Combination of Arsenic Trioxide and Valproic Acid Efficiently Inhibits Growth of Lung Cancer Cells via G2/M-Phase Arrest and Apoptotic Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
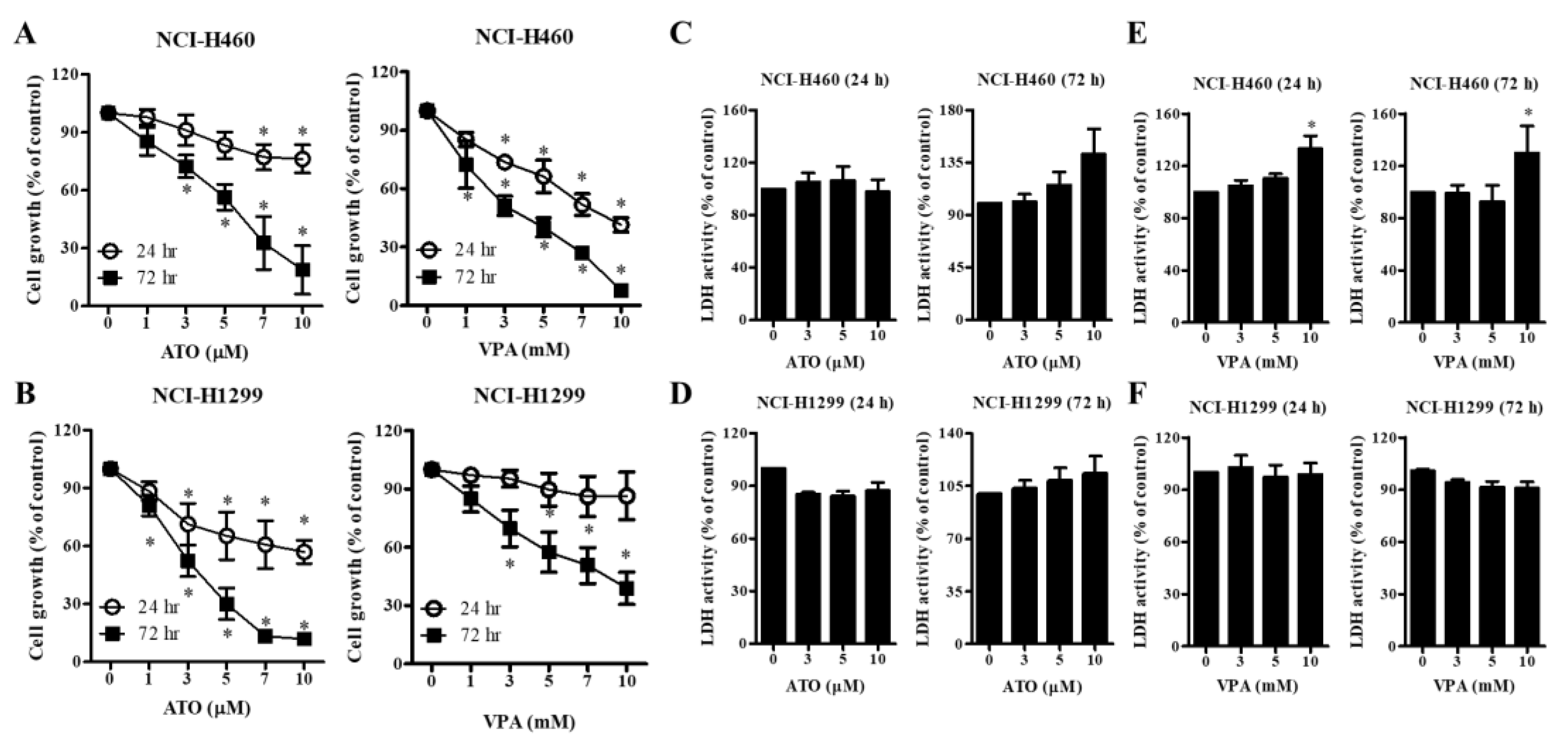
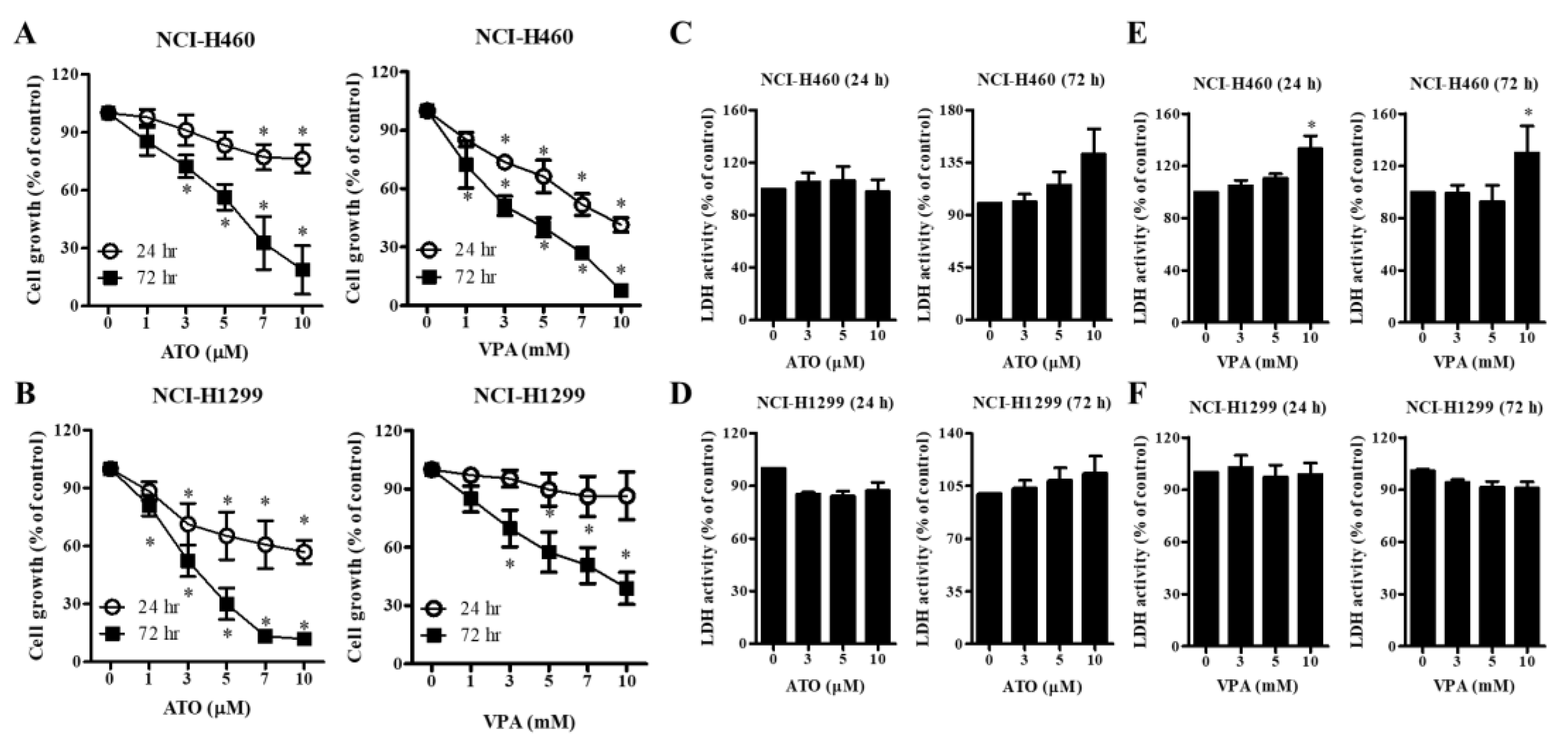
2.1. Effects of ATO or VPA on Cell Growth and Necrotic Cell Death in Lung Cancer Cells
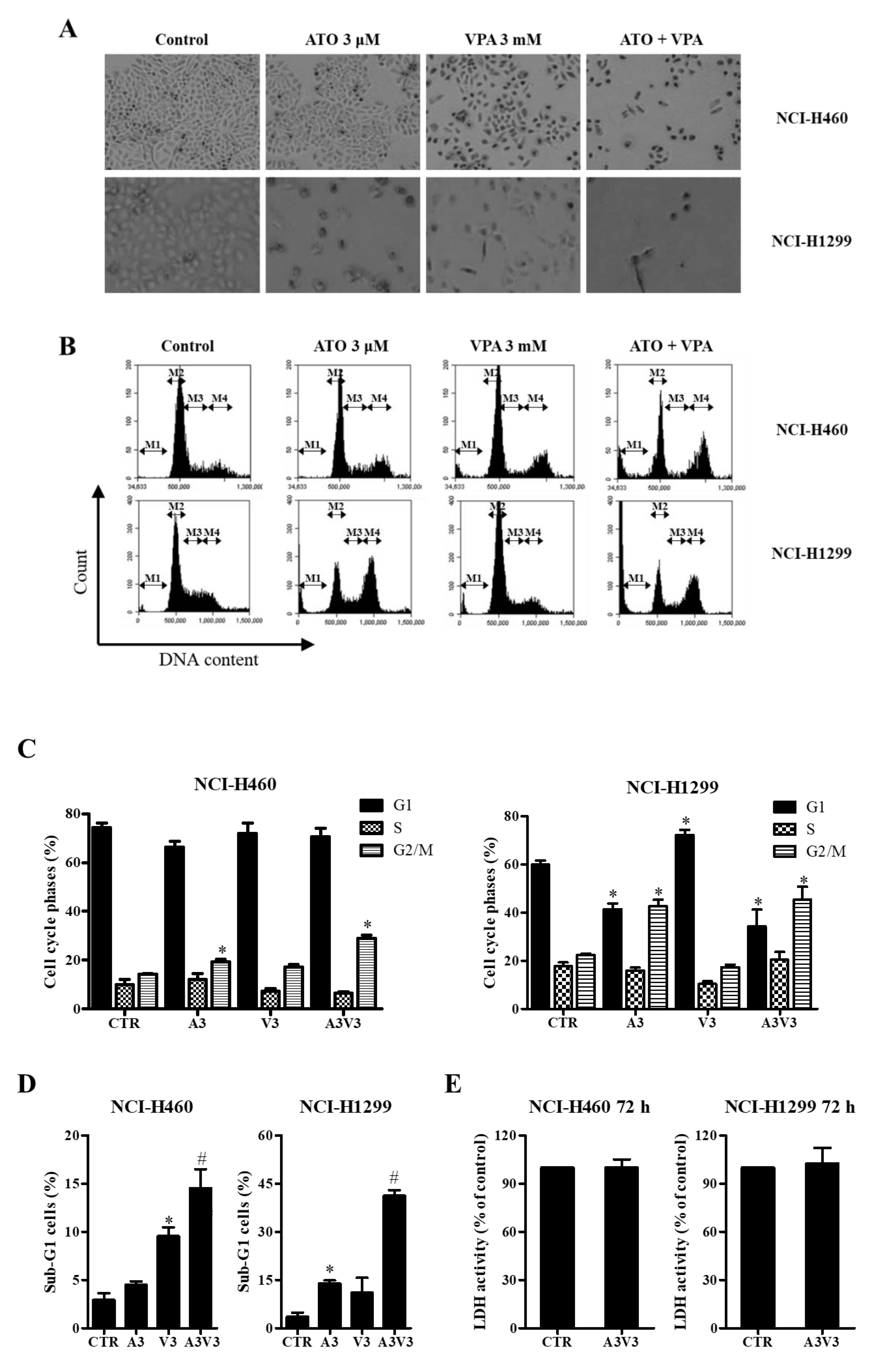
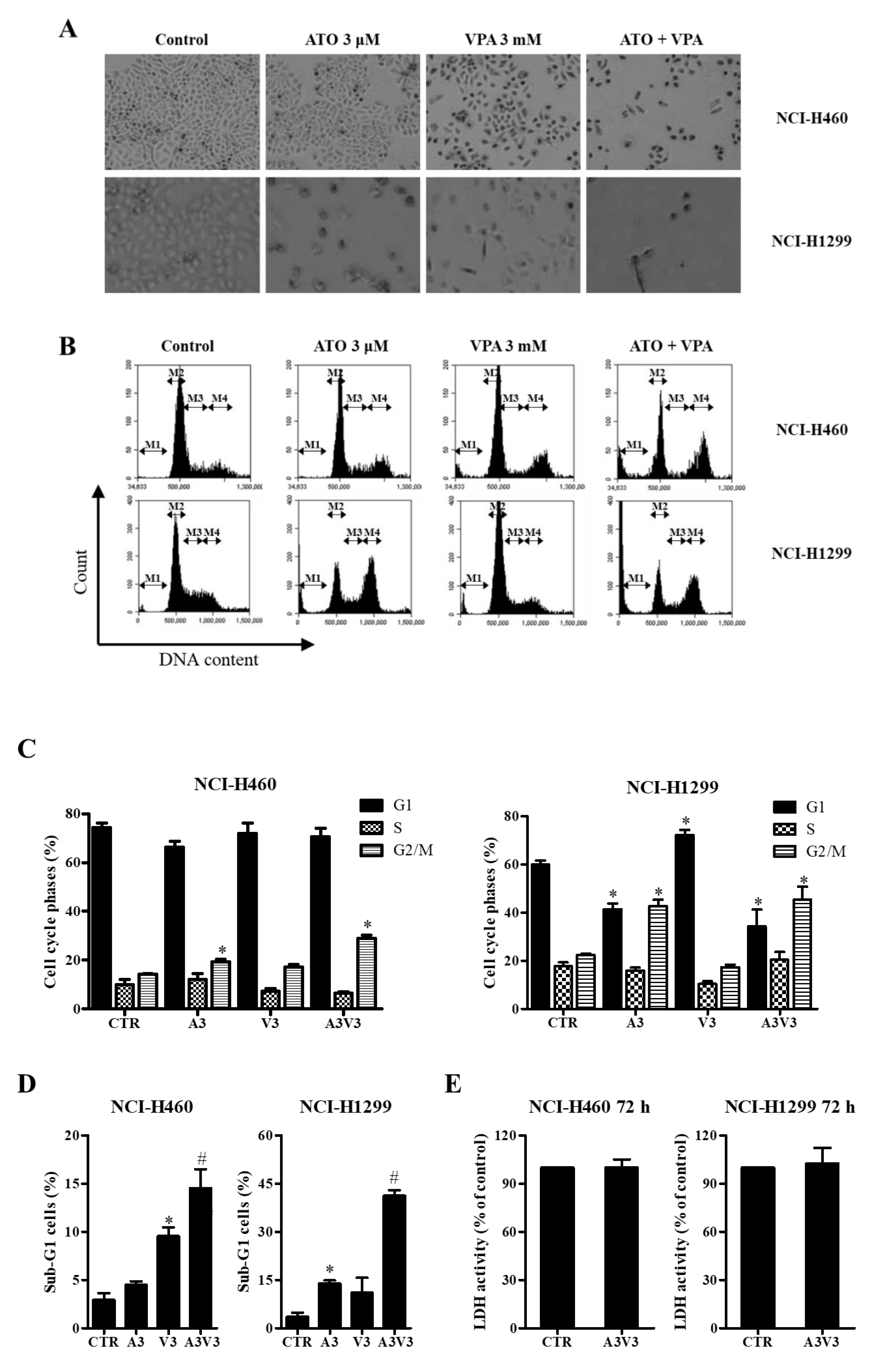
2.2. Effects of ATO and VPA Alone and in Combination on Cell Morphology and Cell Cycle Distributions
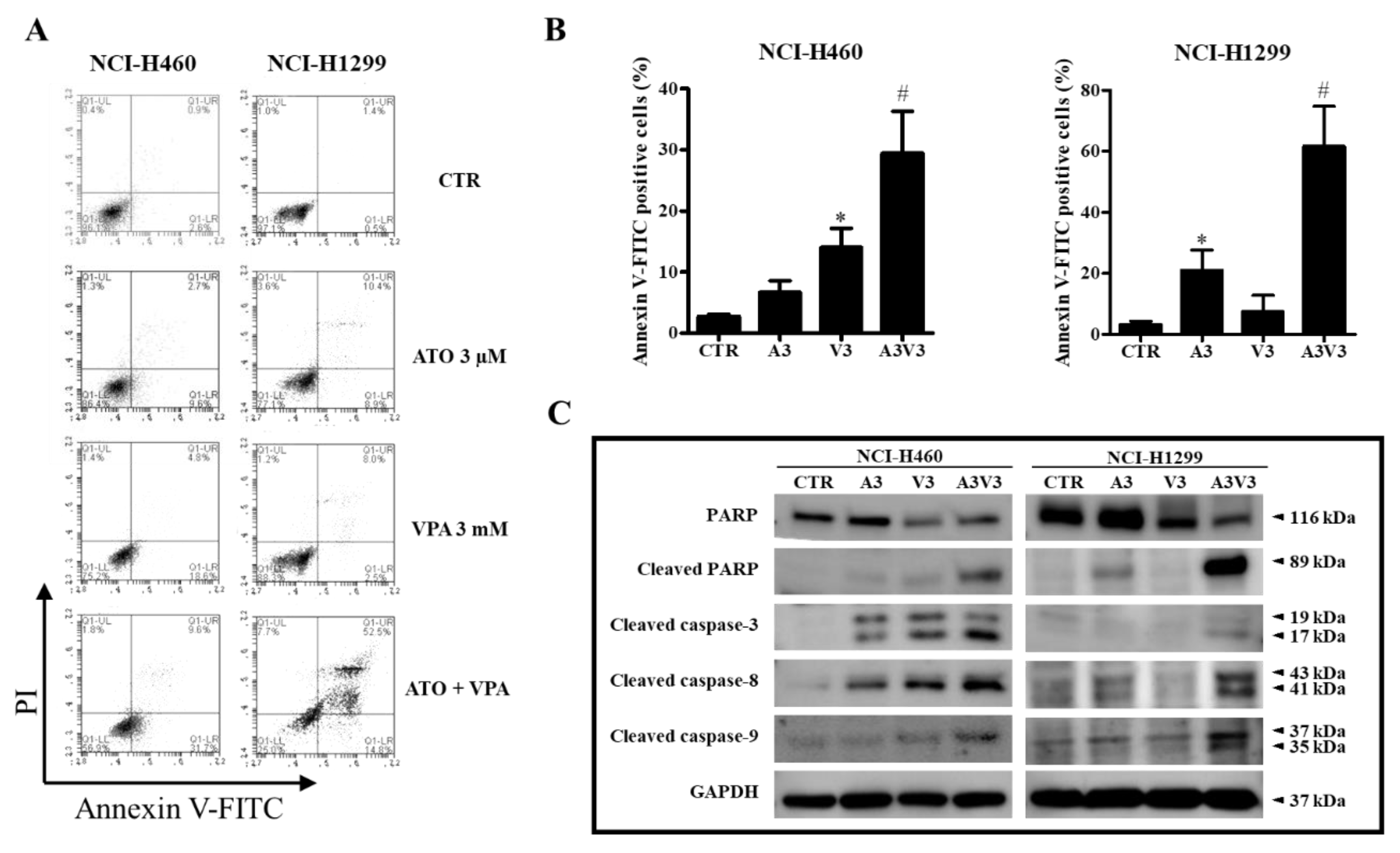
2.3. Effects of ATO and VPA Alone and in Combination on Cell Death, LDH Release, and Apoptosis
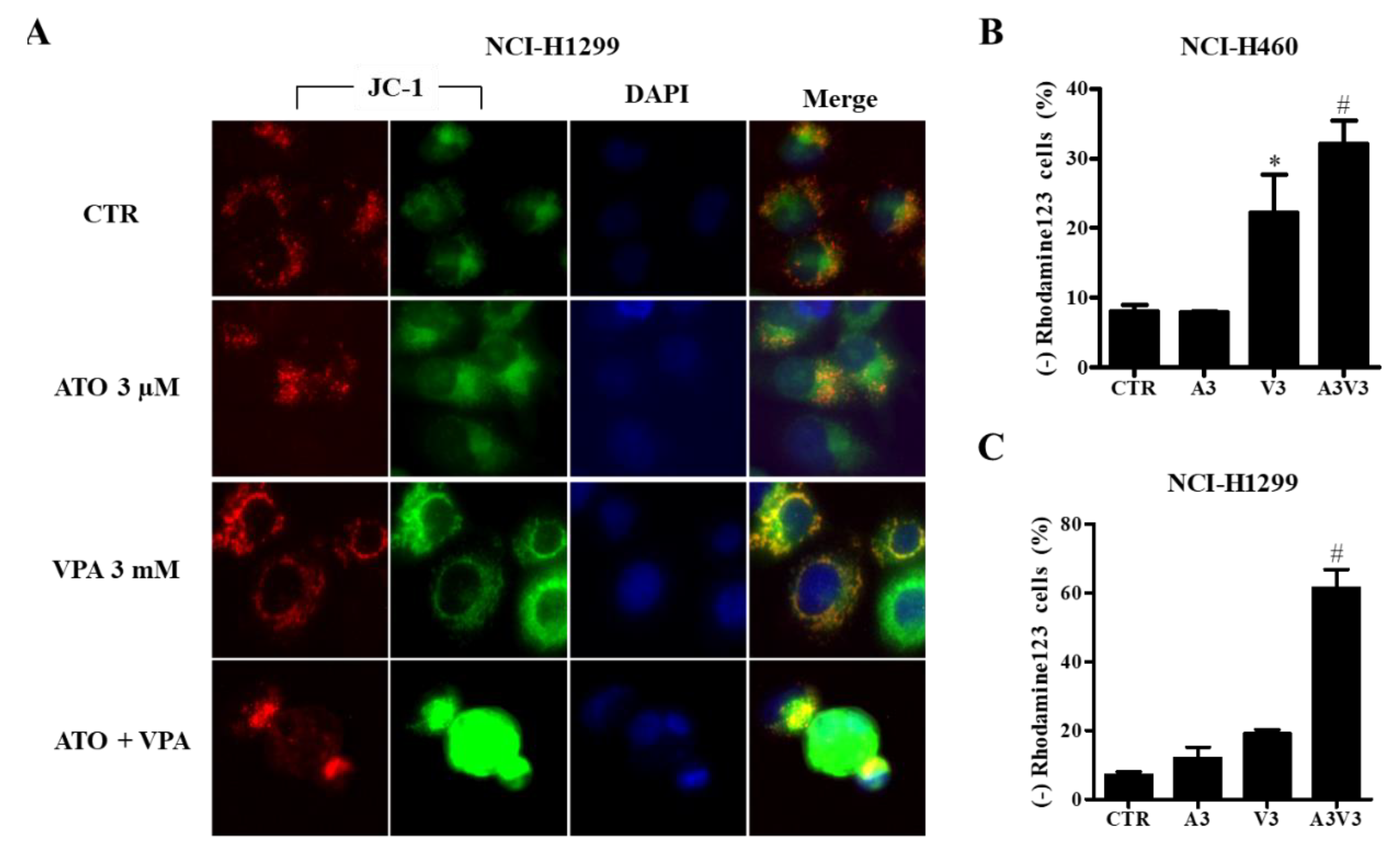
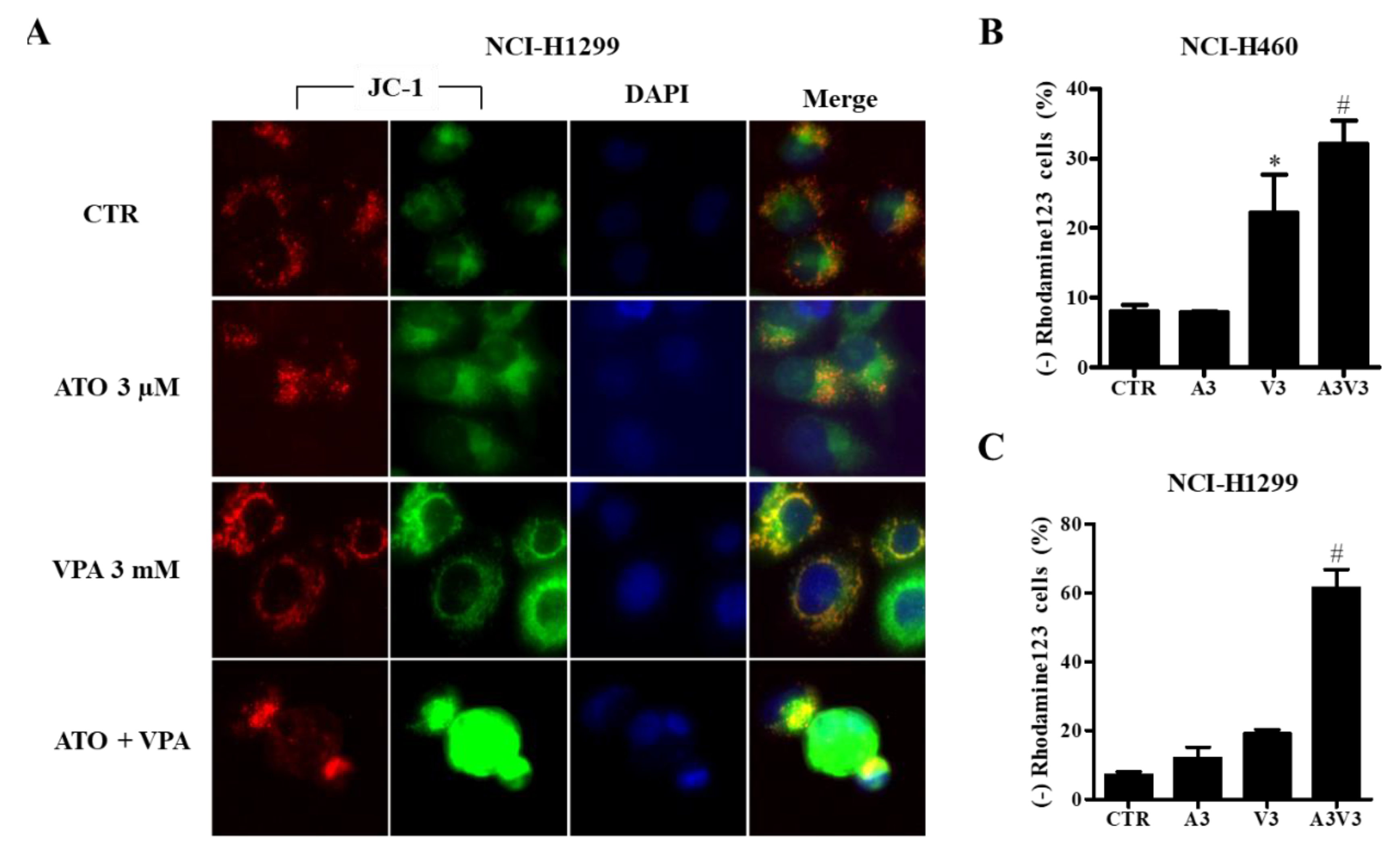
2.4. Effects of ATO and VPA Alone and in Combination on MMP (∆Ψm)
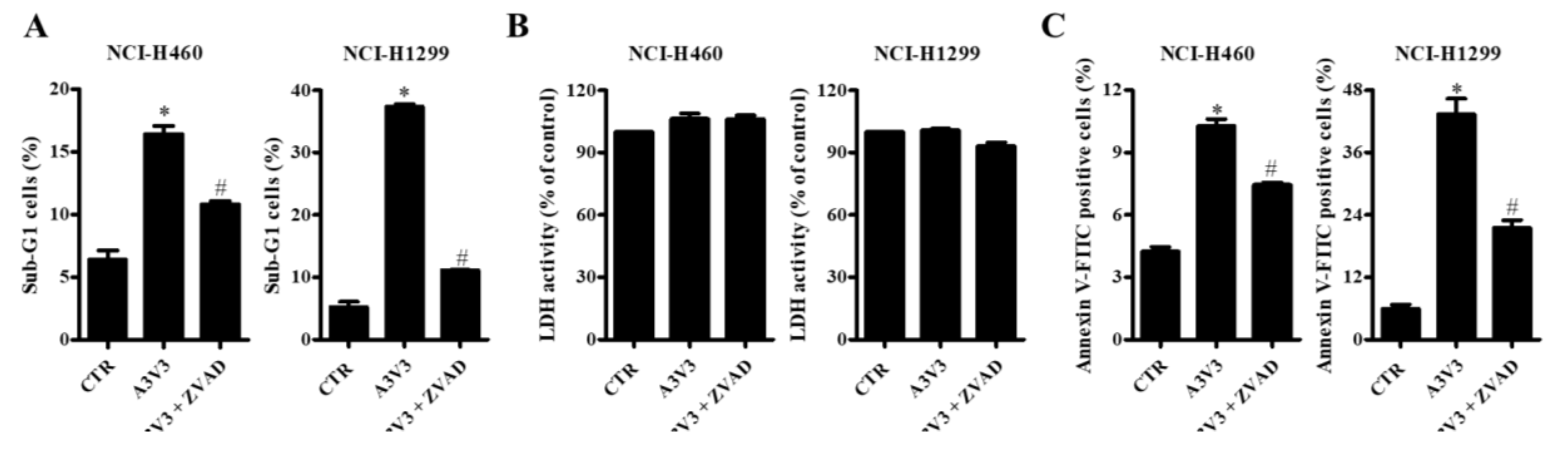
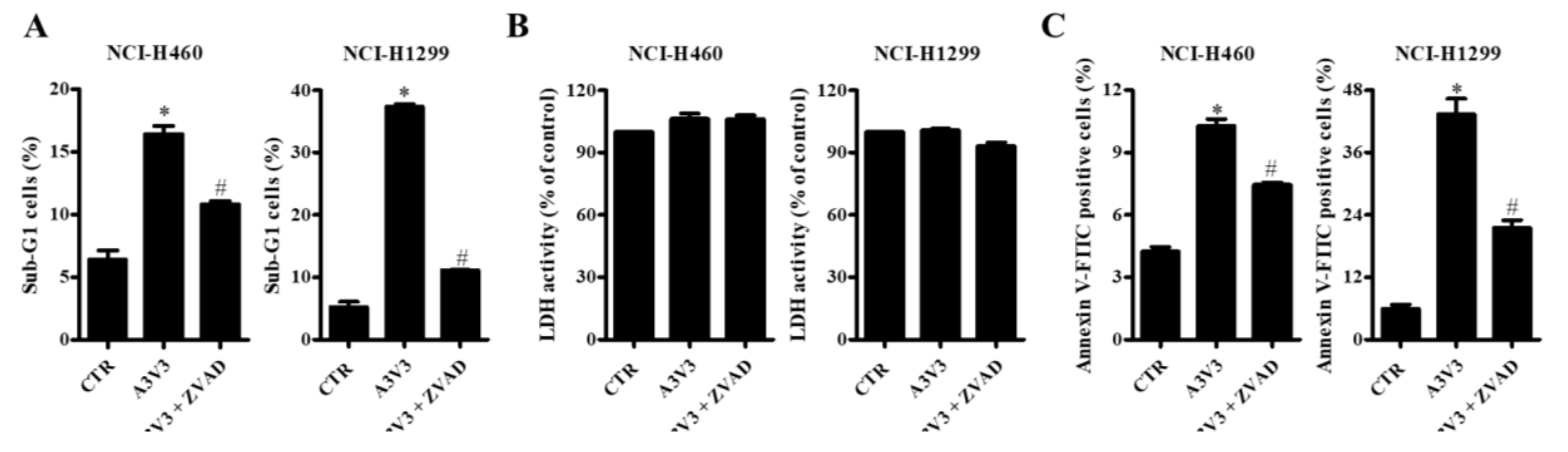
2.5. Effect of Z-VAD on Cell Death in Lung Cancer Cells
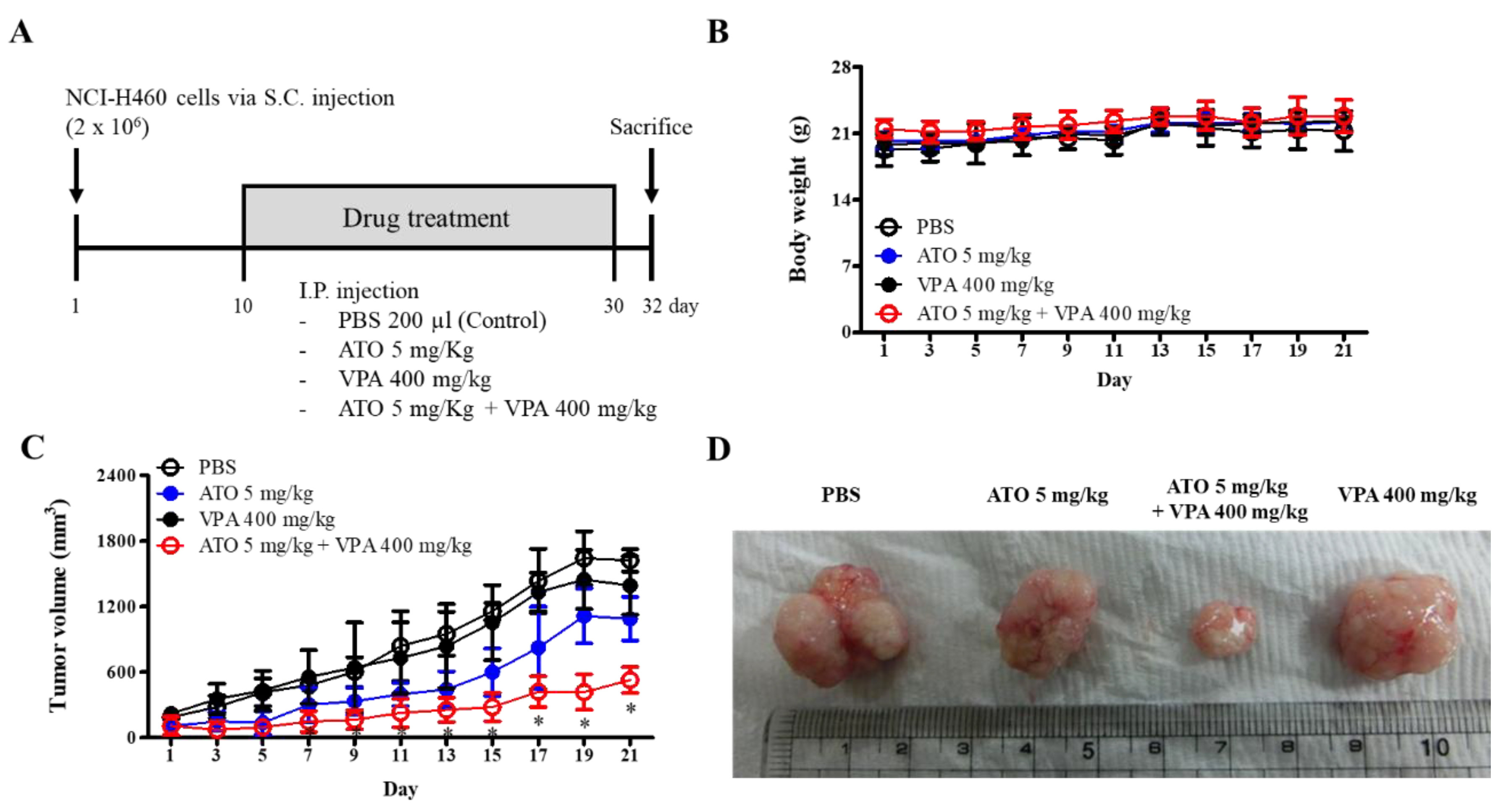
2.6. Anti-Tumor Effect of ATO and VPA Alone and in Combination on NCI-H460 Xenograft Nude Mice
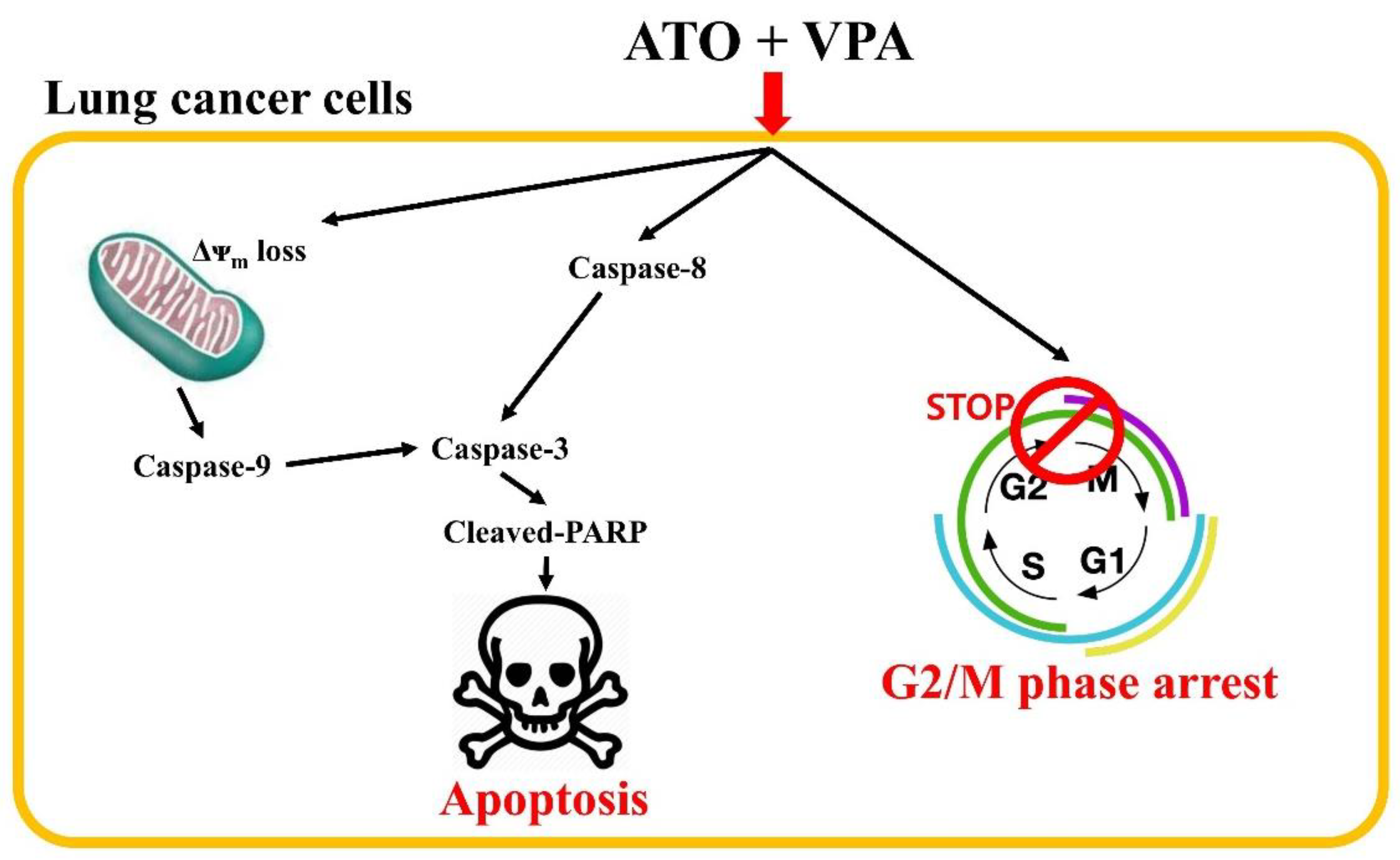
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Cell Growth Inhibition Assay
4.4. Lactate Dehydrogenase (LDH) Cytotoxicity Assay
4.5. Observation of Cell Morphologic Changes
4.6. Cell Cycle Distribution Analysis
4.7. Annexin V/PI Staining for Apoptosis Detection
4.8. Western Blot Analysis
4.9. Measurement of MMP (∆Ψm) Levels
4.10. Lung Cancer Xenograft Model
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATO | arsenic trioxide |
| VPA | valproic acid |
| ATO/VPA | arsenic trioxide and valproic acid; NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer |
| APL | acute promyelocytic leukemia |
| PARP-1 | anti-poly ADP ribose polymerase-1; FITC, fluorescein isothiocyanate |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| Z-VAD | benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone |
| MMP | mitochondrial membrane potential (∆Ψm) |
References
- Boloker, G.; Wang, C.; Zhang, J. Updated statistics of lung and bronchus cancer in United States (2018). J. Thorac. Dis. 2018, 10, 1158–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Cruz, C.S.D.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [Green Version]
- Grigoroiu, M.; Tagett, R.; Draghici, S.; Dima, S.; Nastase, A.; Florea, R.; Sorop, A.; Ilie, V.; Bacalbasa, N.; Tica, V.; et al. Gene-expression Profiling in Non-small Cell Lung Cancer with Invasion of Mediastinal Lymph Nodes for Prognosis Evaluation. Cancer Genom. Proteom. 2015, 12, 231–242. [Google Scholar]
- Shen, Z.X.; Chen, G.Q.; Ni, J.H.; Li, X.S.; Xiong, S.M.; Qiu, Q.Y.; Zhu, J.; Tang, W.; Sun, G.L.; Yang, K.Q.; et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APE). 2. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997, 89, 3354–3360. [Google Scholar] [CrossRef] [PubMed]
- Soignet, S.L.; Maslak, P.; Wang, Z.G.; Jhanwar, S.; Calleja, E.; Dardashti, L.J.; Corso, D.; DeBlasio, A.; Gabrilove, J.; Scheinberg, D.A.; et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 1998, 339, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.H.; Schipper, H.M.; Lee, J.S.; Singer, J.; Waxman, S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002, 62, 3893–3903. [Google Scholar]
