Pharmacological Inhibition and Activation of the Ca2+ Activated Cl− Channel TMEM16A

 and
and
Abstract
:1. Introduction
2. Results
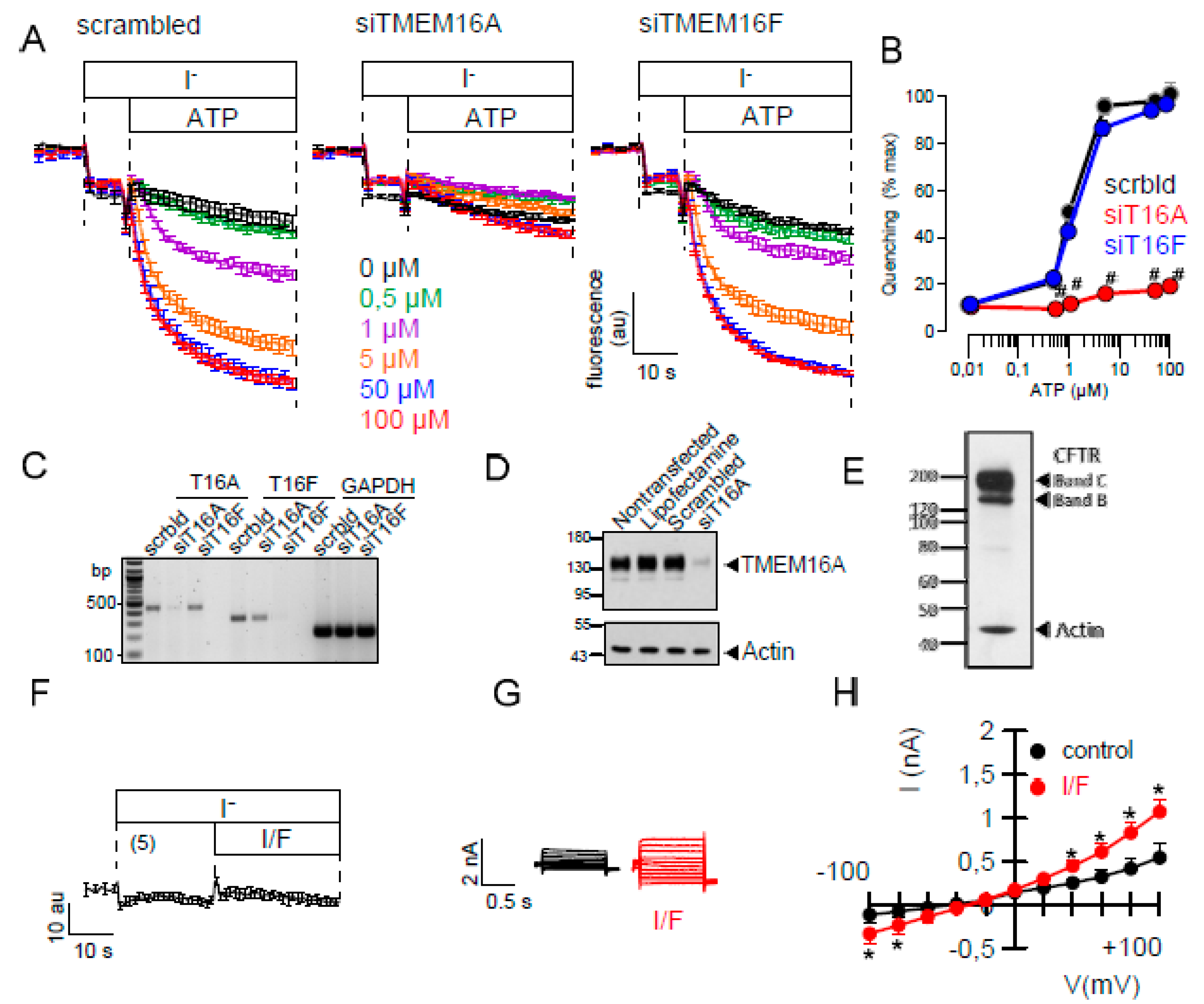
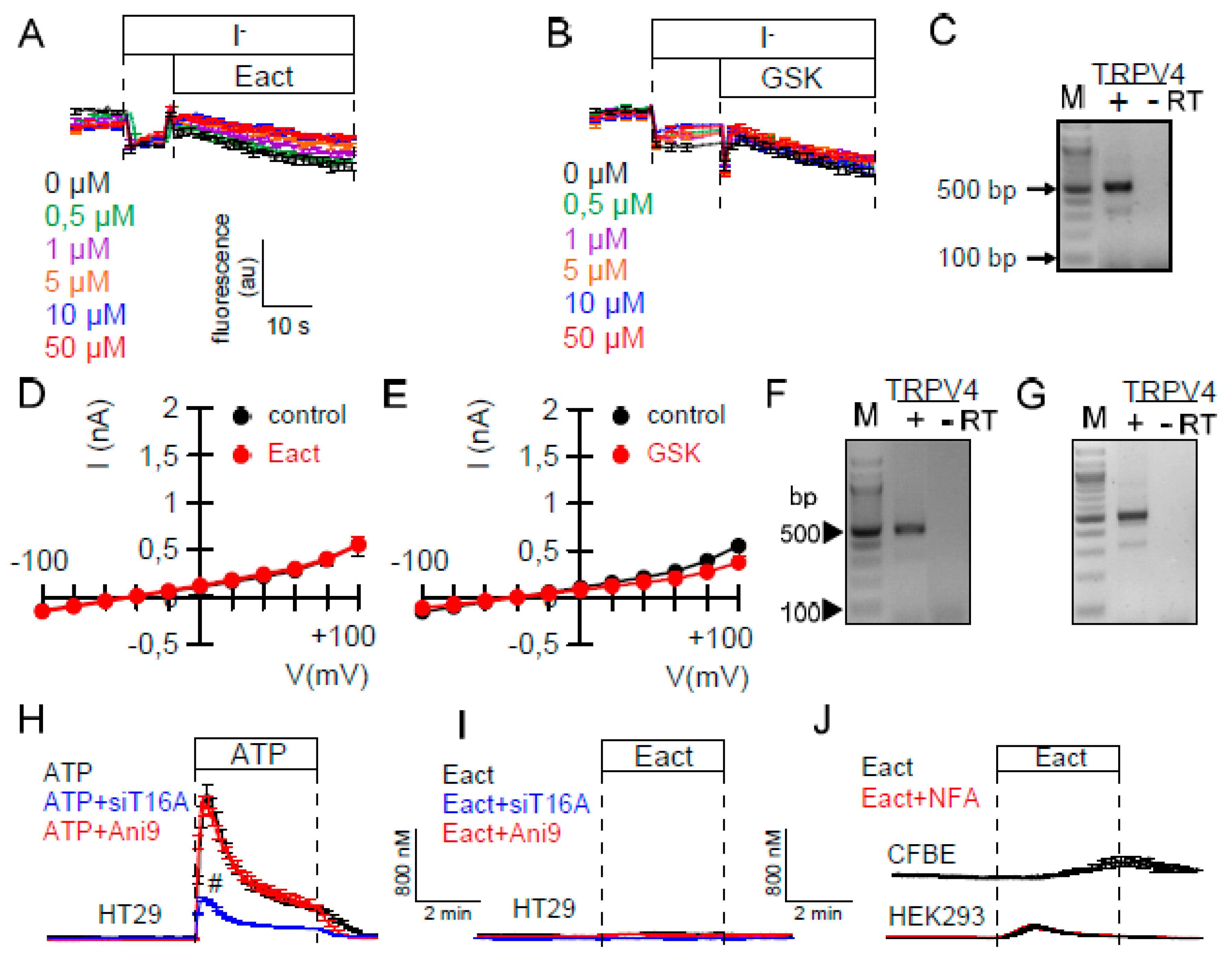
2.1. Ca2+-Dependent Cl− Transport in Colonic Epithelial Cells is not Activated by Eact
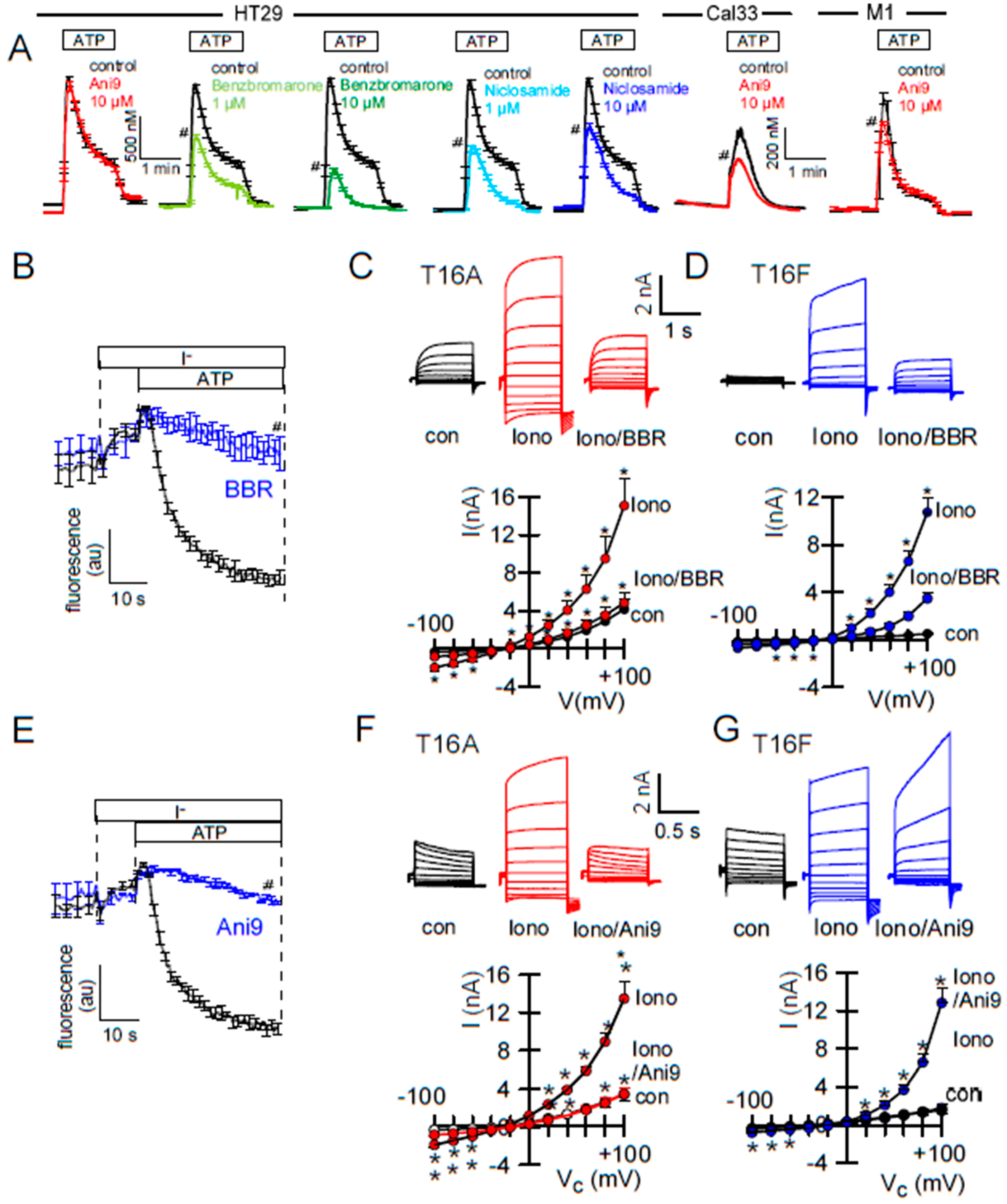
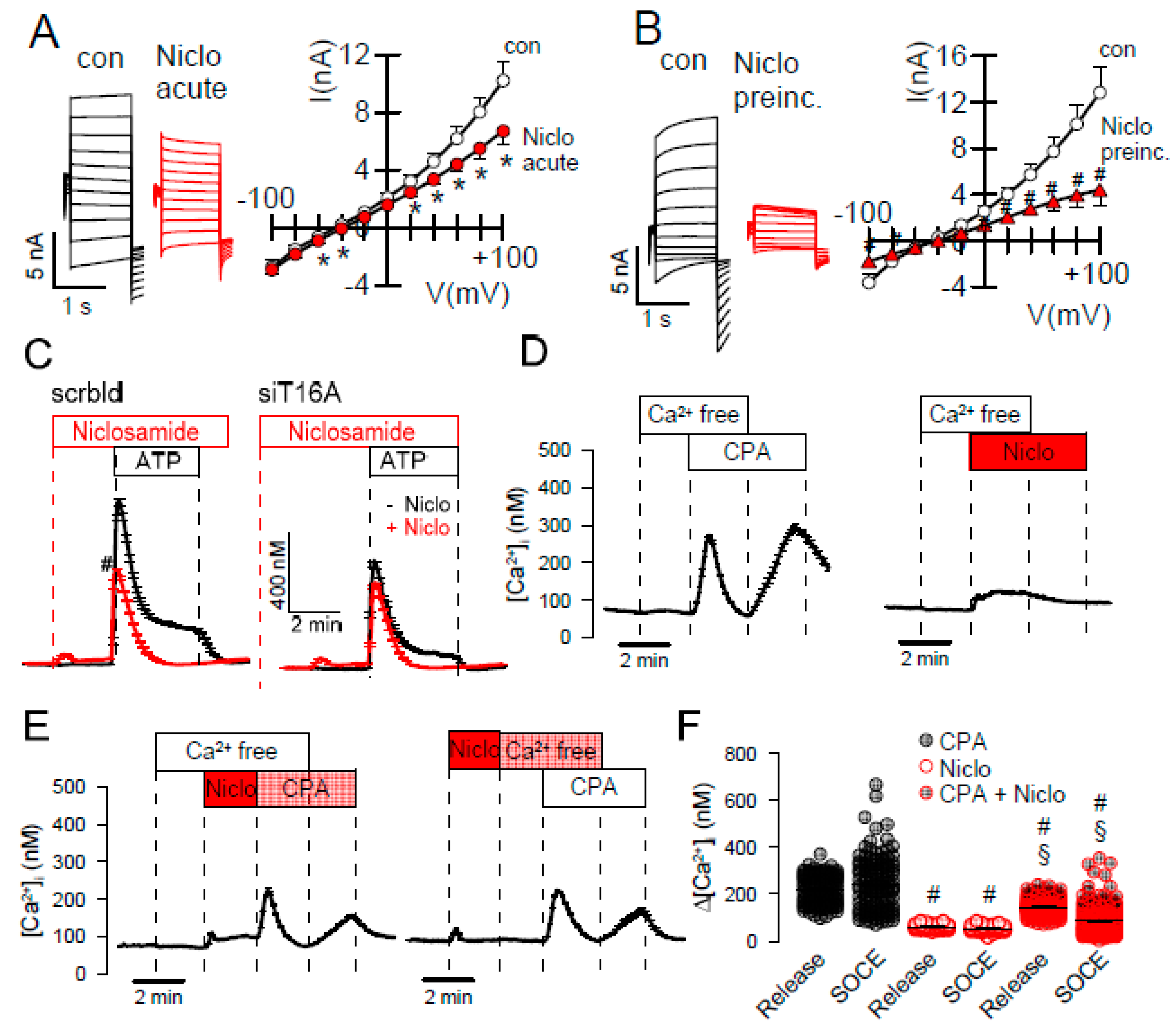
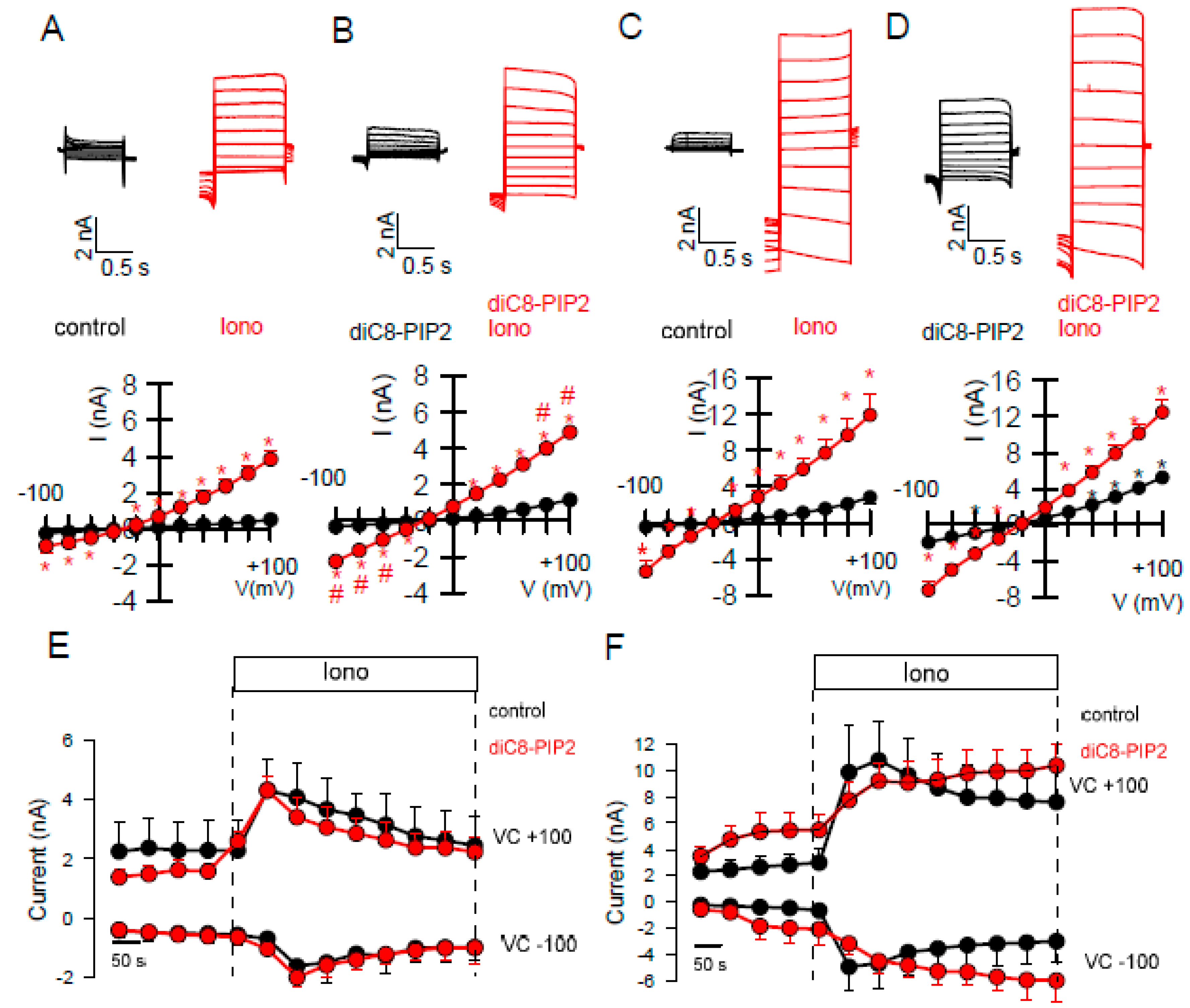
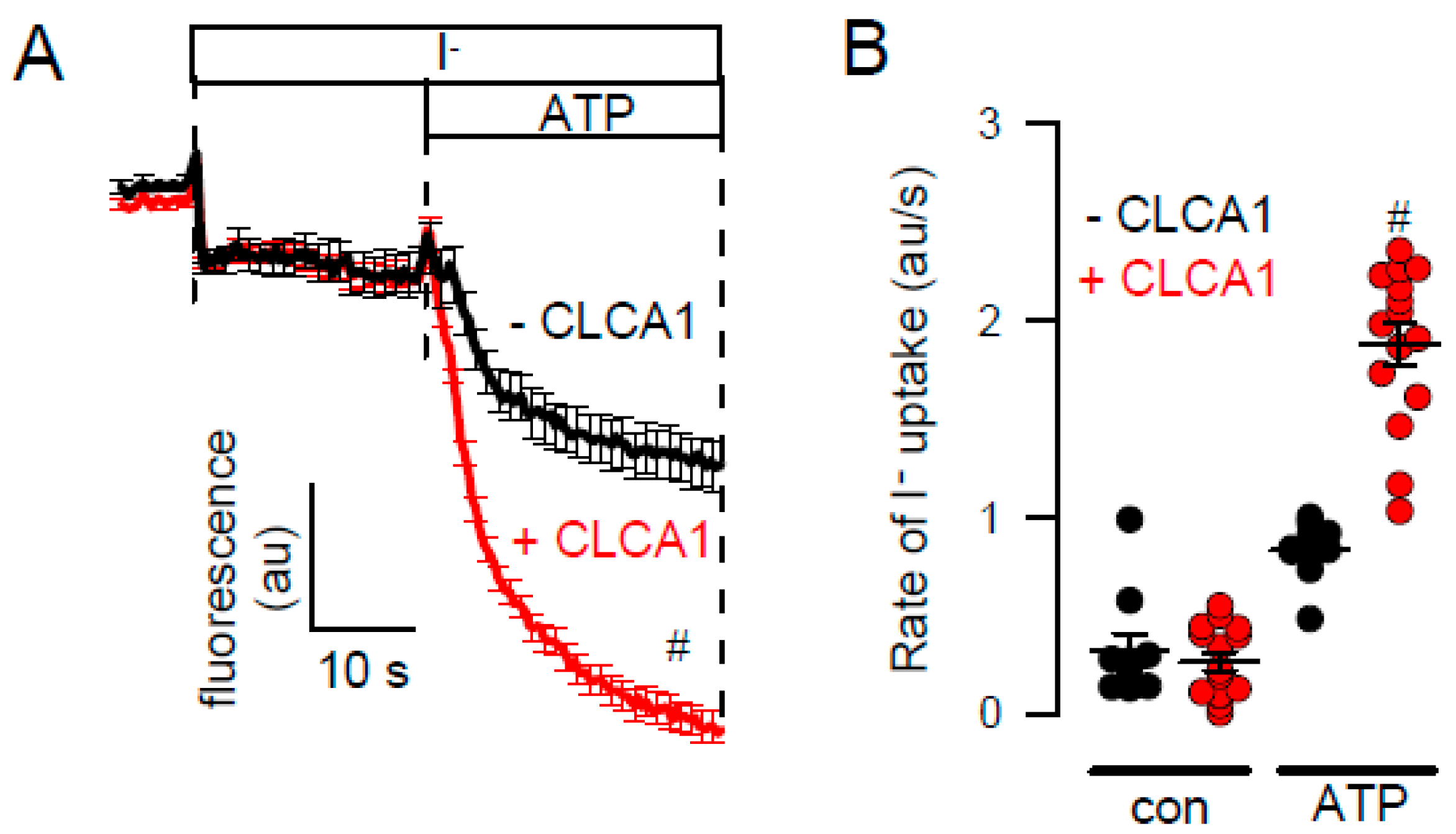
2.2. Effects of Inhibitors of TMEM16A on Whole Cell Currents and Intracellular Ca2+
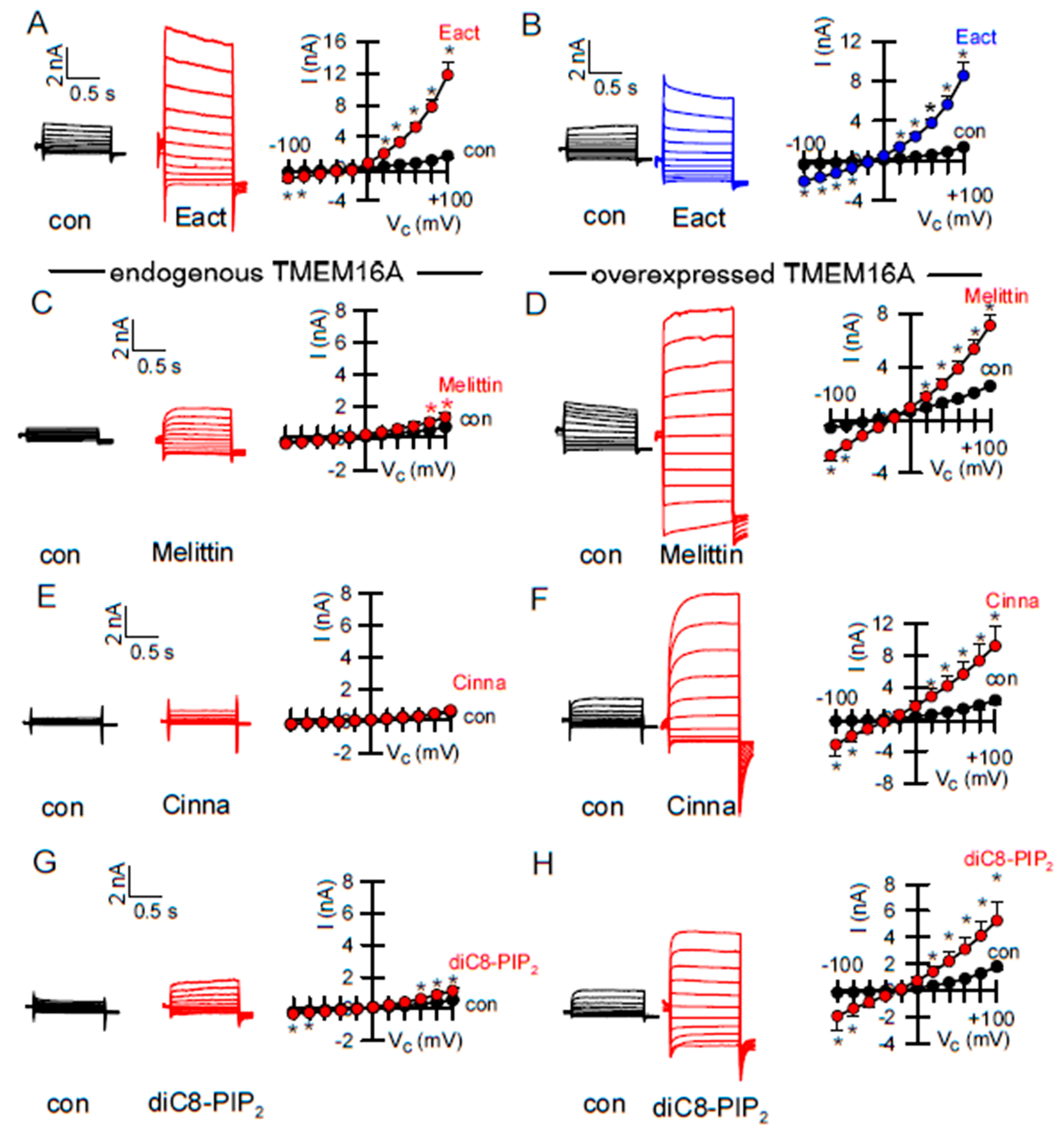
2.3. Activators of TMEM16A Act Differently on Endogenous and Overexpressed TMEM16A
3. Discussion
4. Material and Methods
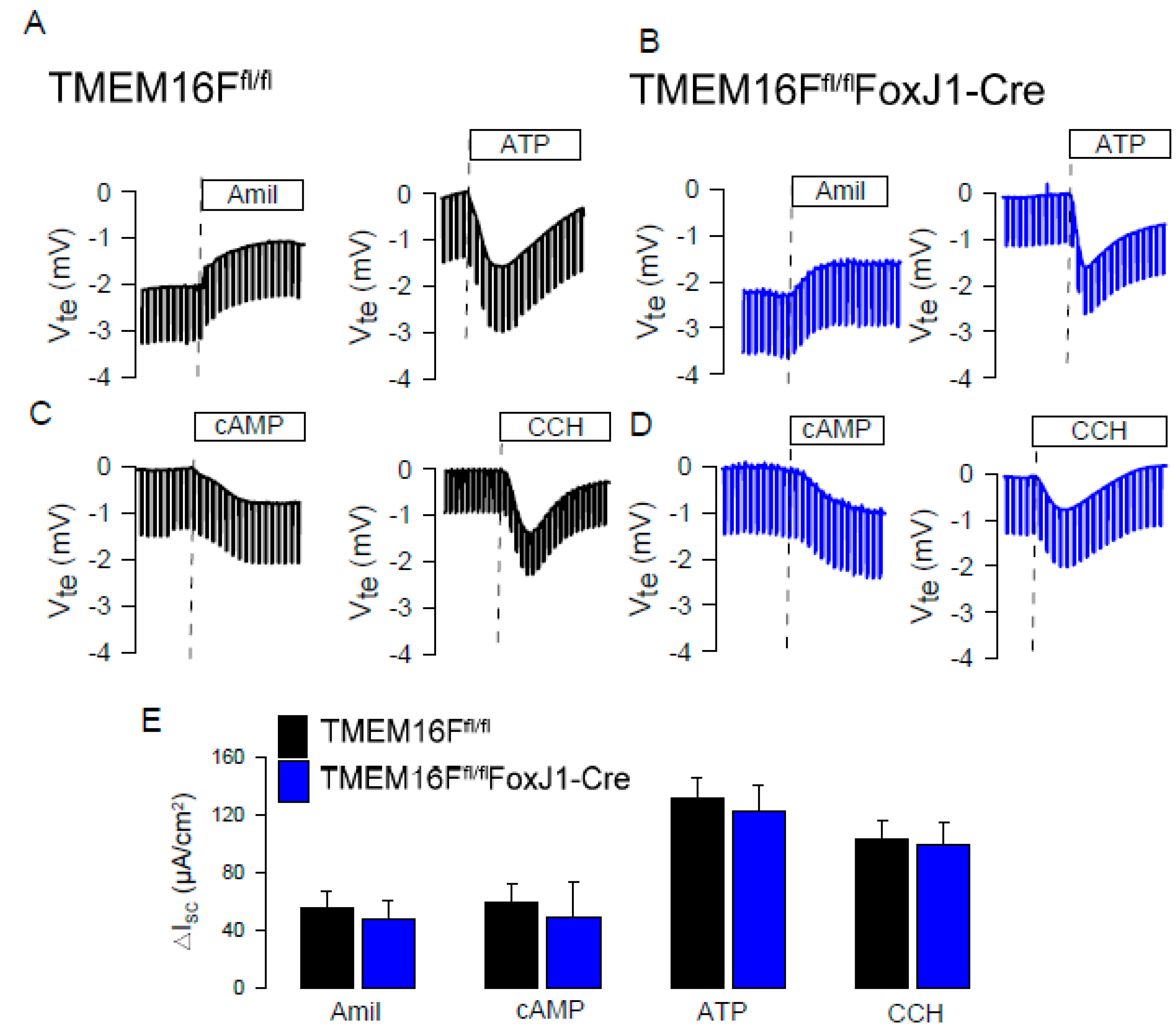
4.1. Knockout Animals
4.2. Cell Culture
4.3. RT-PCR
4.4. Western Blotting
4.5. Patch Clamping
4.6. Iodide-Sensitive YFP-Quenching Assay of Anion Conductance/TMEM16A Activity
4.7. Ca2+ Measurements
4.8. Data and Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pedemonte, N.; Galietta, L.J. Structure and Function of TMEM16 Proteins (Anoctamins). Physiol. Rev. 2014, 94, 419–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzelmann, K.; Ousingsawat, J.; Cabrita, I.; Doušová, T.; Bähr, A.; Janda, M.; Schreiber, R.; Benedetto, R. TMEM16A in Cystic Fibrosis: Activating or Inhibiting? Front. Pharmacol. 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallos, G.; Remy, K.E.; Danielsson, J.; Funayama, H.; Fu, X.W.; Chang, H.Y.; Yim, P.D.; Xu, D.; Emala, C.W., Sr. Functional Expression of the TMEM16 Family of Calcium Activated Chloride Channels in Airway Smooth Muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L625–L634. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, R.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019, 33, 4502–4512. [Google Scholar] [CrossRef]
