Mitochondrial Protection by PARP Inhibition

1
Department of Biochemistry and Medical Chemistry, University of Pecs Medical School, 7624 Pecs, Hungary
2
Szentagothai Research Centre, University of Pecs, 7624 Pecs, Hungary
3
HAS-UP Nuclear-Mitochondrial Interactions Research Group, 1245 Budapest, Hungary
*
Author to whom correspondence should be addressed.
†
Deceased on 1 August 2019.
Int. J. Mol. Sci. 2020, 21(8), 2767; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082767
Submission received: 31 March 2020
/
Revised: 11 April 2020
/
Accepted: 14 April 2020
/
Published: 16 April 2020
(This article belongs to the Special Issue Mitochondria-Targeted Approaches in Health and Disease)
{kind=link}
{kind=link}
Abstract
:Inhibitors of the nuclear DNA damage sensor and signalling enzyme poly(ADP-ribose) polymerase (PARP) have recently been introduced in the therapy of cancers deficient in double-strand DNA break repair systems, and ongoing clinical trials aim to extend their use from other forms of cancer non-responsive to conventional treatments. Additionally, PARP inhibitors were suggested to be repurposed for oxidative stress-associated non-oncological diseases resulting in a devastating outcome, or requiring acute treatment. Their well-documented mitochondria- and cytoprotective effects form the basis of PARP inhibitors’ therapeutic use for non-oncological diseases, yet can limit their efficacy in the treatment of cancers. A better understanding of the processes involved in their protective effects may improve the PARP inhibitors’ therapeutic potential in the non-oncological indications. To this end, we endeavoured to summarise the basic features regarding mitochondrial structure and function, review the major PARP activation-induced cellular processes leading to mitochondrial damage, and discuss the role of PARP inhibition-mediated mitochondrial protection in several oxidative stress-associated diseases.
1. Introduction
Inhibitors of the nuclear DNA damage sensor and signalling enzyme poly(ADP-ribose) (PAR) polymerase (PARP) have recently gained considerable interest in tumour therapy [1]. Following the success of clinical trials, the United States Food and Drug Administration (FDA) approved four PARP inhibitors in the therapy of breast cancer gene (BRCA)1/2 mutated malignancies not responding to conventional treatments [2], and further trials are currently ongoing to extend their use to cancers carrying other forms of genomic instability [3]. Additionally, based on their cytoprotective properties demonstrated in many models, PARP inhibitors were suggested to be repurposed for non-oncological diseases such as acute pancreatitis, stroke, lung injury, traumatic brain injury, septic shock, and various degenerative diseases [4].
PARP1, the family’s principal member, is present in a million-copy number in the nucleus of mammalian cells [5]. When PARP1 is incapacitated, PARP2 takes over its role and vice versa. Together, they are assumed to perform more than 90% of DNA strand break-induced PARylation [5]. PARP1- and PARP2-deficient mice are viable and their phenotype does not considerably differ from that of the wild type. However, the PARP1/PARP2 double knock out mutation results in embryonic lethality [5]. PARP1 regulates various cellular processes either by its enzymatic activity or simply by its physical presence via protein–protein interactions [6]. Several of these processes lead to the destabilization of mitochondrial membrane systems and impairment of mitochondrial function that eventually can result in cell death [7]. Therefore, inhibitors of the enzyme protect mitochondrial integrity and function, yet may contribute to PARP inhibitor resistance, which limits cancer therapy [8]. Preclinical evidence for the PARP inhibitors’ potential in non-oncological human diseases were reviewed elsewhere consistently [4]. Therefore, in this review, we focused on the processes involved in the PARP inhibitors’ protective effects. We summarised the basic features of mitochondrial structure and function, reviewed the major PARP activation-induced cellular processes leading to mitochondrial damage, and presented mechanistic aspects of PARP activation-induced mitochondrial damages in several oxidative stress-associated diseases.
2. Mitochondrial Structure and Function
2.1. Mitochondria, Mitochondrial DNA, and Oxidative Phosphorylation
Individual mitochondria have a spherocylindrical shape of approximately 1–2 µm in length and 0.1–0.5 µm in diameter [9]. They are characterised by two membrane systems; the outer membrane encloses the intermembrane space, and the highly folded inner membrane envelops the matrix. The outer membrane’s protein to phospholipid ratio is nearly 1:1 by weight, and it contains a substantial number of pore-forming voltage-dependent anion channels (VDACs), which, in their open state, allow the passage of small molecules across the membrane. The inner membrane contains roughly three times as much protein by weight when compared with the outer one, and is rich in cardiolipin, which makes this membrane essentially impermeable to all substances with the exception of gases [10]. Channels and specific transporters are responsible for trafficking substrates and products of the mitochondrial metabolic processes [11]. The matrix contains several copies of the mitochondrial DNA as well as a substantial number of different enzymes, including those of pyruvate and fatty acid oxidation, and also those of the Krebs cycle [9]. The human mitochondrial genome of 16,569 DNA base pairs contains at least 37 genes, which encodes 13 proteins of the oxidative phosphorylation machinery, 22 tRNAs, and 2 rRNAs [12]. Mitochondrial oxidative phosphorylation produces most of the ATP necessary for different functions of the various tissues. The process’ machinery, the enzyme complexes of the respiratory chain, are located in the inner mitochondrial membrane, where they receive the reduced dinucleotides NADH and FADH2 resulting from the breakdown reactions of the fuel molecules including glucose, amino acids, and lipids. Electrons of the reduced dinucleotides via stepwise redox reactions are transferred along the respiratory complexes I to IV, and, eventually, reduce molecular oxygen. According to the reigning chemiosmotic hypothesis, simultaneously to the electron transport, a proton gradient is built up between the matrix and the intermembrane space, which represents the driving force for ATP synthesis from ADP and phosphate at complex V [13]. Regardless of the exact mechanism, oxidative phosphorylation yields close to eight times as much ATP per glucose molecule when compared to substrate-level phosphorylation [13,14,15].
2.2. Mitochondrial Membrane Potential
Mitochondrial membrane potential (ΔΨm) results from the electrical (charge difference) potential of the ions’ gradient across the mitochondrial inner membrane. In addition to contributing to the proton motive force regarding ATP synthesis, ΔΨm is considered to have a substantial role in mitochondrial translocation of proteins encoded by the nuclear DNA [16], transport of metal ions, such as K+, Ca2+, and Mg2+ [17,18,19], regulation of reactive oxygen species (ROS) production by the mitochondrial electron transport chain [20], regulation of mitochondrial network dynamics [21], and most importantly, regulation of cell death [22,23,24]. These mitochondrial phenomena are intricately interconnected. Mitochondrial biogenesis as well as replacement of proteins, which are continuously damaged by mitochondrially produced ROS, requires the concerted operation of the nuclear and mitochondrial gene expression [25]. Additionally, nearly 50 to 70% of the nuclearly encoded mitochondrial proteins possess positively charged N-terminal mitochondria-targeted sequences, and transport of these proteins via translocases of the inner/outer membrane (TIM/TOM complex) is regulated by ΔΨm [16,26]. K+ via the K+/H+ antiporter and the ATP-sensitive K+ channel prevents excess matrix swelling and contraction, respectively, thereby maintaining activity of the electron transport chain, and regulating mitochondrial ROS production [27,28]. Intracellular Ca2+ enters the matrix along its electrochemical gradient via the Ca2+ uniporter route when its concentration exceeds a submicromolar threshold level [29,30], and the accumulated mitochondrial Ca2+ is released via a Na+/Ca2+ exchanger when the intracellular Ca2+ signal declines [31].
2.3. Mitochondrial Ca2+ and ROS
Elevated mitochondrial Ca2+ causes allosteric stimulation of pyruvate-, isocitrate-, and α-ketoglutarate dehydrogenase complexes [32], as well as the activation of complex V [33], glycerol-3-phosphate dehydrogenase [34], and adenine nucleotide translocase [35]. Eventually, this coordinated activation of the mitochondrial energy-producing metabolic pathways results in increased ATP synthesis [32]. However, the increased metabolic rate is accompanied by higher respiratory chain electron leakage that may result in more mitochondrial ROS generation by complexes I and III [36]. In addition, Ca2+ by stimulating nitric oxide synthase (NOS) [37], can induce the formation of NO˙, which inhibits complex IV [38] that enhances ROS generation at complex III [39]. Comprehensively, increased mitochondrial Ca2+ seemingly promotes ROS production in the presence of respiratory chain inhibition while attenuates it under normal conditions [40]. The predominant mitochondrial ROS produced by the respiratory chain is O2‾˙ that a spontaneous chemical reaction or the enzymatic action of superoxide dismutase can convert to H2O2, which can be transformed to OH˙ by metal ions in the Fenton reaction [41]. Mitochondrial ROS generation is strongly dependent on ΔΨm since mild uncoupling was demonstrated to reduce mitochondrial ROS production in various systems [42,43,44,45].