- Hyun Park, W.; Hee Cho, Y.; Won Jung, C.; Oh Park, J.; Kim, K.; Hyuck Im, Y.; Lee, M.H.; Ki Kang, W.; Park, K. Arsenic trioxide inhibits the growth of A498 renal cell carcinoma cells via cell cycle arrest or apoptosis. Biochem. Biophys. Res. Commun. 2003, 300, 230–235. [Google Scholar] [CrossRef]
- Seol, J.G.; Park, W.H.; Kim, E.S.; Jung, C.W.; Hyun, J.M.; Kim, B.K.; Lee, Y.Y. Effect of arsenic trioxide on cell cycle arrest in head and neck cancer cell line PCI-1. Biochem. Biophys. Res. Commun. 1999, 265, 400–404. [Google Scholar] [CrossRef]
- Uslu, R.; Sanli, U.A.; Sezgin, C.; Karabulut, B.; Terzioglu, E.; Omay, S.B.; Goker, E. Arsenic trioxide-mediated cytotoxicity and apoptosis in prostate and ovarian carcinoma cell lines. Clin. Cancer Res. 2000, 6, 4957–4964. [Google Scholar]
- Oketani, M.; Kohara, K.; Tuvdendorj, D.; Ishitsuka, K.; Komorizono, Y.; Ishibashi, K.; Arima, T. Inhibition by arsenic trioxide of human hepatoma cell growth. Cancer Lett. 2002, 183, 147–153. [Google Scholar] [CrossRef]
- Woo, S.H.; Park, I.C.; Park, M.J.; Lee, H.C.; Lee, S.W.J.; Chun, Y.J.; Lee, S.H.; Hong, S.I.; Rhee, C.H. Arsenic trioxide induces apoptosis through a reactive oxygen species-dependent pathway and loss of mitochondrial membrane potential in HeLa cells. Int. J. Oncol. 2002, 21, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.C.; Cao, E.H.; Li, J.F.; Ma, W.; Qin, J.F. Induction of apoptosis and inhibition of human gastric cancer MGC-803 cell growth by arsenic trioxide. Eur. J. Cancer 1999, 35, 1258–1263. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a G2 arrest of the cell cycle and apoptosis accompanied with the depletion of GSH. Cancer Lett. 2008, 270, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Li, J.X.; Shen, Y.Q.; Cai, B.Z.; Zhao, J.; Bai, X.; Lu, Y.J.; Li, X.Q. Arsenic trioxide induces the apoptosis in vascular smooth muscle cells via increasing intracellular calcium and ROS formation. Mol. Biol. Rep. 2010, 37, 1569–1576. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. Arsenic trioxide induces human pulmonary fibroblast cell death via increasing ROS levels and GSH depletion. Oncol. Rep. 2012, 28, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Cha, Y.; Park, D.W.; Lee, C.H.; Baek, S.H.; Kim, S.Y.; Kim, J.R.; Kim, J.H. Arsenic trioxide induces apoptosis in human colorectal adenocarcinoma HT-29 cells through ROS. Cancer Res. Treat. 2006, 38, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Ahronian, L.G.; Corcoran, R.B. Strategies for monitoring and combating resistance to combination kinase inhibitors for cancer therapy. Genome Med. 2017, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- He, H.; An, R.; Hou, J.; Fu, W. Arsenic trioxide induced rhabdomyolysis, a rare but severe side effect, in an APL patient: A case report. Front. Med. 2017, 11, 284–286. [Google Scholar] [CrossRef]
- Vineetha, V.P.; Raghu, K.G. An Overview on Arsenic Trioxide-Induced Cardiotoxicity. Cardiovasc. Toxicol. 2019, 19, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Induction of apoptosis in arsenic trioxide-treated lung cancer A549 cells by buthionine sulfoximine. Mol. Cells 2008, 26, 158–164. [Google Scholar] [PubMed]
- Zhang, L.; Zou, Y.; Chen, Y.; Guo, Y.; Yang, W.; Chen, X.; Wang, S.; Liu, X.; Ruan, M.; Zhang, J.; et al. Role of cytarabine in paediatric acute promyelocytic leukemia treated with the combination of all-trans retinoic acid and arsenic trioxide: A randomized controlled trial. BMC Cancer 2018, 18, 374. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.; Xu, L.; Xu, M.; Zhang, L.; Zhang, Y.; Zhang, K.; Zhang, L.; Liang, C. Combination of Arsenic trioxide and Everolimus (Rad001) synergistically induces both autophagy and apoptosis in prostate cancer cells. Oncotarget 2017, 8, 11206–11218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petta, V.; Gkiozos, I.; Strimpakos, A.; Syrigos, K. Histones and lung cancer: Are the histone deacetylases a promising therapeutic target? Cancer Chemother. Pharm. 2013, 72, 935–952. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Mohankumar, D.; Saha, B.; Colin, C.M.; Lee, J.Y.; Martin, M.W.; Zheng, X.; Coppola, D.; Chellappan, S. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 2020, 10, 4722. [Google Scholar] [CrossRef]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef]
- You, B.R.; Park, W.H. Suberoylanilide hydroxamic acid induces thioredoxin1-mediated apoptosis in lung cancer cells via up-regulation of miR-129-5p. Mol. Carcinog. 2017, 56, 2566–2577. [Google Scholar] [CrossRef]
- Cinatl, J., Jr.; Cinatl, J.; Scholz, M.; Driever, P.H.; Henrich, D.; Kabickova, H.; Vogel, J.U.; Doerr, H.W.; Kornhuber, B. Antitumor activity of sodium valproate in cultures of human neuroblastoma cells. Anticancer Drugs 1996, 7, 766–773. [Google Scholar] [CrossRef]
- DeVane, C.L. Pharmacokinetics, drug interactions, and tolerability of valproate. Psychopharmacol. Bull. 2003, 37 (Suppl. 2), 25–42. [Google Scholar]
- Han, B.R.; You, B.R.; Park, W.H. Valproic acid inhibits the growth of HeLa cervical cancer cells via caspase-dependent apoptosis. Oncol. Rep. 2013, 30, 2999–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, A.; Keane, P.E.; Meldrum, B.S.; Simiand, J.; Vernieres, J.C. Mechanism of anticonvulsant action of valproate. Prog. Neurobiol. 1982, 19, 315–359. [Google Scholar] [CrossRef]