- Zhang, C.H.; Li, Y.; Zhao, W.; Lifshitz, L.M.; Li, H.; Harfe, B.D.; Zhu, M.S.; ZhuGe, R. The transmembrane protein 16A Ca(2+)-activated Cl− channel in airway smooth muscle contributes to airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2013, 187, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Caci, E.; Scudieri, P.; Di Carlo, E.; Morelli, P.; Bruno, S.; De, F.I.; Bragonzi, A.; Gianotti, A.; Sondo, E.; Ferrera, L.; et al. Upregulation of TMEM16A Protein in Bronchial Epithelial Cells by Bacterial Pyocyanin. PLoS ONE 2015, 10, e0131775. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Wang, H.; Jiao, J.; Li, Y.; Fan, E.; Zhang, L.; Bachert, C. TMEM16A-Mediated Mucin Secretion in IL-13-Induced Nasal Epithelial Cells From Chronic Rhinosinusitis Patients. Allergy Asthma Immunol. Res. 2015, 7, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Miner, K.; Labitzke, K.; Liu, B.; Elliot, R.; Wang, P.; Henckels, K.; Gaida, K.; Elliot, R.; Chen, J.J.; Liu, L.; et al. Drug Repurposing: The Anthelmintics Niclosamide and Nitazoxanide Are Potent TMEM16A Antagonists That Fully Bronchodilate Airways. Front. Pharmacol. 2019, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Fajac, I.; De Boeck, K. New horizons for cystic fibrosis treatment. Pharmacol. Ther. 2017, 170, 205–211. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, W.; Sun, J.; Tao, T.; Chen, X.; Zheng, Y.Y.; Zhang, C.H.; Chen, Z.; Gao, Y.Q.; She, F.; et al. Inflammatory mediators mediate airway smooth muscle contraction through a G protein-coupled receptor-transmembrane protein 16A-voltage-dependent Ca(2+) channel axis and contribute to bronchial hyperresponsiveness in asthma. J. Allergy Clin. Immunol. 2018, 141, 1259–1268.e1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, B.D.; Lane, S.J.; van Beek, E.J.; Dodd, J.D.; Costello, R.W.; Tiddens, H.A. Asthma and cystic fibrosis: A tangled web. Pediatr. Pulmonol. 