2.4. Mitochondrial Permeability Transition
Mitochondrial Ca2+ overload, ROS, and elevated inorganic phosphate initiates mitochondrial permeability transition (mPT). During the process, the permeability transition pore (PTP), a high conductance cyclosporine A sensitive channel of the inner mitochondrial membrane opens and allows rapid unselective passage of water and solutes up to 1.5 kDa of size across the inner membrane [22,46]. The assumed physiological opening of the PTP allows the rapid exchange of solutes, such as Ca2+ and ROS, between the matrix and the cytosol [47]. This mechanism was proposed to supplement the mitochondrial Na+/Ca2+ exchanger in removing excess Ca2+ from the matrix once the cytosolic Ca2+ decreases below its submicromolar threshold level after a transient elevation [48]. However, the long-term opening of the PTP results in dissipation of ΔΨm, the reversal of ATP synthase’s operation, in an attempt to maintain ΔΨm [49], excess ROS production, release of Ca2+ from the matrix and of proapoptotic proteins from the intermembrane space leading to apoptotic cell death, and influx of water into the matrix, which causes mitochondrial swelling and rupture of the inner membrane resulting in necrotic cell death [50]. Operation of mPT in this manner was implicated in the pathomechanism of various diseases including myocardial infarct [51], stroke [52], neurodegenerative diseases [53], and muscular dystrophy [54].
2.5. Mitochondrial Fission and Fusion
Mitochondria of a cell continually oscillate between the forms of individual entities and the fused network of multiple organelles by the processes of fusion and fission [55]. These processes have roles in such fundamental functions as mitochondrial biogenesis [56], cellular propagation of mitochondria, and mitochondrial quality control [57,58]. Fragmentation of the mitochondrial network occurs in response to impairment of oxidative phosphorylation once the mitochondrial inner membrane potential is abnormal [59,60]. However, fragmentation of mitochondria does not necessarily evoke apoptosis [55]. The mitochondrial outer membrane fission protein (FIS)-1, the limiting factor in the fission process, recruits dynamin-like protein (DRP)-1 to punctuate structures on the surface of the mitochondria. DRP-1 homo-oligomers perform the fission by forming a ring around the mitochondrial tubule, and generate mechanical force via conformational changes leading to membrane constriction, similar to dynamin [61]. In contrast to fission, fusion needs separate dynamin-like large GTPases for the sequential fusion of the outer and the inner mitochondrial membranes [62]. Fusion of the outer membrane is mediated by the resident large GTPases mitofusin (Mfn)1 and 2 [63], both of which were found to be essential regarding embryonic development [64]. Mfn1 is expressed ubiquitously, and it has a higher GTPase activity when compared to Mfn2 [65]. Expression of Mfn2 is tissue-specific [66], and it was reported to have a role in tethering mitochondria and endoplasmic reticulum [67]. Fusion of the inner membrane is mediated by another dynamin-like large GTPase, optic atrophy (OPA)1 [62]. The enzyme exists in two forms, an inner membrane-anchored long form required for fusion, and a soluble short form associated with fission rather than fusion of the inner membrane. Although differential splicing can account for the existence of the two forms [68], activation of the metalloendopeptidase OMA1 under stress conditions can lead to complete conversion of the long to the short form of OPA1, which prevents mitochondrial fusion until the de novo synthesis of new OPA1 [69].
3. Interplay of PARP with Akt-Mediated Mitochondrial Protection
3.1. Akt’s Effects on Outer Mitochondrial Membrane Permeabilisation-Associated Processes
The phosphatidylinositol-3 kinase (PI3K)-protein kinase B/Akt pathway mediates proliferation-inducing and cytoprotective effects of cAMP, hypoxia and cytokines, as well as various growth factors [70]. The activation of the pathway can protect the mitochondria via various mechanisms including preservation of the outer mitochondrial membrane’s integrity [71]. Pro-apoptotic and anti-apoptotic members of the B-cell lymphoma (Bcl)-2 protein family have opposite effects on the outer membrane. Heterodimerisation of pro-apoptotic members, such as Bcl-2-associated X (Bax) and Bcl-2 homologous antagonist/killer (Bak), especially in the presence of Bcl-2 homology domain (BH)3-only proteins, such as Bcl-2-associated agonist of cell death (Bad), Bcl-2-like protein 11 (Bim), BH3 interacting-domain death agonist (Bid) and p53 upregulated modulator of apoptosis (PUMA) permeabilises the outer membrane via pore formation, which allows cytochrome C release from the mitochondrial intermembrane space leading to caspase-9 activation that eventually results in apoptotic cell death [71,72]. Anti-apoptotic members, such as Bcl-2, Bcl-xL, and myeloid leukemia cell differentiation protein (Mcl)-1, antagonise the pro-apoptotic family members’ effect thereby preserving the outer membrane’s integrity and promoting cell survival [73]. Akt phosphorylates Bad, which forestalls the heterodimer formation between Bad and other pro-apoptotic Bcl-2 family members. Rather, phosphorylated Bad, by forming a complex with the cytoplasmic scaffolding protein 14-3-3, is eliminated from the balance between the pro-and anti-apoptotic Bcl-2 family members, resulting in the prevention of cytochrome C release [74]. Additionally, Akt directly phosphorylates, and thereby inactivates caspase-9 at its Ser196, which contributes to Akt’s effect of blocking the intrinsic apoptotic pathway [75].
3.2. Akt’s Effects on Glycogen Synthase Kinase-3β-Mediated Processes
Due to its extensive participation in signalling pathways (in addition to its metabolic role of regulating glycogen synthesis), glycogen synthase kinase (GSK)-3β is one of Akt’s most prominent downstream targets in mediating mitochondrial protection [76]. In hypoxia, GSK-3β is activated by phosphorylation of its Tyr216 [77], and contributes to the hypoxia or ischemia-induced tissue injury. It represses expression, nuclear translocation, and binding to antioxidant response element (ARE) DNA sequence, of nuclear factor erythroid 2-related factor 2 (Nrf2) [78]. The diminished binding of the transcription factor results in a reduced expression of Nrf2/ARE-regulated genes encoding proteins of the antioxidant defence system, such as superoxide dismutase, peroxidase, catalase, and enzymes of glutathione synthesis and reactivation [78]. The resulting oxidative stress damages the mitochondria, which significantly contributes to hypoxia or ischemia-induced mitochondrial impairments and tissue injury [76]. Furthermore, the activation of GSK-3β diminishes nuclear translocation of the transcription factor cAMP response element-binding protein (CREB), thereby diminishing CREB’s binding to the co-activator CREB-binding protein (CBP) [79]. This process causes altered interaction with the pro-inflammatory transcription factor nuclear factor (NF)κB leading to increased inflammatory response [80]. The oxidative stress accompanying the inflammatory response contributes to mitochondrial damages [76]. Akt counteracts these harmful effects of GSK-3β by phosphorylating its Ser9, thereby inhibiting the enzyme [81]. In addition to preventing GSK-3β’s diminishing effect on CREB activation [79], Akt directly phosphorylates CREB’s Ser133, which promotes CREB’s binding to CBP and enhances expression of CREB-regulated genes critical for mitochondrial protection and survival [82].
3.3. Akt’s Effects on Mechanistic Target of Rapamycin and Forkhead Transcription Factor-Mediated Processes
The other major node of the PI3K-Akt pathway is mechanistic (previously mammalian) target of rapamycin (mTOR), a downstream target of Akt, mTOR complex (mTORC)1, and an upstream activator of Akt, mTORC2. Akt activates mTOR by direct phosphorylation, which induces transcription factors associated with growth and cell survival, as well as factors regulating translation initiation, hypoxia, and angiogenesis [83]. Furthermore, the mTOR pathway activates peroxisome proliferator–activated receptor γ coactivator-1α (PGC-1α), the major transcription factor of mitochondrial biogenesis, thereby modulating the mitochondrial copy number and mitochondrial function [84,85]. In addition to phosphorylating its cytoplasmic targets, activated Akt translocates to the nucleus, and regulates various transcription factors by phosphorylating them [86]. Forkhead family transcription factors induce the expression of genes, which encode various growth factors, proteins involved in the stress response, and synthetic enzymes of carbohydrate and lipid metabolism [87]. Additionally, Bcl-2 family members can be transactivated by forkhead transcriptional factors. Two functional forkhead response elements were reported to be present within the sequence of Bim promoter [88,89]. When they are phosphorylated by Akt, forkhead transcriptional factors do not translocate to the nucleus, rather, they form a complex with the cytoplasmic 14-3-3 protein and are subjected to proteosomal degradation [87].
