Hypoxia, Inflammation and Necrosis as Determinants of Glioblastoma Cancer Stem Cells Progression

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
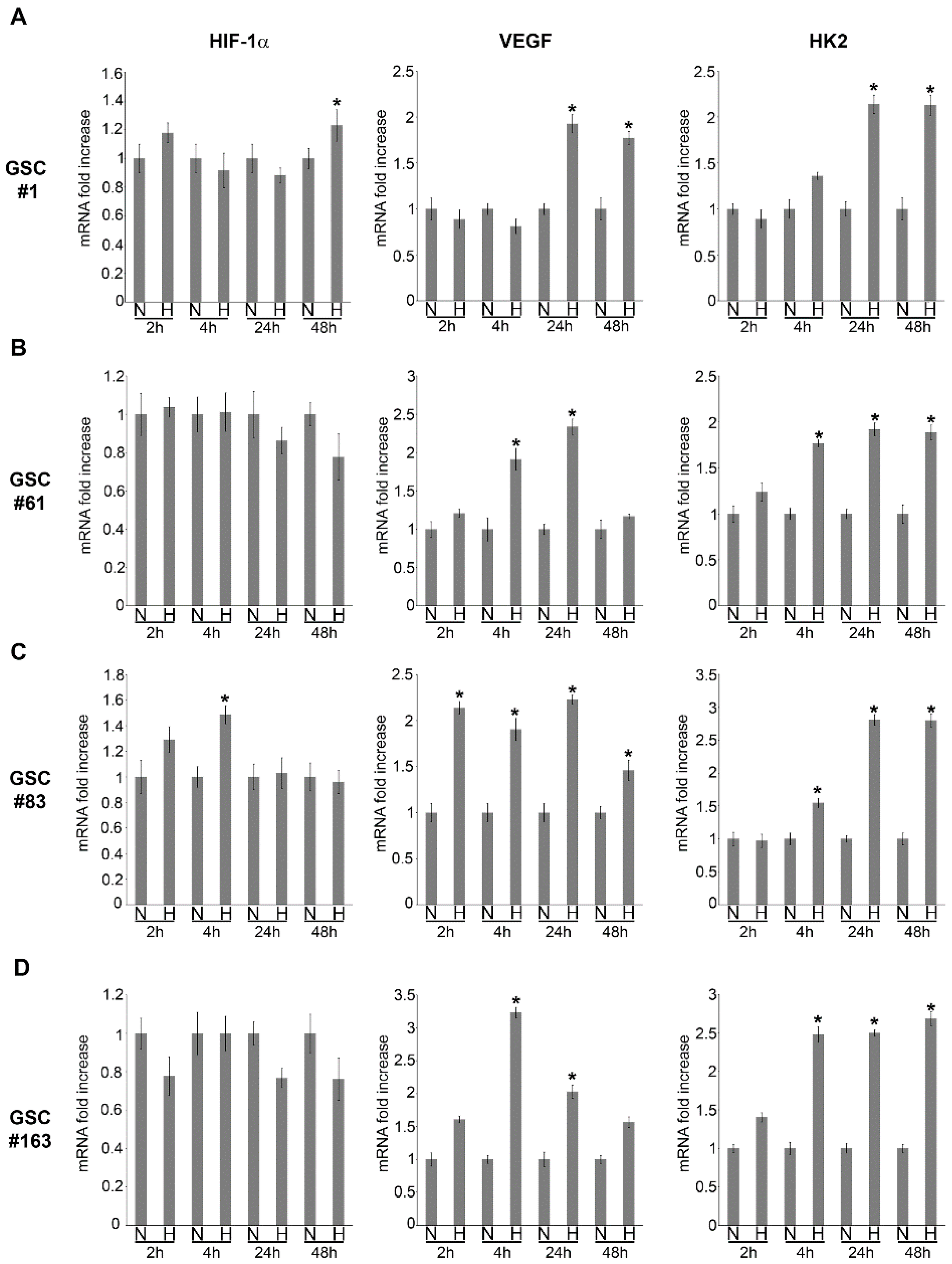
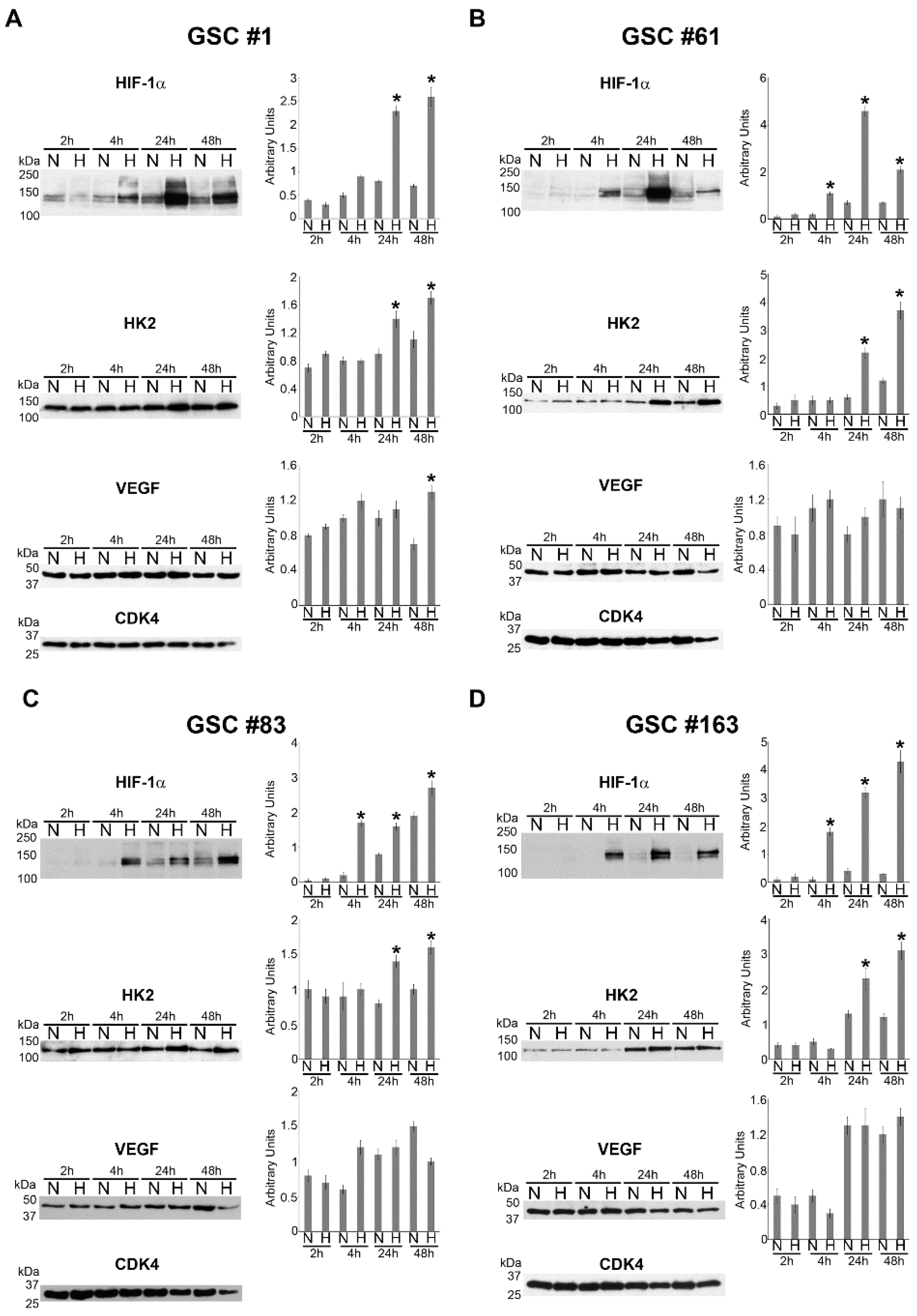
2.1. HIF-1α and HIF-1α-Dependent Gene Expression in GSCs
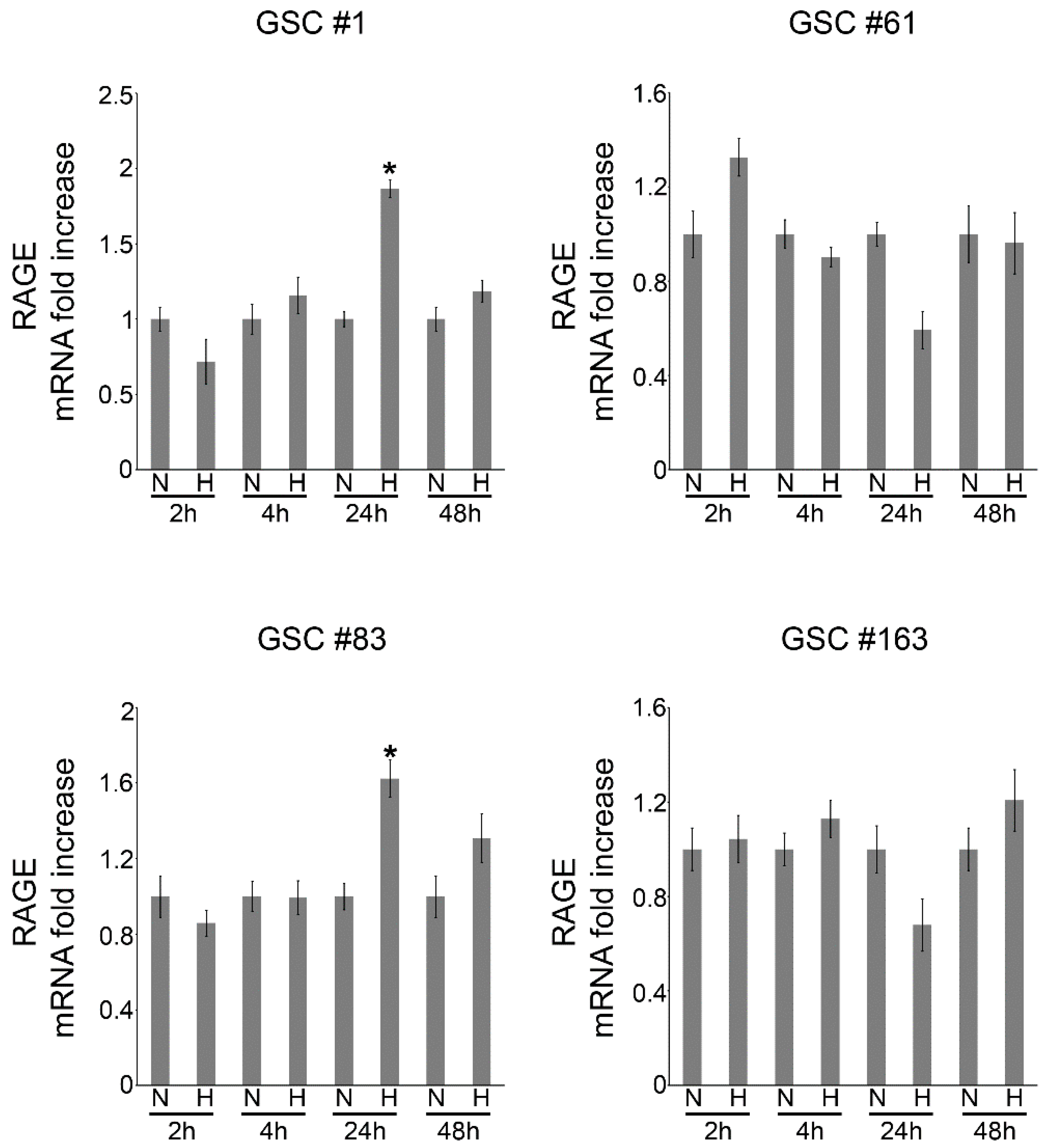
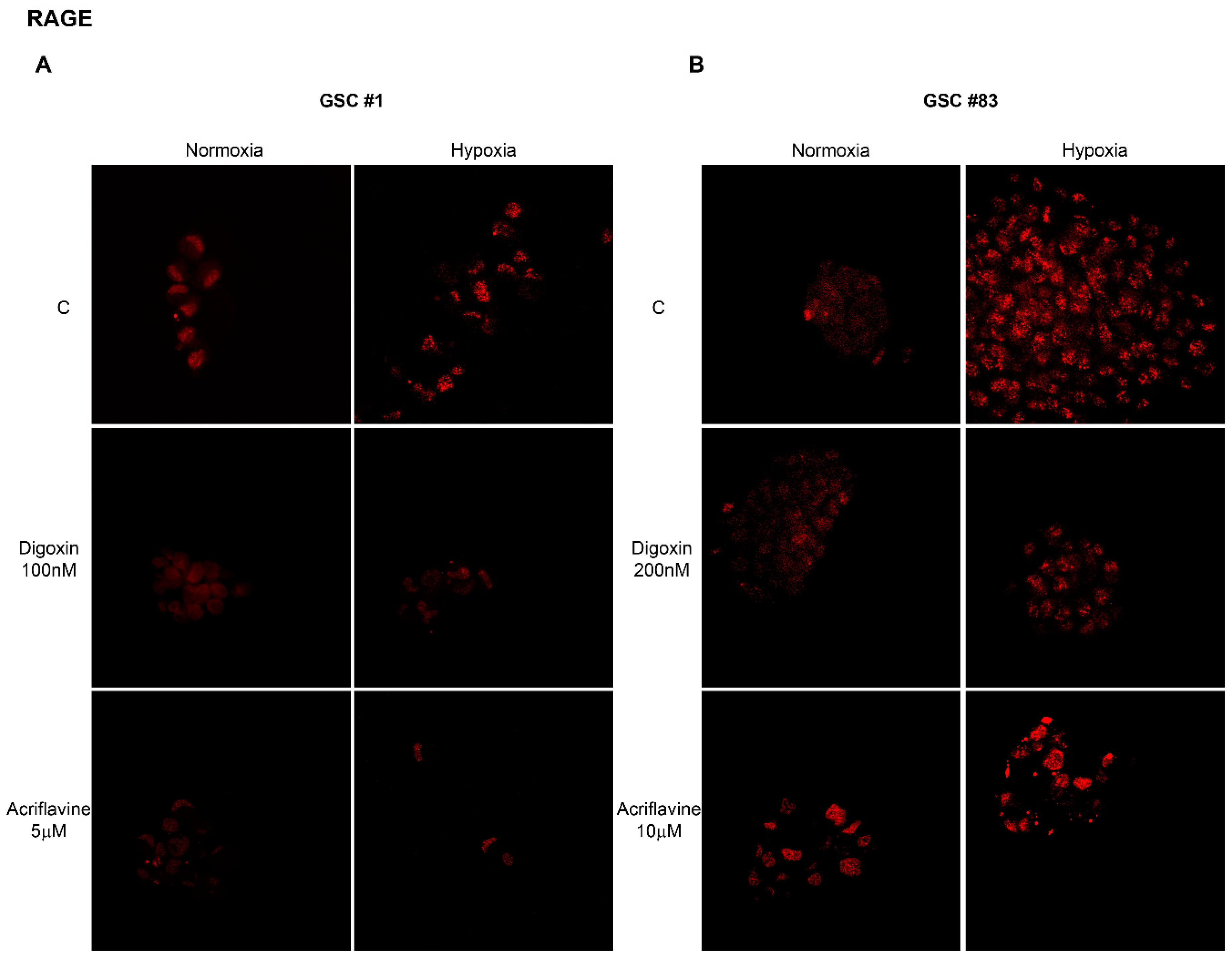
2.2. Hypoxia-Dependent Expression of the Alarmin Receptor RAGE
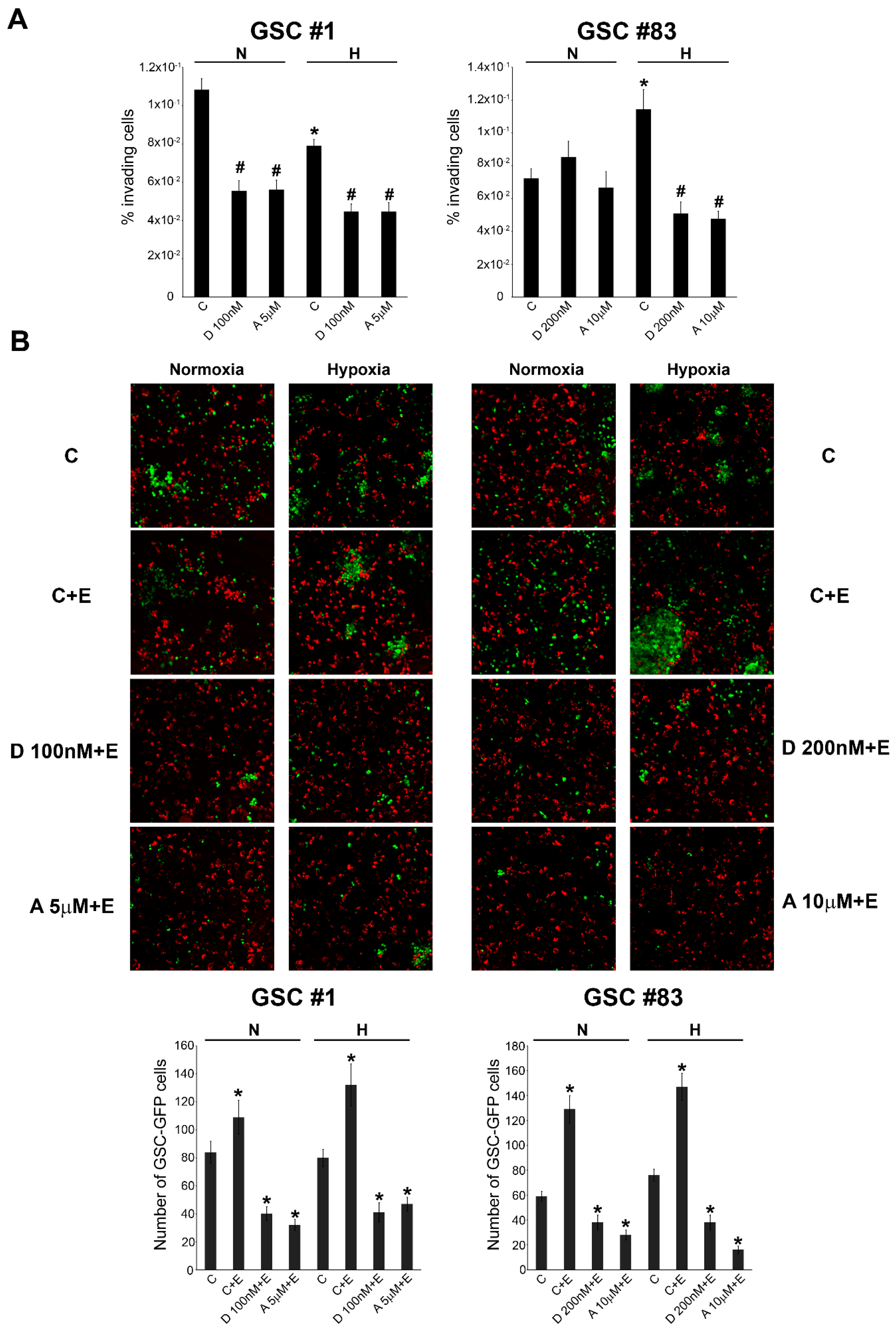
2.3. GSC Migration and iInvasion in the Presence of Necrotic Extracts
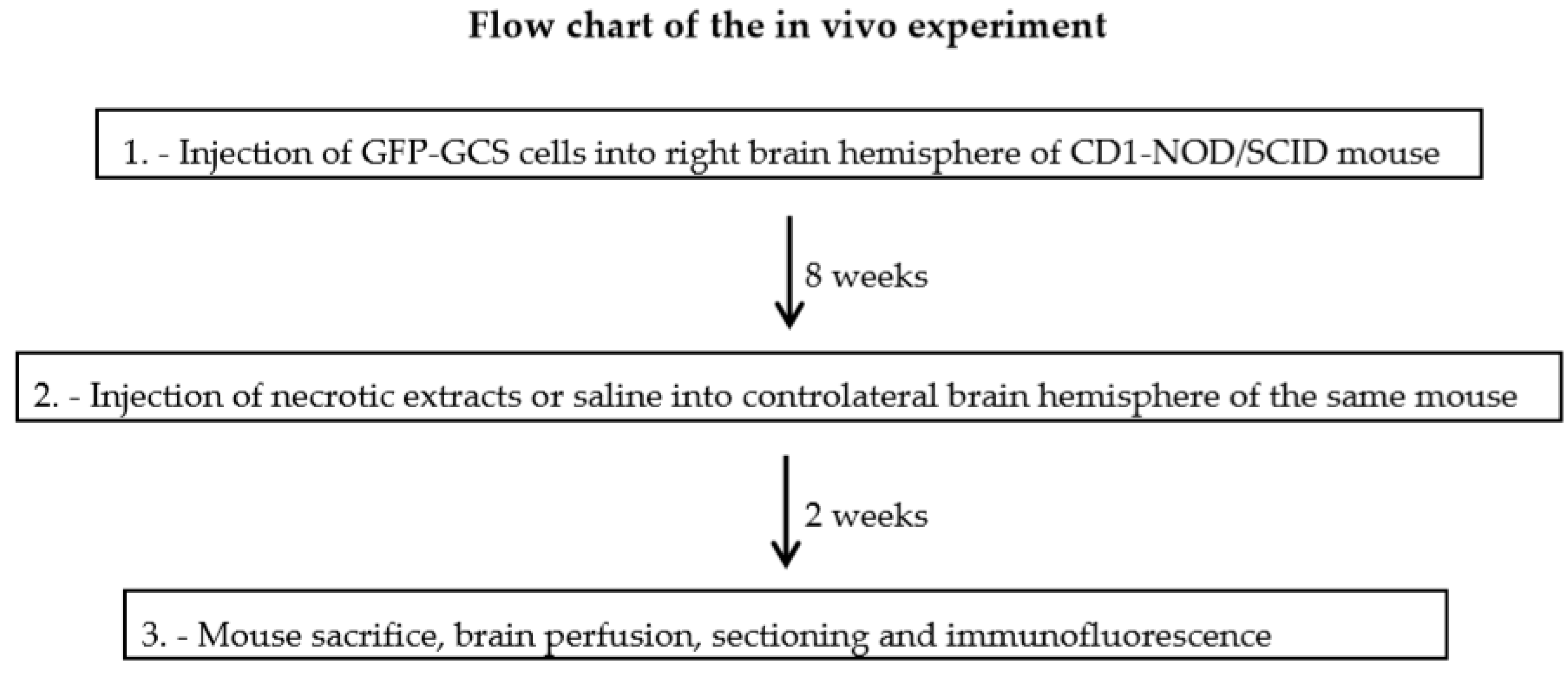
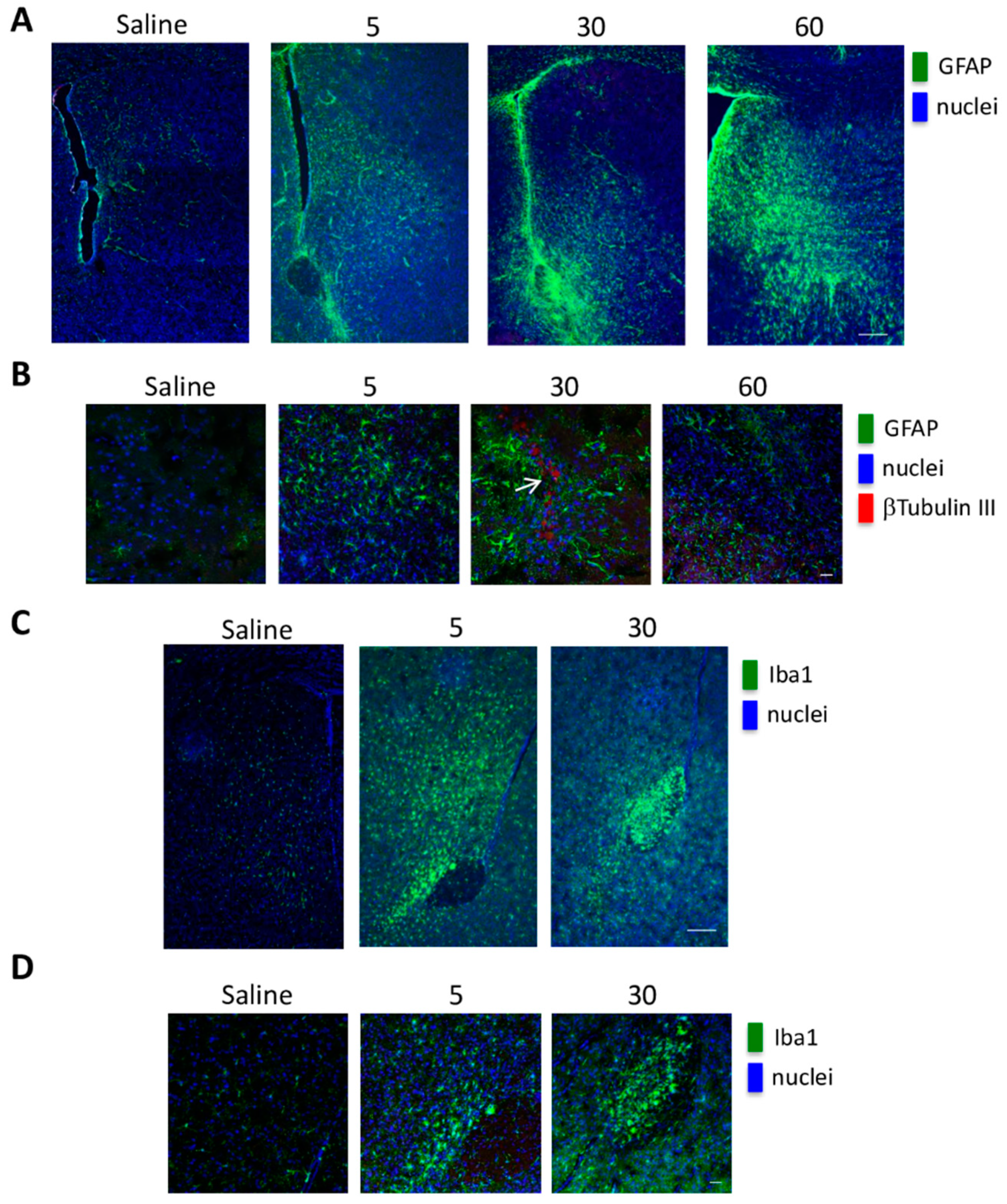
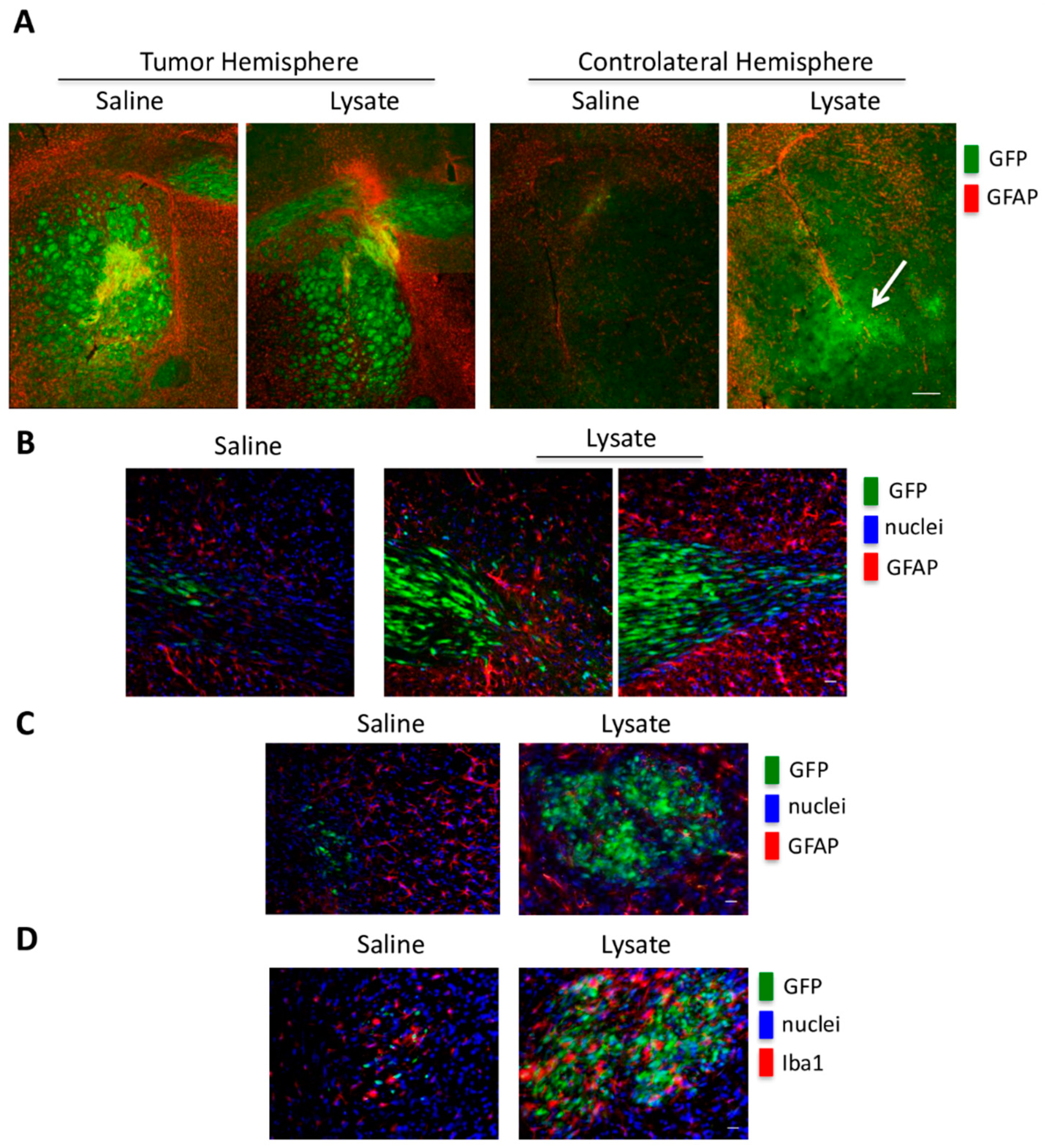
2.4. In Vivo GSC Migration
3. Discussion
Limitations and Considerations
4. Materials and Methods
4.1. Cell Lines and Hypoxia Treatments
4.2. Necrotic Cell Extracts
4.3. SDS-PAGE and Western Blot Analysis
4.4. Real-Time PCR Analysis
4.5. Immunofluorescence Assays
4.6. Cellular Invasion
4.7. Co-Culture Experiments
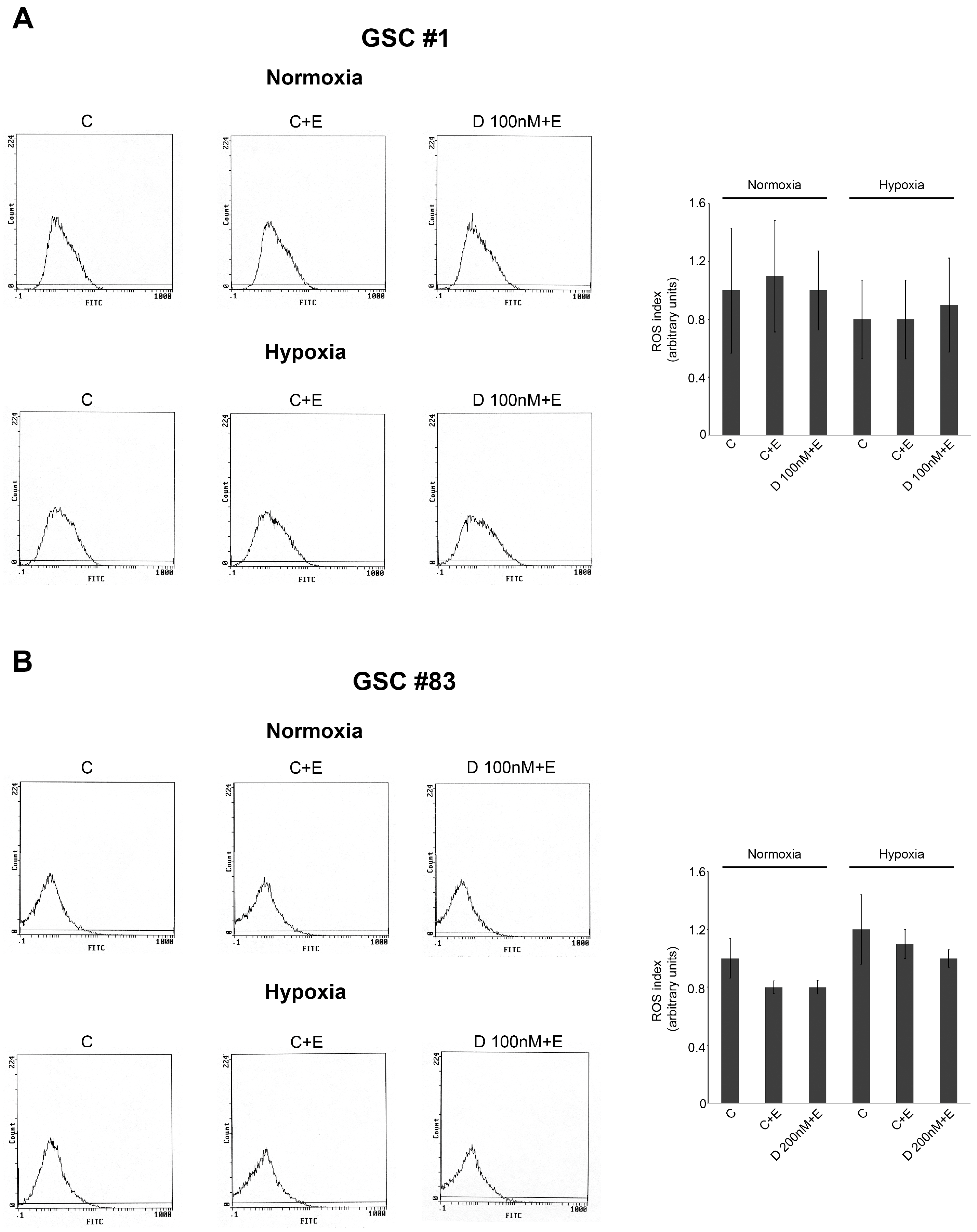
4.8. Measurement of Intracellular ROS
4.9. In Vivo Experiments
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CSC | Cancer stem cell |
| GCS | glioblastoma stem-like cell |
| HIF | hypoxia inducible factor |
| PDH | prolyl hydroxylase |
| VHL | von Hippel-Lindau |
| ROS | Reactive Oxygen Species |
| HRE | Hypoxia Response Element |
| DAMP | damage associated molecular pattern |
| HMGB1 | High Mobility Group Box 1 |
| RAGE | Receptor for advanced glycation end products |
| TLR | Toll-like receptor |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| VEGF | vascular endothelial growth factor |
| HK2 | Hexokinase 2 |
| GFAP | Glial fibrillary acidic protein |