2014, 49, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Lee, Y.H.; Kang, M.J.; Lee, H.J.; Oh, R.; Min, H.J.; Namkung, W.; Choi, J.Y.; Lee, S.N.; Kim, C.H.; et al. Synergistic mucus secretion by histamine and IL-4 through TMEM16A in airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Jiang, Y.; Li, L.; Liu, Y.; Tang, H.; Jiang, D. TMEM16A mediates the hypersecretion of mucus induced by Interleukin-13. Exp. Cell Res. 2015, 334, 260–269. [Google Scholar] [CrossRef]
- Namkung, W.; Yao, Z.; Finkbeiner, W.E.; Verkman, A.S. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 2011, 25, 4048–4062. [Google Scholar] [CrossRef] [Green Version]
- Genovese, M.; Borrelli, A.; Venturini, A.; Guidone, D.; Caci, E.; Viscido, G.; Gambardella, G.; di Bernardo, D.; Scudieri, P.; Galietta, L.J.V. TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J. Physiol. 2019. [Google Scholar] [CrossRef]
- Danahay, H.L.; Lilley, S.; Fox, R.; Charlton, H.; Sabater, J.; Button, B.; McCarthy, C.; Collingwood, S.P.; Gosling, M. TMEM16A Potentiation: A Novel Therapeutic Approach for the Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- De Jesus-Perez, J.J.; Cruz-Rangel, S.; Espino-Saldana, A.E.; Martinez-Torres, A.; Qu, Z.; Hartzell, H.C.; Corral-Fernandez, N.E.; Perez-Cornejo, P.; Arreola, J. Phosphatidylinositol 4,5-bisphosphate, cholesterol, and fatty acids modulate the calcium-activated chloride channel TMEM16A (ANO1). Biochim. Biophys. Acta 2017, 1863, 299–312. [Google Scholar] [CrossRef]
- Le, S.C.; Jia, Z.; Chen, J.; Yang, H. Molecular basis of PIP2-dependent regulation of the Ca(2+)-activated chloride channel TMEM16A. Nat. Commun. 2019, 10, 3769. [Google Scholar] [CrossRef]
- Tian, Y.; Schreiber, R.; Wanitchakool, P.; Kongsuphol, P.; Sousa, M.; Uliyakina, I.; Palma, M.; Faria, D.; Traynor-Kaplen, A.E.; Fragata, J.I.; et al. Control of TMEM16A by INO-4995 and other inositolphosphates. Br. J. Pharmacol. 2013, 168, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.; Ousingsawat, J.; Wanitchakool, P.; Sirianant, L.; Benedetto, R.; Reiss, K.; Kunzelmann, K. Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca2+ and plasma membrane lipid. J. Physiol. (Lond.) 2018, 596, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Shah, S.; Liu, Y.; Zhang, H.; Lees, M.; Fu, Z.; Lippiat, J.D.; Beech, D.J.; Sivaprasadarao, A.; Baldwin, S.A.; et al. Activation of the Cl− Channel ANO1 by Localized Calcium Signals in Nociceptive Sensory Neurons Requires Coupling with the IP3 Receptor. Sci. Signal. 2013, 6, ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrita, I.; Benedetto, R.; Fonseca, A.; Wanitchakool, P.; Sirianant, L.; Skryabin, B.V.; Schenk, L.K.; Pavenstadt, H.; Schreiber, R.; Kunzelmann, K. Differential effects of anoctamins on intracellular calcium signals. FASEB J. 2017, 31, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Shah, S.; Du, X.; Zhang, H.; Gamper, N. Activation of Ca2+-activated Cl− channel ANO1 by localized Ca2+ signals. J. Physiol. 2016, 594, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. Niclosamide repurposed for the treatment of inflammatory airway disease. JCI insight 2019, 8, 128414. [Google Scholar] [CrossRef]
- Schreiber, R.; Faria, D.; Skryabin, B.V.; Rock, J.R.; Kunzelmann, K. Anoctamins support calcium-dependent chloride secretion by facilitating calcium signaling in adult mouse intestine. Pflügers Arch. 2015, 467, 1203–1213. [Google Scholar] [CrossRef]
- Shimizu, T.; Iehara, T.; Sato, K.; Fujii, T.; Sakai, H.; Okada, Y. TMEM16F is a component of a Ca2+-activated Cl− channel but not a volume-sensitive outwardly rectifying Cl− channel. Am. J. Physiol. Cell. Physiol. 2013, 304, C748–C759. [Google Scholar] [CrossRef] [Green Version]
- Grubb, S.; Poulsen, K.A.; Juul, C.A.; Kyed, T.; Klausen, T.K.; Larsen, E.H.; Hoffmann, E.K. TMEM16F (Anoctamin 6), an anion channel of delayed Ca2+ activation. J. Gen. Physiol. 2013, 141, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Ousingsawat, J.; Wanitchakool, P.; Kmit, A.; Romao, A.M.; Jantarajit, W.; Schreiber, S.; Kunzelmann, K. Anoctamin 6 mediates effects essential for innate immunity downstream of P2 × 7-receptors in macrophages. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, H.K.; Park, J.; Jeon, D.K.; Jo, S.; Jo, M.; Namkung, W. Ani9, A Novel Potent Small-Molecule ANO1 Inhibitor with Negligible Effect on ANO2. PLoS ONE 2016, 11, e0155771. [Google Scholar] [CrossRef] [Green Version]
- Kadri, H.; Lambourne, O.A.; Mehellou, Y. Niclosamide, a Drug with Many (Re)purposes. ChemMedChem 2018, 13, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, S.; Ren, S.; Chen, Y.; Zhan, Y.; An, H. The Natural Compound Cinnamaldehyde is a Novel Activator of Calcium-Activated Chloride Channel. J. Membr. Biol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Arreola, J.; Hartzell, H.C. Wasted TMEM16A channels are rescued by phosphatidylinositol 4,5-bisphosphate. Cell Calcium 2019, 84, 102103. [Google Scholar] [CrossRef] [PubMed]
- Tembo, M.; Wozniak, K.L.; Bainbridge, R.E.; Carlson, A.E. Phosphatidylinositol 4,5-bisphosphate (PIP2) and Ca(2+) are both required to open the Cl(−) channel TMEM16A. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Sala-Rabanal, M.; Yurtsever, Z.; Nichols, C.G.; Brett, T.J. Secreted CLCA1 modulates TMEM16A to activate Ca(2+)-dependent chloride currents in human cells. Elife 2015, 4, e05875. [Google Scholar] [CrossRef]
- Sala-Rabanal, M.; Yurtsever, Z.; Berry, K.N.; Nichols, C.G.; Brett, T.J. Modulation of TMEM16A channel activity by the von Willebrand factor type A (VWA) domain of the calcium-activated chloride channel regulator 1 (CLCA1). J. Biol. Chem. 2017, 292, 9164–9174. [Google Scholar] [CrossRef] [Green Version]
- Berry, K.N.; Brett, T.J. Structural and Biophysical Analysis of the CLCA1 VWA Domain Suggests Mode of TMEM16A Engagement. Cell Rep. 2020, 30, 1141–1151.e1143. [Google Scholar] [CrossRef] [Green Version]
