; Yang, H.; Zhu, L. Disulfiram chelated with copper promotes apoptosis in human breast cancer cells by impairing the mitochondria functions. Scanning 2016, 38, 825–836. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, L.; Zhu, L.; Li, P.-z.; Wang, G.; Cai, H.; Song, J.; Long, D.; Berman, Z.; Lin, L.; Cheng, X.; et al. Dysbacteriosis-Derived Lipopolysaccharide Causes Embryonic Osteopenia through Retinoic-Acid-Regulated DLX5 Expression. Int. J. Mol. Sci. 2020, 21, 2518. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072518
You L, Zhu L, Li P-z, Wang G, Cai H, Song J, Long D, Berman Z, Lin L, Cheng X, et al. Dysbacteriosis-Derived Lipopolysaccharide Causes Embryonic Osteopenia through Retinoic-Acid-Regulated DLX5 Expression. International Journal of Molecular Sciences. 2020; 21(7):2518. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072518
Chicago/Turabian StyleYou, Lingsen, Liwei Zhu, Pei-zhi Li, Guang Wang, Hongmei Cai, Jinhuan Song, Denglu Long, Zachary Berman, Li Lin, Xin Cheng, and et al. 2020. "Dysbacteriosis-Derived Lipopolysaccharide Causes Embryonic Osteopenia through Retinoic-Acid-Regulated DLX5 Expression" International Journal of Molecular Sciences 21, no. 7: 2518. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072518