| IBA1 | Ionized calcium binding adaptor molecule 1 |
| GFP | green fluorescent protein |
| IRR | inflammatory reparative response |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, R.D.; Lanzen, J.L.; Snyder, S.A.; Dewhirst, M.W. Comparison of tumor and normal tissue oxygen tension measurements using OxyLite or microelectrodes in rodents. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2533–H2544. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer. 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef] [Green Version]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, A.; Agnihotri, S.; Wilson, M.R.; Burrell, K.E.; Tonge, P.D.; Alamsahebpour, A.; Jalali, S.; Taccone, M.S.; Mansouri, S.; Golbourn, B.; et al. Targeting hexokinase 2 enhances response to radio-chemotherapy in glioblastoma. Oncotarget. 2016, 7, 69518–69535. [Google Scholar] [CrossRef] [Green Version]
- Baek, J.H.; Mahon, P.C.; Oh, J.; Kelly, B.; Krishnamachary, B.; Pearson, M.; Chan, D.A.; Giaccia, A.J.; Semenza, G.L. OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Mol Cell. 2005, 17, 503–512. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xie, Y.; Sun, X.; Zeh, H.J., 3rd; Kang, R.; Lotze, M.T.; Tang, D. DAMPs, ageing, and cancer: The ‘DAMP Hypothesis’. Ageing Res. Rev. 2015, 24, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Patel, S. Danger-Associated Molecular Patterns (DAMPs): The Derivatives and Triggers of Inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; Yamamoto, H.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Vetter, S.W. The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim. Biophys. Acta 2015, 1852, 2706–2711. [Google Scholar] [CrossRef] [Green Version]