- Ousingsawat, J.; Martins, J.R.; Schreiber, R.; Rock, J.R.; Harfe, B.D.; Kunzelmann, K. Loss of TMEM16A causes a defect in epithelial Ca2+ dependent chloride transport. J. Biol. Chem. 2009, 284, 28698–28703. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; O’Neal, W.K.; Gabriel, S.E.; Randell, S.H.; Harfe, B.D.; Boucher, R.C.; Grubb, B.R. Transmembrane protein 16A (TMEM16A) is a Ca2+ regulated Cl−—Secretory channel in mouse airways. J. Biol. Chem. 2009, 284, 14875–14880. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, R.; Ousingsawat, J.; Wanitchakool, P.; Zhang, Y.; Holtzman, M.J.; Amaral, M.; Rock, J.R.; Schreiber, R.; Kunzelmann, K. Epithelial Chloride Transport by CFTR Requires TMEM16A. Sci. Rep. 2017, 7, 12397. [Google Scholar] [CrossRef] [Green Version]
- Greger, R.; Kunzelmann, K. Simultaneous recording of the cell membrane potential and properties of the cell attached membrane of HT29 colon carcinoma and CF-PAC cells. Pflügers Arch. 1991, 419, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Sirianant, L.; Ousingsawat, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Cellular Volume regulation by Anoctamin 6:Ca2+, phospholipase A2,osmosensing. Pflügers Arch. 2015, 468, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Vajanaphanich, M.; Schultz, C.; Rudolf, M.T.; Wasserman, M.; Enyedi, P.; Craxton, A.; Shears, S.B.; Tsien, R.Y.; Barrett, K.E.; Traynor-Kaplan, A.E. Long-term uncoupling of chloride secretion from intracellular calcium levels by Ins(3,4,5,6)P4. Nature 1994, 371, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, H.A.; Leblanc, N.; Albert, A.P.; Greenwood, I.A. Inhibitory role of phosphatidylinositol 4,5 bisphosphate on TMEM16A encoded calcium-activated chloride channels in rat pulmonary artery. Br. J Pharmacol. 2014, 171, 4311–4321. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Feng, J.; Luo, J.; Yang, P.; Brett, T.J.; Hu, H. Eact, a small molecule activator of TMEM16A, activates TRPV1 and elicits pain- and itch-related behaviors. Br. J. Pharmacol. 