- Shan, Z.; Feng-Nian, R.; Jie, G.; Ting, Z. Effects of valproic acid on proliferation, apoptosis, angiogenesis and metastasis of ovarian cancer in vitro and in vivo. Asian Pac. J. Cancer Prev. 2012, 13, 3977–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.C.; Bellodi-Privato, M.; Kubrusly, M.S.; Molan, N.A.; Tharcisio, T., Jr.; de Oliveira, E.R.; D’Albuquerque, L.A. Valproic acid inhibits human hepatocellular cancer cells growth in vitro and in vivo. J. Exp. Ther. Oncol. 2011, 9, 85–92. [Google Scholar]
- Hubaux, R.; Vandermeers, F.; Crisanti, M.C.; Kapoor, V.; Burny, A.; Mascaux, C.; Albelda, S.M.; Willems, L. Preclinical evidence for a beneficial impact of valproate on the response of small cell lung cancer to first-line chemotherapy. Eur. J. Cancer 2010, 46, 1724–1734. [Google Scholar] [CrossRef]
- Eyvani, H.; Moghaddaskho, F.; Kabuli, M.; Zekri, A.; Momeny, M.; Tavakkoly-Bazzaz, J.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Arsenic trioxide induces cell cycle arrest and alters DNA methylation patterns of cell cycle regulatory genes in colorectal cancer cells. Life Sci. 2016, 167, 67–77. [Google Scholar] [CrossRef]
- Moghaddaskho, F.; Eyvani, H.; Ghadami, M.; Tavakkoly-Bazzaz, J.; Alimoghaddam, K.; Ghavamzadeh, A.; Ghaffari, S.H. Demethylation and alterations in the expression level of the cell cycle-related genes as possible mechanisms in arsenic trioxide-induced cell cycle arrest in human breast cancer cells. Tumour Biol. 2017, 39, 1010428317692255. [Google Scholar] [CrossRef] [Green Version]