- Tóbon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Tafani, M.; Di Vito, M.; Frati, A.; Pellegrini, L.; De Santis, E.; Sette, G.; Eramo, A.; Sale, P.; Mari, E.; Santoro, A.; et al. Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J. Neuroinflammat. 2011, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.A.; Sansone, L.; Carnevale, I.; Limana, F.; Runci, A.; Polletta, L.; Perrone, G.A.; De Santis, E.; Tafani, M. One Special Question to Start with: Can HIF/NFkB be a Target in Inflammation? Endocr. Metab. Immu. Disord. Drug Targets 2015, 15, 171–185. [Google Scholar] [CrossRef]
- Rider, P.; Kaplanov, I.; Romzova, M.; Bernardis, L.; Braiman, A.; Voronov, E.; Apte, R.N. The transcription of the alarmin cytokine interleukin-1 alpha is controlled by hypoxia inducible factors 1 and 2 alpha in hypoxic cells. Front. Immunol. 2012, 3, 290. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. Bioessays 2016, 38 (Suppl. 1), S65–S74. [Google Scholar] [CrossRef]
- Nandy, S.B.; Lakshmanaswamy, R. Cancer Stem Cells and Metastasis. Prog. Mol. Biol. Transl. Sci. 2017, 151, 137–176. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- O’Flaherty, J.D.; Gray, S.; Richard, D.; Fennell, D.; O’Leary, J.J.; Blackhall, F.H.; O’Byrne, K.J. Circulating tumour cells, their role in metastasis and their clinical utility in lung cancer. Lung Cancer 2012, 76, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Ahmad, R.; Srivastava, A.S.; Singh, A.B. Cancer stem cells and colorectal cancer: An overview. Curr. Top Med. Chem. 2011, 11, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.K. Ovarian cancer stem cells: Molecular concepts and relevance as therapeutic targets. Mol. Aspects Med. 2014, 39, 110–125. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stemcell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 349–354. [Google Scholar] [CrossRef]