2016, 173, 1208–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzelmann, K.; Ousingsawat, J.; Benedetto, R.; Cabrita, I.; Schreiber, R. Contribution of Anoctamins to Cell Survival and Cell Death. Cancers 2019, 19, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill, A.; Hall, M.L.; Borawski, J.; Hodgson, C.; Jenkins, J.; Piechon, P.; Popa, O.; Rothwell, C.; Tranter, P.; Tria, S.; et al. Small molecule-facilitated degradation of ANO1 protein: A new targeting approach for anticancer therapeutics. J. Biol. Chem. 2014, 289, 11029–11041. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Z.; Li, F.Y.; Lv, X.F.; Ma, M.M.; Li, X.Y.; Lin, C.X.; Wang, G.L.; Guan, Y.Y. Endophilin A2 regulates calcium-activated chloride channel activity via selective autophagy-mediated TMEM16A degradation. Acta Pharmacol. Sin. 2020, 41, 208–217. [Google Scholar] [CrossRef]
- Benedetto, R.; Ousingsawat, J.; Cabrita, I.; Pinto, M.; Lerias, J.; Wanitchakool, P.; Schreiber, R.; Kunzelmann, K. Plasma membrane localized TMEM16 Proteins are Indispensable for expression of CFTR. J. Mol. Med. 2019, 97, 711–722. [Google Scholar] [CrossRef]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Kunzelmann, K.; Tilmann, M.; Hansen, C.P.; Greger, R. Inhibition of epithelial chloride channels by cytosol. Pflügers Arch. 1991, 418, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Schenk, L.K.; Ousingsawat, J.; Skryabin, B.V.; Schreiber, R.; Pavenstadt, H.; Kunzelmann, K. Regulation and Function of TMEM16F in Renal Podocytes. Int. J. Mol. Sci. 2018, 19, 1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Huang, G.; Shornick, L.P.; Roswit, W.T.; Shipley, J.M.; Brody, S.L.; Holtzman, M.J. A transgenic FOXJ1-Cre system for gene inactivation in ciliated epithelial cells. Am. J. Respir. Cell Mol. Biol. 2007, 36, 515–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Endogenous | Expressed | ||
|---|---|---|---|---|