3.4. Mechanism for Akt-Mediated Protective Effect of PARP Inhibition
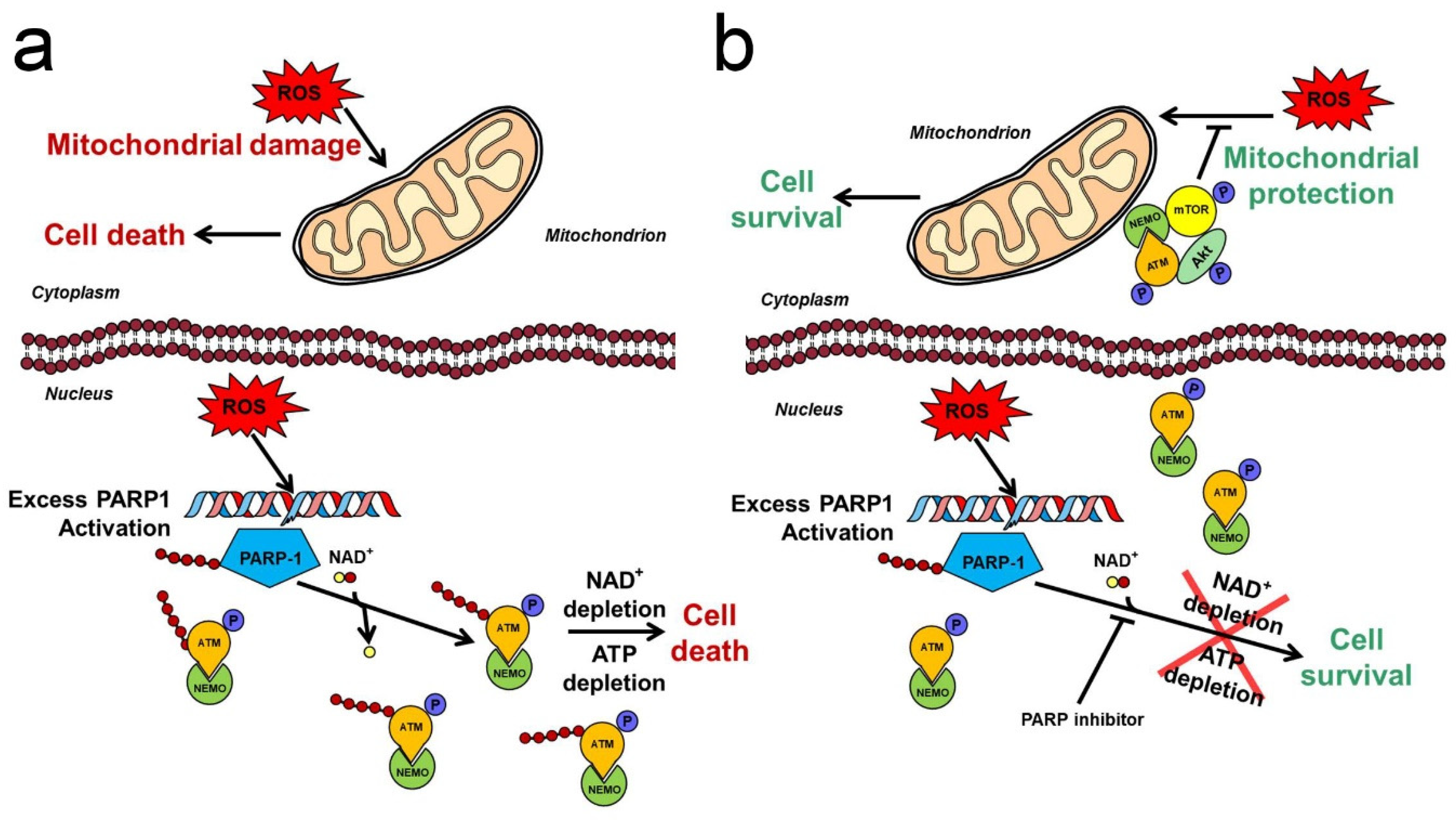
Negative regulation of the PI3K-Akt pathway is mediated by the phosphatase and tensin homologue deleted on chromosome 10 (PTEN) [90], an oxidation-sensitive SH enzyme [91]. Due to PTEN’s rapid oxidative inactivation, most oxidative stress situations activate Akt for a period of time, although this activation is usually insufficient to protect the cells against the oxidative stress-induced mitochondrial and cellular damages [92,93]. Via generating DNA strand breaks, oxidative stress activates the PARP, which counteracts the Akt activation and potentiates the ROS-induced impairment of cellular and mitochondrial macromolecules. Therefore, PARP inhibitors possess a potential to protect the macromolecules against oxidative damages [94]. As it was demonstrated in a number of in vitro, ex vivo and in vivo experiments, nuclear PARP inhibition achieved either by pharmacological means or by gene silencing, activates Akt [92,95,96]. This activation could become so substantial that it significantly contributes to the mitochondria- and cytoprotective effects of PARP inhibitors in experimental or pathological oxidative stress situations [97,98,99,100,101]. Recently, we presented a mechanism (Figure 1) for PARP inhibition-induced Akt activation [102]. It proposes that the oxidative stress-generated DNA strand breaks activate PARP1. The enzyme PARylates itself, which forms a scaffold, and recruits ataxia telangiectasia mutated kinase (ATM) and nuclear inhibitor of NF-κB kinase-γ (NEMO) [103]. The attachment of ATM to the PAR scaffold results in its activation and phosphorylation of itself and of NEMO [104]. However, when a pharmacological inhibitor or genetic manipulation precludes excess PARP activation, the unPARylated ATM complexes with NEMO and translocates to the cytoplasm, where the complex binds to the outer mitochondrial membrane, at least partially. There, the ATM–NEMO complex forms a signallosome with Akt and mTOR that activates Akt, which, in turn, can exert its mitochondria- and cytoprotective effects via its downstream targets (Figure 1) [102]. This model contains several elements already reported by others. ATM was reported to mediate Akt activation in cancer cell lines, in insulin-treated myocytes, and under cellular stress conditions [105,106,107]. The pathomechanism of diabetes and neurodegenerative diseases was found to involve impaired ATM-related Akt activation [107]. Furthermore, as it was formerly published by independent groups, PARP1 can PARylate ATM directly in different oxidative stress situations [108,109]. Finally, the process of ATM–NEMO complex formation followed by its nuclear-to-cytosolic translocation in response to PARP activity has already been published as a fundamental part of the mechanism of genotoxicity-induced NFκB-regulated apoptosis [103,110]. To complement the elements others formerly described, we demonstrated how silencing any component of the proposed ATM–NEMO–Akt–mTOR signallosome has abolished the PARP inhibitor’s protective effect, and also, that the signallosome is localised to the mitochondria, at least partially (Figure 1) [102]. We also demonstrated that, although PARP1 inhibition causes synthetic lethality in ATM-deficient cells [111], transdominant transfection of these cells with continuously active Akt rescued them, emphasising the significance of Akt activation in the protective effect of PARP inhibition [102]. This protective effect is beneficial regarding the therapy of non-malignant diseases, yet impairs the therapeutic efficacy of the PARP inhibitors in cancer [112]. The latter concept is confirmed by independent observations, in which Akt inhibitors increase toxicity of PARP inhibitors in cancer cells [113,114,115].
4. Interplay of PARP with MAPK-Mediated Mitochondrial Damage
4.1. Mitogen-Activated Protein Kinases and Their Activation in Oxidative Stress
Mitogen-activated protein kinase (MAPK) cascades transmit extracellular signals to intracellular targets thereby regulating a number of various cellular processes including proliferation, differentiation, apoptosis and stress responses [116]. The cascades consist of 2-3 layers of upstream activating kinases, the six MAPK groups (extracellular signal-regulated kinase (ERK)1/2, the c-Jun N-terminal kinases (JNK), the p38 MAPKs, ERK3/4, ERK5 and ERK7/8), and their downstream targets. In most systems, the apoptosis signal-regulating kinase (ASK)1/ MAPK kinase kinase (MEKK)1/2—dual specificity MAPK kinases 4 and 7 (MKK4/7)—JNK and ASK1/dual leucine zipper-bearing kinase (DLK)/MEKK3/4—MKK3/6—p38 MAPK pathways are related to stress responses and apoptosis, while the rapidly accelerated fibrosarcoma protein (Raf)—MAPK kinase (MEK)—ERK signalling pathways are associated with proliferation and differentiation [117]. Depending on the tissue type, a number of different signals can activate the MAPK cascades. However, they are readily activated by ROS in most systems [118]. Various mechanisms induce this activation. Phosphatases, responsible for inactivation of the upstream activating kinases, are oxidation-sensitive and rapidly break down in oxidative stress. The ROS can act directly on growth receptors in the absence of their ligands, thereby inducing Ras activation. Furthermore, ROS is capable of activating Ras in a receptor-independent manner [119], and stimulating ERK1/2 activation even in Ras negative cells via non-receptor protein tyrosine kinase c-Src [120]. Oxidative stress and reactive nitrogen species (RNS) activate JNK and p38 pathways through ASK1 and MEKK1. The ROS oxidises and/or the RNS nitrosylates reduced thioredoxin, which inhibits ASK1 by binding to its N-terminal. The oxidised/nitrosylated thioredoxin disassociates, which enables ASK1 oligomerisation and autophosphorylation resulting in activation [121]. Additionally, pro-inflammatory cytokines, such as tumour necrosis factor-α can also activate JNK and p38 MAPK pathways [122].