- Lang, S.H.; Frame, F.M.; Collins, A.T. Prostate cancer stem cells. J. Pathol. 2009, 217, 299–306. [Google Scholar] [CrossRef]
- Malaguarnera, R.; Morcavallo, A.; Giuliano, S.; Belfiore, A. Thyroid cancer development and progression: Emerging role of cancer stem cells. Minerva Endocrinol. 2012, 37, 103–115. [Google Scholar] [PubMed]
- Lorico, A.; Bratbak, D.; Meyer, J.; Kunke, D.; Krauss, S.; Plott, W.E.; Solodushko, V.; Baum, C.; Fodstad, O.; Rappa, G. Gamma-glutamylcysteine synthetase and L-buthionine-(S,R)-sulfoximine: A new selection strategy for gene-transduced neural and hematopoietic stem/progenitor cells. Hum. Gene Ther. 2005, 16, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Pallini, R.; Ricci-Vitiani, L.; Banna, G.L.; Signore, M.; Lombardi, D.; Todaro, M.; Stassi, G.; Martini, M.; Maira, G.; Larocca, L.M.; et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 8205–8212. [Google Scholar] [CrossRef] [Green Version]
- Marziali, G.; Signore, M.; Buccarelli, M.; Grande, S.; Palma, A.; Biffoni, M.; Rosi, A.; D’Alessandris, Q.G.; Martini, M.; Larocca, L.M.; et al. Metabolic/Proteomic Signature Defines Two Glioblastoma Subtypes With Different Clinical Outcome. Sci. Rep. 2016, 6, 21557. [Google Scholar] [CrossRef]
- Günther, H.S.; Schmidt, N.O.; Phillips, H.S.; Kemming, D.; Kharbanda, S.; Soriano, R.; Modrusan, Z.; Meissner, H.; Westphal, M.; Lamszus, K. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 2008, 27, 2897–2909. [Google Scholar] [CrossRef]