- Das, C.M.; Aguilera, D.; Vasquez, H.; Prasad, P.; Zhang, M.; Wolff, J.E.; Gopalakrishnan, V. Valproic acid induces p21 and topoisomerase-II (alpha/beta) expression and synergistically enhances etoposide cytotoxicity in human glioblastoma cell lines. J. Neurooncol. 2007, 85, 159–170. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, W.; Shi, C.; Ma, W.; Liu, J.; Wang, Y.; Jiang, G. The G1 phase arrest and apoptosis by intrinsic pathway induced by valproic acid inhibit proliferation of BGC-823 gastric carcinoma cells. Tumour Biol. 2011, 32, 335–346. [Google Scholar] [CrossRef]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [Green Version]
- Mehmet, H. Caspases find a new place to hide. Nature 2000, 403, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Kito, M.; Matsumoto, K.; Wada, N.; Sera, K.; Futatsugawa, S.; Naoe, T.; Nozawa, Y.; Akao, Y. Antitumor effect of arsenic trioxide in murine xenograft model. Cancer Sci. 2003, 94, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Y.; Yang, S.; Chen, Y.R.; Li, P.C.; Dou, M.M.; Zhang, J. Resveratrol and arsenic trioxide act synergistically to kill tumor cells in vitro and in vivo. PLoS ONE 2014, 9, e98925. [Google Scholar] [CrossRef] [PubMed]
- Sidana, A.; Wang, M.; Shabbeer, S.; Chowdhury, W.H.; Netto, G.; Lupold, S.E.; Carducci, M.; Rodriguez, R. Mechanism of growth inhibition of prostate cancer xenografts by valproic acid. J. Biomed. Biotechnol. 2012, 2012, 180363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirsath, N.; Rathos, M.; Chaudhari, U.; Sivaramakrishnan, H.; Joshi, K. Potentiation of anticancer effect of valproic acid, an antiepileptic agent with histone deacetylase inhibitory activity, by the cyclin-dependent kinase inhibitor P276-00 in human non-small-cell lung cancer cell lines. Lung Cancer 2013, 82, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ren, Q.; Zuo, W.; Jia, R.; Xie, L.; Lin, R.; Zhao, H.; Chen, J.; Lei, Y.; Wang, P.; et al. Valproic acid exhibits anti-tumor activity selectively against EGFR/ErbB2/ErbB3-coexpressing pancreatic cancer via induction of ErbB family members-targeting microRNAs. J. Exp. Clin. Cancer Res. 2019, 38, 150. [Google Scholar] [CrossRef]
- Park, W.H. Hydrogen peroxide inhibits the growth of lung cancer cells via the induction of cell death and G1phase arrest. Oncol. Rep. 2018, 40, 1787–1794. [Google Scholar]
- Park, W.H. Treatment with a JNK inhibitor increases, whereas treatment with a p38 inhibitor decreases, H2O2-induced calf pulmonary arterial endothelial cell death. Oncol. Lett. 2017, 14, 1737–1744. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.K.; Han, B.R.; Park, W.H. Combination of Arsenic Trioxide and Valproic Acid Efficiently Inhibits Growth of Lung Cancer Cells via G2/M-Phase Arrest and Apoptotic Cell Death. Int. J. Mol. Sci. 2020, 21, 2649. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072649
Park HK, Han BR, Park WH. Combination of Arsenic Trioxide and Valproic Acid Efficiently Inhibits Growth of Lung Cancer Cells via G2/M-Phase Arrest and Apoptotic Cell Death. International Journal of Molecular Sciences. 2020; 21(7):2649. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072649
Chicago/Turabian StylePark, Hyun Kyung, Bo Ram Han, and Woo Hyun Park. 2020. "Combination of Arsenic Trioxide and Valproic Acid Efficiently Inhibits Growth of Lung Cancer Cells via G2/M-Phase Arrest and Apoptotic Cell Death" International Journal of Molecular Sciences 21, no. 7: 2649. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072649