4.2. MAPK-Mediated Mitochondrial Processes
When activated, ERK1/2 supports ATP synthesis, preserves ΔΨm, forestalls cytochrome C release, and inactivates Bad by activating p90 ribosomal S6 kinase, which phosphorylates Ser155 of Bad, thereby inactivating it [123,124]. In contrast to these mitochondria protecting effects, ERK was reported to mediate oxidative damage, such as vacuolation, mitochondrial translocation of Bax, and outer membrane permeabilisation to neuronal mitochondria, seen in neurodegenerative diseases [125]. Also, activated ERK1/2 is targeted to the mitochondria, where it associates with the outer mitochondrial membrane, and seemingly enters the intermembrane space to be in position to exert its benign or malign effects [126]. In contrast to ERK1/2, JNK and p38 MAPK activation are implicated nearly exclusively to be involved in mediating mitochondrial damage in various oxidative stress situations. They facilitate mitochondrial Bax translocation, transcriptional regulation of TR-3, an apoptosis-initiating steroid receptor-like protein, cytochrome C and the second mitochondria-derived activator of caspase (Smac) release, and activating phosphorylation of Bad [127]. JNK also phosphorylates the anti-apoptotic Bcl-2 family member Bcl-xL on its Thr47 and Ser62, which decreases Bcl-xL’s binding to Bax thereby impairing anti-apoptotic function of Bcl-xL [128]. JNK but not p38 MAPK signalling is seemingly associated with mitochondrial ROS generation. The elevated mitochondrial ROS together with Ca2+ overload and ATP depletion induces mPT, which may aggravate JNK activation-mediated mitochondrial dysfunction [129]. Similarly to ERK1/2, mitochondrial targeting of JNK may likely be essential for exerting its effects [130]. Recently, a mitochondrial SH3-domain-binding protein 5 (SAB) was reported to operate as the only mitochondrial docking site for JNK [127]. SAB is localised to the outer membrane and binds JNK with its N-terminal SH3 domain, which is in the mitochondrial intermembrane space. SAB depletion prevents mitochondrial translocation of JNK completely. However, it does not interfere with p38 MAPK’s association with the mitochondria [131]. In addition to its apoptosis-inducing effects, mitochondrial JNK regulates mitochondrial bioenergetics, decreases respiration rates and ATP production, and by phosphorylating pyruvate dehydrogenase, shifting the metabolism from aerobic toward anaerobic [129].
4.3. Mechanism for MAPK Phosphatase-1-Mediated Protective Effect of PARP Inhibition
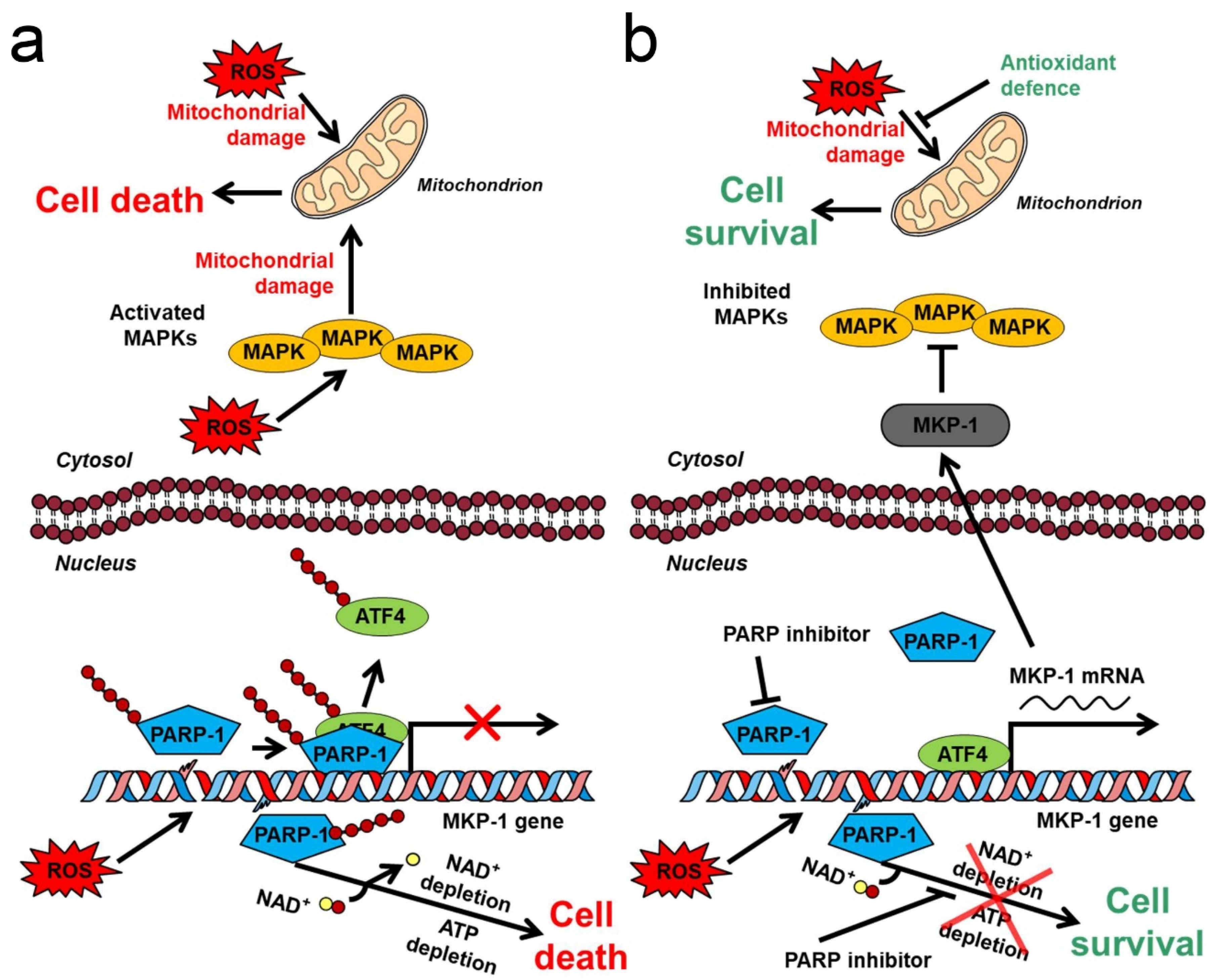
As it was previously reported, ROS or alkylating agent-induced PARP activation leads to the collapse of ΔΨm, induction of mPT, and, inevitably, cell death [132,133,134,135]. Notably, these detrimental effects are often mediated by JNK and p38 MAPK activation [134,135,136]. Furthermore, activation of MAPK phosphatase (MKP)-1, which dephosphorylates thereby inactivates at least all classical MAPK groups such as ERK1/2, JNKs and p38 MAPKs, was reported to ameliorate the damages due to oxidative stress [137,138]. Accordingly, PARP inhibition reduces the detrimental effects of oxidative stress, and upregulates MKP-1 expression as we previously discovered [138]. Since MKP-1 expression is regulated by heat shock and cAMP-mediated mechanisms [139,140], after systematically investigating how silencing all pertinent heat shock factors and CREB transcription factors affects PARP inhibition-induced MKP-1 expression, we determined that activating transcription factor (ATF)4/CREB2 is responsible for mediating the MKP-1-upregulating effect of PARP inhibition [141]. Accordingly, we proposed a mechanism (Figure 2) consistent with all experimental data regarding the mitochondria- and cytoprotective effects of PARP inhibition in oxidative stress [141]. Accordingly, ROS-induced DNA strand breaks activate PARP1, which PARylates ATF4 transcription factor. PARylation reduces ATF4′s affinity toward its responsive elements and is replaced to self-PARylated PARP-1. The resulting diminished MKP-1 expression fails to compensate for the MAPK-activating effect of oxidative stress that leads to MAPK activation, which causes mitochondrial damage and ultimately, cell death. Inhibition of PARP either by a pharmacological agent or genetic manipulation, turns the entire process around resulting in mitochondrial protection and cell survival (Figure 2) [141]. All steps of the model were extensively validated. ROS-induced PARP activation was shown to cause massive PARylation and reduced binding of ATF4, while increased binding of PARylated PARP to ATF4′s target DNA sequence. The PARP inhibitor PJ-34 turned all these effects around, while neither ATF4 nor PARP1 demonstrated affinity toward a mutated CRE sequence under any of the experimental conditions, indicating specificity of the DNA binding. Furthermore, PJ-34 strengthened ATF4 binding to MKP-1′s initiation site. Accordingly, inhibition, or silencing of PARP-1 upregulated MKP-1 expression both in the nucleus and the cytoplasm, inactivated JNK and p38 MAPK, preserved ΔΨm, and reduced the cell death. Most importantly, ATF4 and/or MKP-1 silencing forestalled these effects of the PARP inhibitor [141]. Similarly to the case of Akt activation, JNK and p38 MAPK inhibition by the PARP inhibitor, and the resulting mitochondria- and cytoprotection are beneficial in the therapy of non-malignant diseases, yet impairs the PARP inhibitors’ therapeutic efficacy in cancer [112].