- D’Alessandris, Q.G.; Biffoni, M.; Martini, M.; Runci, D.; Buccarelli, M.; Cenci, T.; Signore, M.; Stancato, L.; Olivi, A.; De Maria, R.; et al. The clinical value of patient-derived glioblastoma tumorspheres in predicting treatment response. Neuro Oncol. 2017, 19, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Papale, M.; Ferretti, E.; Battaglia, G.; Bellavia, D.; Mai, A.; Tafani, M. EZH2, HIF-1, and Their Inhibitors: An Overview on Pediatric Cancers. Front. Pediatr. 2018, 6, 328. [Google Scholar] [CrossRef]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobre, A.R.; Entenberg, D.; Wang, Y.; Condeelis, J.; Aguirre-Ghiso, J.A. The Different Routes to Metastasis via Hypoxia-Regulated Programs. Trends Cell Biol. 2018, 28, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid. Med. Cell Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Chen, W.W.; Zhang, X. Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible factors on therapeutic approaches. Oncol Lett. 2016, 12, 2283–2288. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Kiebish, M.A.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Metabolic management of brain cancer. Biochim. Biophys. Acta 2011, 1807, 577–594. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chen, L.; He, H.; Huang, W.; Zhang, R.; Li, P.; Meng, Y.; Jiang, X. Up-regulation of microRNA-16 in Glioblastoma Inhibits the Function of Endothelial Cells and Tumor Angiogenesis by Targeting Bmi-1. Anticancer Agents Med. Chem. 2016, 16, 609–620. [Google Scholar] [CrossRef]
- Xiao, B.; Zhou, X.; Ye, M.; Lv, S.; Wu, M.; Liao, C.; Han, L.; Kang, C.; Zhu, X. MicroRNA-566 modulates vascular endothelial growth factor by targeting Von Hippel-Landau in human glioblastoma in vitro and in vivo. Mol. Med. Rep. 2016, 13, 379–385. [Google Scholar] [CrossRef]