| Activation | Potentiation | Activation | Potentiation | |
| Eact | No (50 µM; n = 5) | No (50 µM; n = 5) | Yes (10 µM; n = 5) | Yes (10 µM; n = 5) |
| GSK1016790A | No (50 µM; n = 5) | No (50 µM; n = 5) | ||
| Melittin | Yes (2 µM; n = 5) | Yes (2 µM; n = 5) | Yes (0.5 µM; n = 5) | Yes (0.5 µM; n = 5) |
| Cinnamaldehyde | No (1 µM; n = 5) | No (1 µM; n = 5) | Yes (1 µM; n = 5) | Yes (0.5 µM; n = 5) |
| diC8-PIP2-PL | No (50 µM; n = 5) | No (50 µM; n = 5) | Yes (50 µM; n = 5) | Yes (50 µM; n = 5) |
| CLCA1 | No | Yes | ||
| Target | Primers | PCR Product |
|---|---|---|
| TMEM16A | forward: 5’-CGACTACGTGTACATTTTCCG | 445 bp |
| reverse: 5´-GATTCCGATGTCTTTGGCTC | ||
| TMEM16F | forward: 5´-GGAGTTTTGGAAGCGACGC | 325 bp |
| reverse: 5´-GTATTTCTGGATTGGGTCTG | ||
| TRPV4 | forward: 5´-GCCCCACATTGTCAACTACC | 490 bp |
| reverse: 5´-CCGCAGCAGTTCATTGATGG | ||
| GAPDH | forward: 5´-GTATTGGGCGCCTGGTCAC | 200 bp |
| reverse: 5´-CTCCTGGAAGATGGTGATGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Centeio, R.; Cabrita, I.; Benedetto, R.; Talbi, K.; Ousingsawat, J.; Schreiber, R.; Sullivan, J.K.; Kunzelmann, K. Pharmacological Inhibition and Activation of the Ca2+ Activated Cl− Channel TMEM16A. Int. J. Mol. Sci. 2020, 21, 2557. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072557
Centeio R, Cabrita I, Benedetto R, Talbi K, Ousingsawat J, Schreiber R, Sullivan JK, Kunzelmann K. Pharmacological Inhibition and Activation of the Ca2+ Activated Cl− Channel TMEM16A. International Journal of Molecular Sciences. 2020; 21(7):2557. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072557
Chicago/Turabian StyleCenteio, Raquel, Inês Cabrita, Roberta Benedetto, Khaoula Talbi, Jiraporn Ousingsawat, Rainer Schreiber, John K. Sullivan, and Karl Kunzelmann. 2020. "Pharmacological Inhibition and Activation of the Ca2+ Activated Cl− Channel TMEM16A" International Journal of Molecular Sciences 21, no. 7: 2557. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072557