5. Interplay of PARPs with Mitochondrial Homeostasis
5.1. Role of NAD+ Metabolism in Mediating PARP Activation-Induced Damages
Oxidative stress situations causing excessive DNA damage activate nuclear PARP, which leads to the collapse of ΔΨm, reduced activity of respiratory complex I, diminished mitochondrial oxidation and ATP production, enhanced O2‾˙ production, and destruction of mitochondrial architecture [142,143]. The elevated ROS, Ca2+ overload, and ATP shortage open the mitochondrial PTP, which results in the release of the mitochondrial content, swelling due to water influx into the matrix, and even disruption of the mitochondria [144]. Short mPT events can result in cytochrome C and Smac release from the intermembrane space. In the cytosol, cytochrome-C associates with apoptotic peptidase-activating factor (Apaf)1 and procaspase-9 that activates caspase-9 by proteolytic cleavage, which induces effector caspases such as caspase-3 leading to caspase-mediated apoptosis [145]. However, the short-term opening of the PTP more often causes translocation of apoptosis-inducing factor (AIF) and endonuclease G from the mitochondria to the nucleus leading to parthanatos, a special type of programmed necrosis [146]. In the so-called ‘suicide hypothesis’ presented more than 35 years ago, cell death resulted from PARP overactivation was attributed to energy failure. According to the hypothesis, rapid consumption of the substrate NAD+ by the copious PARP enzyme depletes cellular NAD+ pools, then ATP is consumed in a failed attempt to re-synthesise NAD+ [147]. Indeed, the mitochondrial NAD+ pool can represent 40% to 70% of the cell’s total NAD+ content depending on the cell type. However, the mitochondrial inner membrane is impermeable for NAD+, therefore the mitochondrial pool is inaccessible for the nuclear PARP1, unless mPT or necrotic disruption of the mitochondria occur [148]. Factually, in ischemia–reperfusion of isolated heart tissue where massive NAD+ loss was observed, cyclosporin A, an mPT inhibitor was reported to prevent the NAD+ loss as well as the cardiomyocyte damages indicating the pivotal role of PTP opening in the detrimental consequences of PARP activation in this system [142,149]. On the other hand, in neuronal systems, nuclear PARP1 activation may induce mitochondrial dysfunction without the depletion of the mitochondrial NAD+ pool. The glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase requires NAD+ for its operation. Therefore, reduction of cytosolic NAD+ pool by excess PARP1 activation can forestall the entry of glucose-derived acetyl-coenzyme A to the matrix [148]. It implies an insufficient supply of the key substrate for the citrate cycle, resulting in energy starvation and mitochondrial depolarisation, since the brain relies on glucose as a fuel in support of mitochondrial ATP production [150]. During the process of parthanatos, following PAR polymer production by excess activation of PARP1, PAR fragments produced by PAR glycohydrolase and ADP ribosyl-acceptor hydrolase can act directly on the mitochondrial membranes to induce AIF release independent of NAD+ depletion [151]. Further degradation of the PAR fragments by ADP-ribose pyrophosphatase produces AMP, which can inhibit adenine nucleotide translocase, further impeding mitochondrial energy production [143]. High AMP may activate AMP-activated protein kinase (AMPK), which can phosphorylate, and thereby activate PARP1. Since PARP1 also activates AMPK, together, they form a feed-forward loop [152].
5.2. Metabolic Changes in Ischemia
In cultured neurons and astrocytes, PARP overactivation was demonstrated to cause ΔΨm collapse, AIF release and nuclear translocation, and cell death, which all could be prevented by normalizing cytosolic NAD+ levels [153,154]. PARP activation is seemingly not essential for the observed ΔΨm collapse, AIF release, and cell death, since NAD+ depletion alone can induce all these effects. [154]. However, considering the copiousness of PARP1, normally its over-activation is most likely the cause of NAD+ depletion. This situation may likely occur in the penumbra after cerebral or myocardial ischemia, where, in an attempt to rescue the cells [155], reverse operation of the ATP synthase consumes ATP to maintain ΔΨm, and substrate-level phosphorylation provides the necessary ATPs by utilising non-glucose substrates [156,157]. However, the said mechanism has a limited capacity, providing a relatively narrow time-window to overcome the detrimental effects of PARP activation [155]. In addition to PARPs, cyclic ADP-ribose hydrolase (CD38) and sirtuins (SIRTs) use NAD+ as a substrate. However, sirtuins are unlikely to participate in the NAD+ depletion process due to their low affinity for the substrate. On the other hand, when PARP inhibition limits NAD+ consumption, the resulting SIRT1 activation enhances PGC-1α expression, thereby increasing mitochondrial biogenesis, metabolism, and antioxidant capacity [7]. The expression level of PGC-1α is seemingly crucial in resisting hypoxic conditions and oxidative stress situations, as it was revealed when comparing oxidative resistance of melanomas expressing PGC-1α in high and low levels [158].
5.3. Coupling Glycolysis and Oxidative Phosphorylation
The PI3K-Akt pathway can also regulate mitochondrial metabolism by directly phosphorylating hexokinase II, the mitochondrial form of this glycolytic enzyme. Phosphorylated hexokinase II associates with VDAC of the outer mitochondrial membrane coupling glycolysis to oxidative phosphorylation, which enables the cell to cope with low oxygen conditions better [159]. Proximity to the mitochondria enables glycolysis to utilise mitochondria-produced ATP for the priming reactions of the pathway (hexokinase and phosphofructokinase) and channel pyruvate directly to the mitochondria in support of ATP production. The result aids in maintaining ΔΨm and VDAC opening [160]. Akt also regulates the process by phosphorylating, and thereby inactivating GSK-3β, which phosphorylates hexokinase II, resulting in disruption of its association with VDAC [161]. PAR fragments also can uncouple glycolysis and oxidative phosphorylation by dissociating hexokinase from the outer mitochondrial membrane [162]. Disruption of this coupling decreases the efficacy of glycolysis, and changes the potential of the outer membrane, leading to cytochrome C release and apoptosis induction, as well as VDAC pore closing, which causes mitochondrial swelling and -rupture [163]. Additionally, excess PARP activation interferes with mitochondrial quality control processes such as mitophagy and unfolded protein response [164].
6. Therapeutic Implications
6.1. Sepsis and Septic shock
Sepsis is a dysregulated host response to severe infection resulting in multi-organ failure and a mortality of 20 to 50%, despite advances in intensive care [165]. In sepsis, due to elevated ROS production and reduced antioxidant capacity, there is a substantial oxidative stress. Within the cells, the reduced/oxidised glutathione ratio drops [166]. Elevated ROS and pro-inflammatory mediators directly damage mitochondrial DNA, lipids, and proteins, and interfere with the machinery of oxidative phosphorylation [167]. Large amounts of NO˙ are produced by the inducible NOS that inhibits complex IV [166]. The elevated ROS and RNS induces DNA strand break-mediated PARP activation causing reduction in the cellular NAD+ level, oxidative phosphorylation abnormalities, and mPT induction [168]. All these processes lead to the release of various small molecules, such as ATP and succinate, and macromolecules, or their fragments, such as mitochondrial DNA, RNA, transcription factor, cytochrome C, cardiolipin and N-formyl peptides, collectively referred to as mitochondria-derived damage-associated molecular patterns (mtDAMPs) [169]. The mtDAMPs are recognised by their associated pattern recognition receptors that induce various inflammatory processes, overactivation of which is responsible for the development of sepsis and septic shock [170]. Due to mitochondrion’s prokaryotic origin, mtDAMPs are similar to molecular patterns of most pathogens, and are likely to trigger excessive inflammatory reactions, which can aggravate sepsis to multi-organ dysfunction syndrome and death [169]. Activation of the inflammatory processes by mtDAMP can further damage mitochondria, forming a vicious circle that is a potential target regarding therapy. Inflammasome and mtDAMP receptor antagonists are suggested for this purpose [170], although repurposing of FDA approved PARP inhibitors for sepsis therapy may be justified [4]. Results on the various experimental sepsis and endotoxemia models in animals are certainly promising, since PARP inhibitors in these models improved survival and diminished multi-organ failure [171].
6.2. Atherosclerosis and Myocardial Infarction
In atherosclerosis, the chronic inflammation, cell damage, and arterial vascular endothelial dysfunction initiated by a physical or chemical injury is exaggerated by vascular cell adhesion molecules-induced adherence of neutrophils, lymphocytes, and monocytes to the endothelial cells [172]. The resulting elevated permeability of the vascular endothelium to endothelial cells and smooth muscle cells is caused by endothelial glycocalyx disruption and inhibition of voltage-sensitive K+, and Ca2+-activated K+ channels. Gradual accumulation of lipid and smooth muscle cells in the vessel intima leads to formation of atherosclerotic plaques, which produce pro-inflammatory factors inducible NOS, and ROS, thereby aggravating the disease [173]. ONOO− formed from NO˙ and O2−˙ enters the nucleus of endothelial cells, and induces DNA strand break-mediated PARP1 activation [174]. In addition to contributing to the vascular inflammation and endothelial dysfunction, PARP1 can facilitate atherosclerotic plaque disruption by activating matrix metalloproteinases (MMPs) [174]. Moderate PARP activation leads to AIF or caspase-mediated apoptosis while overactivation causes atherosclerotic plaque disruption and necrotic cell death [172]. PARP inhibitors were demonstrated to ameliorate all these effects in various experimental systems [172,173,174].