- Grande, S.; Palma, A.; Ricci-Vitiani, L.; Luciani, A.M.; Buccarelli, M.; Biffoni, M.; Molinari, A.; Calcabrini, A.; D’Amore, E.; Guidoni, L.; et al. Metabolic Heterogeneity Evidenced by MRS among Patient-Derived Glioblastoma Multiforme Stem-Like Cells Accounts for Cell Clustering and Different Responses to Drugs. Stem Cells Int. 2018, 2018, 3292704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudin, M.J.; Weaver, V.M. Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Giannoni, E.; Comito, G.; Chiarugi, P. Microenvironment and tumor cell plasticity: An easy way out. Cancer Lett. 2013, 341, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer-a Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- Tafani, M.; Schito, L.; Pellegrini, L.; Villanova, L.; Marfe, G.; Anwar, T.; Rosa, R.; Indelicato, M.; Fini, M.; Pucci, B.; et al. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-{kappa}B. Carcinogenesis 2011, 32, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Gopal, P.; Gosker, H.R.; Theije, C.C.; Eurlings, I.M.; Sell, D.R.; Monnier, V.M.; Reynaert, N.L. Effect of chronic hypoxia on RAGE and its soluble forms in lungs and plasma of mice. Biochim. Biophys. Acta 2015, 1852, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Tafani, M.; Russo, A.; Di Vito, M.; Sale, P.; Pellegrini, L.; Schito, L.; Gentileschi, S.; Bracaglia, R.; Marandino, F.; Garaci, E.; et al. Up-regulation of pro-inflammatory genes as adaptation to hypoxia in MCF-7 cells and in human mammary invasive carcinoma microenvironment. Cancer Sci. 2010, 101, 1014–1023. [Google Scholar] [CrossRef]
- Pichiule, P.; Chavez, J.C.; Schmidt, A.M.; Vannucci, S.J. Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J. Biol. Chem. 2007, 282, 36330–36340. [Google Scholar] [CrossRef] [Green Version]
- Franses, J.W.; Drosu, N.C.; Gibson, W.J.; Chitalia, V.C.; Edelman, E.R. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int. J. Cancer 2013, 133, 1334–1344. [Google Scholar] [CrossRef] [Green Version]
- Mamlouk, S.; Wielockx, B. Hypoxia-inducible factors as key regulators of tumor inflammation. Int. J. Cancer 2013, 132, 2721–2729. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammation and oncogenesis: A vicious connection. Curr. Opin. Genet. Dev. 2010, 20, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Li, Y.; Yang, Z.; Xu, W.; Yang, Y.; Tan, X. ROS mediated EGFR/MEK/ERK/HIF-1α Loop Regulates Glucose metabolism in pancreatic cancer. Biochem. Biophys. Res. Commun. 2018, 500, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Guidoni, L.; Ricci-Vitiani, L.; Rosi, A.; Palma, A.; Grande, S.; Luciani, A.M.; Pelacchi, F.; di Martino, S.; Colosimo, C.; Biffoni, M.; et al. 1H NMR detects different metabolic profiles in glioblastoma stem-like cells. NMR Biomed. 2014, 27, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflammat. 2015, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.C.; Tsai, J.T.; Chao, T.Y.; Ma, H.I.; Liu, W.H. Musashi-1 Enhances Glioblastoma Migration by Promoting ICAM1 Translation. Neoplasia 2019, 21, 459–468. [Google Scholar] [CrossRef]
- Alieva, M.; Margarido, A.S.; Wieles, T.; Abels, E.R.; Colak, B.; Boquetale, C.; Jan Noordmans, H.; Snijders, T.J.; Broekman, M.L.; van Rheenen, J. Preventing inflammation inhibits biopsy-mediated changes in tumor cell behavior. Sci. Rep. 2017, 7, 7529. [Google Scholar] [CrossRef] [Green Version]