Acute myocardial infarction is followed by reperfusion, due to either coronary thrombolytic therapy or surgical intervention [175]. However, the return of oxygen to a respiratory chain that has suffered hypoxic damages during the preceding ischemia results in a substantial perfusion injury characterised by massive ROS and RNS production, upregulated expression of inflammatory cytokines, and PARP activation [176]. Additionally, the oxidative and nitrosative stress leads to endothelial dysfunction, transendothelial migration of neutrophils, JNK and p38 MAPK activation, DNA fragmentation, and myocardial injury [174]. MMP-2 activated by ONOO− cleaves troponin I, which results in impaired myocardial contractility [177]. PARP1 activation is sustained long after the coronary reperfusion in the necrotic zone and the penumbra, resulting in impeded systolic and diastolic function of the left ventricle, Ca2+ overload, and eventual cardiomyocyte death [178]. PARP inhibition achieved either by pharmacological means or by genetic manipulation significantly diminished the said detrimental effects in various experimental models [133,173,174,176]. Hearts of PARP1-deficient mice were reported to resist ischemic myocardial depression as well as reperfusion injury [179]. In Langendorff-perfused isolated rat hearts, PARP inhibitors were demonstrated to improve ATP and creatine phosphate recovery, attenuate ischemia–reperfusion-induced lipid peroxidation, protein oxidation, DNA strand breaks, and inactivation of respiratory complexes [133]. In a porcine myocardial infarction model, the PARP inhibitor PJ34 was reported to reduce infarct size by 25.7% and forestall myocardial PAR formation in the area [180]. In a rat model of arterial restenosis following angioplasty, PJ34 reduces leukocyte infiltration, neointima formation, and carotid artery restenosis, and improves functional recovery [181]. Finally, in a human clinical study of 40 patients of ST segment elevation myocardial infarction, the PARP inhibitor INO-1001 diminishes serum PARP activity and reduced inflammation [182].
6.3. Stroke
Ischemic stroke is caused by occlusion or collapse of one or more arteries of the central nervous system (CNS), resulting in critically reduced blood flow in a region. Although energy demand of the CNS is high, neuron and glia cells can survive without oxygen up to several hours on glycolysis, despite its meagre energy yield of 2 ATP/glucose molecule—a possible scenario in the ischemic penumbra. However, they succumb within minutes once both oxygen and glucose are deprived—the situation in the core of the ischemic area [183]. Since the brain can exclusively utilise glucose regarding energy production, blocking glycolysis results in deprivation of fuel molecules (pyruvate) from the mitochondrial ATP-producing machinery (pyruvate dehydrogenase complex, citrate cycle, oxidative phosphorylation), causing complete energy failure, even in the presence of oxygen [155]. ATP shortage is aggravated by the temporary reverse operation of the ATP synthase, which consumes rather than produces ATP in a futile attempt to maintain ΔΨm until fuel for substrate-level phosphorylation runs out [49]. The resulting ATP depletion collapses the cell membrane potential, enabling the influx of extracellular Ca2+ via the voltage-gated Ca2+ channels and reversing the glutamate uptake transporters responsible for the majority of glutamate release during ischemia [184]. Reperfusion, which is restoration of the blood flow, still may kill the cells by excitotoxicity, oxidative stress, inflammatory processes, apoptosis, or parthanatos [183]. Excitotoxicity is caused by the sustained action of glutamate on primarily the N-methyl-D-aspartate type of glutamate receptors and results in ONOO− and ROS production-mediated DNA damage accumulation, which activate PARP1 [185]. However, PARP1-induced NAD+ depletion affects the mitochondrial NAD+ pool only, if opening of the high conductance PTP allows NAD+ traffic between the matrix and the cytosol [186]. Even the transient opening of the PTP that does not harm the inner and outer membranes permits NAD+ release from the mitochondria, indicating the pivotal importance of mPT in mediating ischemia–reperfusion-induced impairment of mitochondrial energy production [187]. As an experimental confirmation, focal ischemia decreases infarct volume dramatically in animals deficient in cyclophilin D [188], the matrix peptidyl-propyl cis-trans isomerase, which desensitise Ca2+-induced opening of the PTP [189]. To demonstrate their potential in human stroke therapy [4], PARP inhibitors were reported to reduce ROS and RNS production, decrease oxidative DNA damage, protein oxidation, and lipid peroxidation, diminish infarct volume, and improve mitochondrial functions in various animal models regarding hypoglycemia as well as focal and global ischemia [155,186].
6.4. Neuroegenarative Diseases
Alzheimer’s disease is a type of amyloidosis of the CNS, leading to progressive cognitive decline. Senile plaques of β-amyloid peptide and neurofibrillary tangles formed by hyperphosphorylated tau are the pathological hallmarks of the disease [190]. Pathogenesis of the disease involves oxidative mtDNA damage, leading to accumulation of mutations, decreased mtDNA transcription, and mtDNA copy number, eventually resulting in mitochondrial dysfunction [190]. The ensuing ROS production activates the Nrf-2 antioxidant defence pathway while the β-amyloid peptide causes mitochondrial fragmentation via DRP1 [191,192]. Inhibition of DRP1 prevented mitochondrial fragmentation, mitochondrial dysfunction, decreased β-secretase 1 expression and β-amyloid deposition, and improved cognitive function of transgenic mice bearing human familial Alzheimer’s disease genes [193,194].
Parkinson’s disease is characterised by motor dysfunction and dementia. The pathology features the loss of dopaminergic neurons and accumulation of α-synuclein deposits (Lewy bodies) in the substantia nigra [195]. Interaction of α-synuclein with respiratory complex I as well as outer mitochondrial membrane proteins TOM20 and VDAC, contributes to mitochondrial dysfunction, oxidative stress, and the resulting neuropathological changes [195]. The PTEN-induced putative kinase (PINK)1 and Parkin, an E3 ubiquitin ligase, participate in mitochondrial quality control via mitophagy. Their mutation leads to ubiquitinylation, then proteosomal degradation of Mfn, prevention of mitochondrial fusion, and premature mitophagy [196]. The protein deglycase DJ-1 translocates to the mitochondria upon oxidative stress-induced acidification of its Cys106, where it strengthens the antioxidant defence system by stabilising and preventing the degradation of Nrf-2 [197]. Activating mutation of the leucine-rich repeat kinase 2 increases DRP1 Ser616 phosphorylation, thereby activating mitochondrial fission and mitophagy [198]. High-temperature requirement protein A2 participates in the mitochondrial quality control by degrading denatured proteins inside the mitochondria. This intermembrane protease is released upon apoptotic stimuli, binds inhibitor of apoptosis proteins, then cleaves caspases and other proteins leading to apoptotic death [191].
Huntington’s disease, characterised by chorea, dystonia, incoordination, and cognitive decline, is caused by CAG trinucleotide repeat expansion in the huntingtin gene, manifesting in polyglutamine repeats in the huntingtin protein [199]. Mitochondrial respiration and ATP synthesis are limited by impeded operation of respiratory complexes II and III [200]. Mutant huntingtin reduces PGC-1α expression via binding to CREB and transcription factor IID subunit 4-activating transcription factors [201]. Additionally, increased Ca2+ influx activates transglutaminase-2 that reduces PGC-1α expression by stabilising chromosomal structure, resulting in mtDNA lesions and depletion. Mutant huntingtin activates DRP1 and FIS-1 while downregulates Mfn1/2, thereby facilitating mitochondrial fragmentation and mitophagy [192]. Also, it can promote apoptosis by interacting with p53 or CBP transcription factors and PUMA or Bax activation, respectively [202]. Currently, therapy is restricted to ameliorate symptoms and there are no disease-modifying treatments yet available. However, PARP-1 inhibition was reported to protect striatal interneurons in the R6/2 mouse model of Huntington’s disease [203,204].
7. Concluding Remarks
The intended extension of PARP inhibitors in cancer therapy aims at the non-homologous recombination DNA repair systems, signalling pathways, angiogenesis, or immune checkpoint mechanisms, all utilising the function of PARP in DNA repair. However, PARP activation affects multiple cellular processes in addition that may form the basis of repurposing PARP inhibitors for non-oncological indications [4]. In many diseases associated with oxidative stress, PARP activation has nuclear effects such as the NAD+ and ATP depletion-mediated necrosis and various extra-nuclear effects on cytosolic and mitochondrial processes. Although the pathomechanism of oxidative stress-associated diseases is multiform, some mitochondrial processes such as ROS production and mPT seemingly have a significant role in most of these diseases. Unfortunately, the said processes are initiated via multiple mechanisms in addition to PARP activation, therefore, inhibition of the enzyme does not necessarily prevent them. The combination of PARP inhibition with an antioxidant or mPT inhibiting agent may represent a solution, and cyclophilin D-deficient animals could serve as a useful experimental system in this respect. A substantial amount of preclinical data is available in reference to the beneficial effects of PARP inhibitors in cell culture, isolated organ or animal models of various oxidative stress-associated human diseases. However, they often fall outside the interest of investigators in the field of cancer research. Additionally, the translational power of such data is questionable. The models fail to recapture timescale and complexity of the disease, therefore only clinical studies can establish the realistic potentiality of PARP inhibitors in a given disease. Although the FDA approval of PARP inhibitors for human cancer therapy created the opportunity, there is an understandable reluctance to administer to patients a substance that regulates such a fundamental cellular process as DNA repair. However, a better understanding of the disease’s pathomechanisms, and accumulating data regarding combination treatments aiming at a coordinated, or ideally, synergistic effect will certainly aid in introducing PARP inhibitors to oxidative stress-associated diseases. Presenting recent data and models of PARP inhibitor-induced processes protecting the mitochondria against the detrimental effects of oxidative stress in this review may hopefully stimulate further preclinical and clinical research in this field.
Author Contributions
Conceptualization, B.S. and F.G.J.; writing—original draft preparation, F.G.J.; writing—review and editing, F.G.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Hungarian grants GINOP-2.3.3-15-2016-00025, GINOP-2.3.2-15-2016-00049, EFOP-3.6.1-16-2016-00004, and Higher Education Institutional Excellence Program.
Acknowledgments
The authors thank the technical help of Laszlo Giran in preparing the figures. We apologize to those authors whose study we have failed to cite.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| AMPK | AMP-activated protein kinase |
| Apaf | apoptotic peptidase-activating factor |
| ARE | antioxidant response element |
| ASK1 | apoptosis signal-regulating kinase |
| ATF | activating transcription factor |
| ATM | ataxia telangiectasia mutated kinase |
| Bad | Bcl-2 associated agonist of cell death |
| Bak | Bcl-2 homologous antagonist/killer |
| Bax | Bcl-2-associated X |
| Bcl | B-cell lymphoma protein |
| BH | Bcl-2 homology domain |
| Bid | BH3 interacting-domain death agonist |
| Bim | Bcl-2-like protein 11 |
| BRCA | breast cancer gene |
| CBP | CREB binding protein |
| CD38 | cyclic ADP-ribose hydrolase |
| CNS | central nervous system |
| CREB | cAMP response element-binding protein |
| DLK | dual leucine zipper-bearing kinase |
| ΔΨm | mitochondrial membrane potential |
| DRP | dynamin-like protein |
| ERK | extracellular signal-regulated kinase |
| FDA | United States Food and Drug Administration |
| FIS | fission protein |
| GSK | glycogen synthase kinase |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| Mcl | myeloid leukemia cell differentiation protein |
| MEK | MAPK kinase |
| MEKK | MAPK kinase kinase |
| Mfn | mitofusin |
| MKK4/7 | dual specificity MAPK kinases 4 and 7 |
| MKP | MAPK phosphatase |
| MMP | matrix metalloproteinase |
| mPT | mitochondrial permeability transition |
| mtDAMP | mitochondria-derived damage-associated molecular pattern |
| mTOR | mechanistic (previously mammalian) target of rapamycin |
| mTORC | mTOR complex |
| NEMO | nuclear inhibitor of NF-κB kinase-γ |
| NF-κB | nuclear factor-κB |
| NOS | nitric oxide synthase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| OPA | optic atrophy protein |
| OMA | mitochondrial metalloendopeptidase |
| PAR | poly(ADP-ribose) (PAR) polymerase |
| PARP | poly(ADP-ribose) polymerase |
| PGC-1α | peroxisome proliferator–activated receptor γ coactivator-1α |
| PI3K | phosphatidylinositol-3 kinase |
| PINK | PTEN-induced putative kinase |
| PTEN | phosphatase and tensin homologue deleted on chromosome 10 |
| PTP | permeability transition pore |
| PUMA | p53 upregulated modulator of apoptosis |
| Raf | rapidly accelerated fibrosarcoma protein |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SAB | mitochondrial SH3-domain binding protein 5 |
| SIRT | sirtuin; mono-ADP-ribosyltransferase |
| Smac | second mitochondria-derived activator of caspase |
| TIM | translocase of the inner membrane |
| TOM | translocase of the outer membrane |
| VDAC | voltage-dependent anion channel |
References
- Min, A.; Im, S.A. PARP Inhibitors as therapeutics: Beyond modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.H.; Chen, S.H.; Yu, X. Poly-ADP ribosylation in DNA damage response and cancer therapy. Mutat. Res. 2019, 780, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Burkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Bai, P.; de Murcia, J.M.; de Murcia, G. PARP-1, PARP-2 and ATM in the DNA damage response: Functional synergy in mouse development. DNA Repair. 2004, 3, 1103–1108. [Google Scholar] [CrossRef]
- Thomas, C.; Tulin, A.V. Poly-ADP-ribose polymerase: Machinery for nuclear processes. Mol. Aspects Med. 2013, 34, 1124–1137. [Google Scholar] [CrossRef] [Green Version]
- Vida, A.; Marton, J.; Miko, E.; Bai, P. Metabolic roles of poly(ADP-ribose) polymerases. Semin. Cell Dev. Biol. 2017, 63, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Cseh, A.M.; Fabian, Z.; Quintana-Cabrera, R.; Szabo, A.; Eros, K.; Soriano, M.E.; Gallyas, F.; Scorrano, L.; Sumegi, B. PARP inhibitor PJ34 protects mitochondria and induces DNA-Damage mediated apoptosis in combination with cisplatin or temozolomide in B16F10 melanoma cells. Front Physiol. 2019, 10, 538. [Google Scholar] [CrossRef] [Green Version]
- Frey, T.G.; Mannella, C.A. The internal structure of mitochondria. Trends Biochem. Sci. 2000, 25, 319–324. [Google Scholar] [CrossRef]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid. Res. 2014, 55, 1–16. [Google Scholar] [CrossRef]
- Sonveaux, P.; Maechler, P.; Martinou, J.C. Channels and transporters in cell metabolism. Biochim. Biophys. Acta 2016, 1863, 2359–2361. [Google Scholar] [CrossRef] [PubMed]
- Capt, C.; Passamonti, M.; Breton, S. The human mitochondrial genome may code for more than 13 proteins. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 3098–3101. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Bagkos, G.; Koufopoulos, K.; Piperi, C. A new model for mitochondrial membrane potential production and storage. Med. Hypotheses 2014, 83, 175–181. [Google Scholar] [CrossRef]
- Nath, S. Integration of demand and supply sides in the ATP energy economics of cells. Biophys. Chem. 2019, 252, 106208. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Garlid, K.D.; Paucek, P. Mitochondrial potassium transport: The K(+) cycle. Biochim. Biophys. Acta 2003, 1606, 23–41. [Google Scholar] [CrossRef] [Green Version]
- Hansford, R.G.; Zorov, D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol. Cell Biochem. 1998, 184, 359–369. [Google Scholar] [CrossRef]
- Maguire, M.E. Magnesium transporters: Properties, regulation and structure. Front Biosci. 2006, 11, 3149–3163. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.G.; Rudler, D.L.; Rackham, O.; Filipovska, A. Is mitochondrial gene expression coordinated or stochastic? Biochem. Soc. Trans. 2018, 46, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Attardi, G.; Schatz, G. Biogenesis of mitochondria. Annu. Rev. Cell Biol. 1988, 4, 289–333. [Google Scholar] [CrossRef]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Philos. Soc. 1966, 41, 445–502. [Google Scholar] [CrossRef]
- Paucek, P.; Mironova, G.; Mahdi, F.; Beavis, A.D.; Woldegiorgis, G.; Garlid, K.D. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J. Biol. Chem. 1992, 267, 26062–26069. [Google Scholar]
- Nicholls, D.G. Mitochondria and calcium signaling. Cell Calcium 2005, 38, 311–317. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Scanlon, J.M.; Brocard, J.B.; Stout, A.K.; Reynolds, I.J. Pharmacological investigation of mitochondrial Ca(2+) transport in central neurons: Studies with CGP-37157, an inhibitor of the mitochondrial Na(+)-Ca(2+) exchanger. Cell Calcium 2000, 28, 317–327. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Denton, R.M. Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev. Neurosci. 1993, 15, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Harris, D.A. Control of mitochondrial ATP synthase in heart cells: Inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 1990, 24, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Wernette, M.E.; Ochs, R.S.; Lardy, H.A. Ca2+ stimulation of rat liver mitochondrial glycerophosphate dehydrogenase. J. Biol. Chem. 1981, 256, 12767–12771. [Google Scholar] [PubMed]
- Mildaziene, V.; Baniene, R.; Nauciene, Z.; Bakker, B.M.; Brown, G.C.; Westerhoff, H.V.; Kholodenko, B.N. Calcium indirectly increases the control exerted by the adenine nucleotide translocator over 2-oxoglutarate oxidation in rat heart mitochondria. Arch. Biochem. Biophys. 1995, 324, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Sohal, R.S.; Agarwal, A.; Agarwal, S.; Orr, W.C. Simultaneous overexpression of copper- and zinc-containing superoxide dismutase and catalase retards age-related oxidative damage and increases metabolic potential in Drosophila melanogaster. J. Biol. Chem. 1995, 270, 15671–15674. [Google Scholar] [CrossRef] [Green Version]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Cleeter, M.W.; Cooper, J.M.; Darley-Usmar, V.M.; Moncada, S.; Schapira, A.H. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994, 345, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.; Darley-Usmar, V.M. Hypothesis: The mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidative cell signals: An alternative function for cytochrome C oxidase. Free Radic. Biol. Med. 2002, 32, 370–374. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Brand, M.D. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem. Soc. Trans. 2003, 31, 1300–1301. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Penicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Lee, H.C.; Kumar, C.; Chance, B. Comparison of the effect of a mitochondrial uncoupler, 2,4-dinitrophenol and adrenaline on oxygen radical production in the isolated perfused rat liver. Acta Physiol. Scand 1992, 145, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [Green Version]
- Huser, J.; Blatter, L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 1999, 343, 311–317. [Google Scholar] [CrossRef]
- Gunter, T.E.; Buntinas, L.; Sparagna, G.; Eliseev, R.; Gunter, K. Mitochondrial calcium transport: Mechanisms and functions. Cell Calcium 2000, 28, 285–296. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondria as ATP consumers in cellular pathology. Biochim. Biophys. Acta 2010, 1802, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The mitochondrial permeability transition pore: Channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial permeability transition pore: A potential drug target for neurodegeneration. Drug. Discov. Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [Green Version]