- Alieva, M.; van Rheenen, J.; Broekman, M.L.D. Potential impact of invasive surgical procedures on primary tumor growth and metastasis. Clin. Exp. Metastasis 2018, 35, 319–331. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSC #1 | GSC #83 | ||
|---|---|---|---|
| Normoxia (mean fluorescence intensity per cell) | Normoxia (mean fluorescence intensity per cell) | ||
| C | 75.3 ± 6.5 | C | 50.7 ± 5.0 |
| Digoxin 100 nM | 70.7 ± 5.7 | Digoxin 200 nM | 56.7 ± 4.0 |
| Acriflavine 5 µM | 66.3 ± 5.7 | Acriflavine 10 µM | 60.3 ± 2.3 |
| Hypoxia (mean fluorescence intensity per cell) | Hypoxia (mean fluorescence intensity per cell) | ||
| C | 92.3 ± 6.5 | C | 107 ± 9.8 |
| Digoxin 100 nM | 56.0 ± 4.6 * | Digoxin 200 nM | 64.3 ± 4.7 * |
| Acriflavine 5 µM | 32.0 ± 2.6 * | Acriflavine 10 µM | 74.7 ± 4.6 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papale, M.; Buccarelli, M.; Mollinari, C.; Russo, M.A.; Pallini, R.; Ricci-Vitiani, L.; Tafani, M. Hypoxia, Inflammation and Necrosis as Determinants of Glioblastoma Cancer Stem Cells Progression. Int. J. Mol. Sci. 2020, 21, 2660. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082660
Papale M, Buccarelli M, Mollinari C, Russo MA, Pallini R, Ricci-Vitiani L, Tafani M. Hypoxia, Inflammation and Necrosis as Determinants of Glioblastoma Cancer Stem Cells Progression. International Journal of Molecular Sciences. 2020; 21(8):2660. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082660
Chicago/Turabian StylePapale, Marco, Mariachiara Buccarelli, Cristiana Mollinari, Matteo A. Russo, Roberto Pallini, Lucia Ricci-Vitiani, and Marco Tafani. 2020. "Hypoxia, Inflammation and Necrosis as Determinants of Glioblastoma Cancer Stem Cells Progression" International Journal of Molecular Sciences 21, no. 8: 2660. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082660