- Bereiter-Hahn, J.; Voth, M. Dynamics of mitochondria in living cells: Shape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 1994, 27, 198–219. [Google Scholar] [CrossRef]
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium 2008, 44, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Baloh, R.H. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist 2008, 14, 12–18. [Google Scholar] [CrossRef]
- Dagda, R.K.; Chu, C.T. Mitochondrial quality control: Insights on how Parkinson‘s disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J. Bioenerg. Biomembr. 2009, 41, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Danino, D.; Moon, K.H.; Hinshaw, J.E. Rapid constriction of lipid bilayers by the mechanochemical enzyme dynamin. J. Struct Biol. 2004, 147, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Malka, F.; Guillery, O.; Cifuentes-Diaz, C.; Guillou, E.; Belenguer, P.; Lombes, A.; Rojo, M. Separate fusion of outer and inner mitochondrial membranes. Embo Rep. 2005, 6, 853–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [PubMed]
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnaune-Pelloquin, L.; Davezac, N.; Mils, V.; Miguel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernandez-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- Delettre, C.; Griffoin, J.M.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease—The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Z.; Tschopp, O.; Baudry, A.; Dummler, B.; Hynx, D.; Hemmings, B.A. Physiological functions of protein kinase B/Akt. Biochem. Soc. T. 2004, 32, 350–354. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediated mitochondrial protection in the heart: Metabolic and survival pathways to the rescue. J. Bioenerg. Biomembr. 2009, 41, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.A.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi, S.; Alidadiani, N.; Dilaver, N.; Heidari, H.R.; Parvizi, R.; Rahbarghazi, R.; Soleimani-Rad, J.; Baradaran, B. Role of glycogen synthase kinase following myocardial infarction and ischemia-reperfusion. Apoptosis 2017, 22, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Woodgett, J. Glycogen synthase kinase-3 and cancer: Good cop, bad cop? Cancer Cell 2008, 14, 351–353. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, Y.L.; Zhu, J.; Lei, S.P.; Dong, Y.; Li, L.Y.; Jiang, B.B.; Tan, L.; Wu, J.X.; Yu, S.S.; et al. GSK-3 beta downregulates Nrf2 in cultured cortical neurons and in a rat model of cerebral ischemia-reperfusion. Sci. Rep. 2016, 6, 20196. [Google Scholar] [CrossRef] [Green Version]
- Goetschel, F.; Kern, C.; Lang, S.; Sparna, T.; Markmann, C.; Schwager, J.; McNelly, S.; Weizsacker, F.; Laufer, S.; Hecht, A.; et al. Inhibition of GSK3 differentially modulates NF-kappa B, CREB, AP-1 and beta-catenin signaling in hepatocytes, but fails to promote TNF-alpha-induced apoptosis. Exp. Cell Res. 2008, 314, 1351–1366. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Pap, M.; Cooper, G.M. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, K.Y.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- KauffmanZeh, A.; RodriguezViciana, P.; Ulrich, E.; Gilbert, C.; Coffer, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 362. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1 alpha transcriptional complex. Nature 2007, 450, 736-U712. [Google Scholar] [CrossRef]
- Meier, R.; Alessi, D.R.; Cron, P.; Andjelkovic, M.; Hemmings, B.A. Mitogenic activation, phosphorylation, and nuclear translocation of protein kinase B beta. J. Biol. Chem. 1997, 272, 30491–30497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Dijkers, P.F.; Medema, R.H.; Lammers, J.W.J.; Koenderman, L.; Coffer, P.J. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 2000, 10, 1201–1204. [Google Scholar] [CrossRef] [Green Version]
- Obexer, P.; Geiger, K.; Ambros, P.F.; Meister, B.; Ausserlechner, M.J. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007, 14, 534–547. [Google Scholar] [CrossRef] [Green Version]
- Simpson, L.; Parsons, R. PTEN: Life as a tumor suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapodi, A.; Debreceni, B.; Hanto, K.; Bognar, Z.; Wittmann, I.; Gallyas, F., Jr.; Varbiro, G.; Sumegi, B. Pivotal role of Akt activation in mitochondrial protection and cell survival by poly(ADP-ribose)polymerase-1 inhibition in oxidative stress. J. Biol. Chem. 2005, 280, 35767–35775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Clarke, S.J.; Khaliulin, I. The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 2007, 1767, 1007–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef]
- Veres, B.; Gallyas, F., Jr.; Varbiro, G.; Berente, Z.; Osz, E.; Szekeres, G.; Szabo, C.; Sumegi, B. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochem. Pharmacol. 2003, 65, 1373–1382. [Google Scholar] [CrossRef]
- Gallyas, F., Jr.; Sumegi, B.; Szabo, C. Role of Akt activation in PARP inhibitor resistance in cancer. Cancers 2020, 12, 532. [Google Scholar] [CrossRef] [Green Version]
- Felici, R.; Cavone, L.; Lapucci, A.; Guasti, D.; Bani, D.; Chiarugi, A. PARP inhibition delays progression of mitochondrial encephalopathy in mice. Neurotherapeutics 2014, 11, 651–664. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Singh, N.; Lingeshwar, P.; Siddiqui, H.H.; Hanif, K. Poly (ADP-ribose) polymerase-1: An emerging target in right ventricle dysfunction associated with pulmonary hypertension. Pulm. Pharmacol. Ther. 2015, 30, 66–79. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, J.; Wang, X.; Li, H.; Liu, T.; Zhao, W. Poly (ADP-ribose) synthetase inhibitor has a heart protective effect in a rat model of experimental sepsis. Int. J. Clin. Exp. Pathol. 2015, 8, 9824–9835. [Google Scholar]
- Olah, G.; Szczesny, B.; Brunyanszki, A.; Lopez-Garcia, I.A.; Gero, D.; Radak, Z.; Szabo, C. Differentiation-associated downregulation of Poly(ADP-Ribose) Polymerase-1 expression in myoblasts serves to increase their resistance to oxidative stress. PLoS ONE 2015, 10, e0134227. [Google Scholar] [CrossRef] [Green Version]
- Krainz, T.; Lamade, A.M.; Du, L.; Maskrey, T.S.; Calderon, M.J.; Watkins, S.C.; Epperly, M.W.; Greenberger, J.S.; Bayir, H.; Wipf, P.; et al. Synthesis and Evaluation of a Mitochondria-Targeting Poly(ADP-ribose) Polymerase-1 Inhibitor. ACS Chem. Biol. 2018, 13, 2868–2879. [Google Scholar] [CrossRef] [PubMed]
- Tapodi, A.; Bognar, Z.; Szabo, C.; Gallyas, F.; Sumegi, B.; Hocsak, E. PARP inhibition induces Akt-mediated cytoprotective effects through the formation of a mitochondria-targeted phospho-ATM-NEMO-Akt-mTOR signalosome. Biochem. Pharmacol. 2019, 162, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol. Cell 2009, 36, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef] [Green Version]
- Stagni, V.; Manni, I.; Oropallo, V.; Mottolese, M.; Di Benedetto, A.; Piaggio, G.; Falcioni, R.; Giaccari, D.; Di Carlo, S.; Sperati, F.; et al. ATM kinase sustains HER2 tumorigenicity in breast cancer. Nat. Commun. 2015, 6, 6886. [Google Scholar] [CrossRef] [Green Version]
- Halaby, M.-J.; Hibma, J.C.; He, J.; Yang, D.-Q. ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal 2008, 20, 1555–1563. [Google Scholar] [CrossRef]
- Yang, D.-Q.; Halaby, M.-J.; Li, Y.; Hibma, J.C.; Burn, P. Cytoplasmic ATM protein kinase: An emerging therapeutic target for diabetes, cancer and neuronal degeneration. Drug. Discov. Today 2011, 16, 332–338. [Google Scholar] [CrossRef]
- Aguilar-Quesada, R.; Munoz-Gamez, J.A.; Martin-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matinez-Romero, R.; Quiles-Perez, R.; Menissier-de Murcia, J.; de Murcia, G.; Ruiz de Almodovar, M.; et al. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 2007, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Bohrson, C.; Pike, A.M.; Wheelan, S.J.; Greider, C.W. ATM kinase is required for telomere elongation in mouse and human cells. Cell Rep. 2015, 13, 1623–1632. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.-H.; Shi, Y.; Tibbetts, R.S.; Miyamoto, S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 2006, 311, 1141–1146. [Google Scholar] [CrossRef]
- Williamson, C.T.; Muzik, H.; Turhan, A.G.; Zamo, A.; O’Connor, M.J.; Bebb, D.G.; Lees-Miller, S.P. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol. Cancer Ther. 2010, 9, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajawat, J.; Shukla, N.; Mishra, D.P. Therapeutic targeting of Poly(ADP-Ribose) Polymerase-1 (PARP1) in cancer: Current developments, therapeutic strategies, and future opportunities. Med. Res. Rev. 2017, 37, 1461–1491. [Google Scholar] [CrossRef] [PubMed]
- Szanto, A.; Hellebrand, E.E.; Bognar, Z.; Tucsek, Z.; Szabo, A.; Gallyas, F., Jr.; Sumegi, B.; Varbiro, G. PARP-1 inhibition-induced activation of PI-3-kinase-Akt pathway promotes resistance to taxol. Biochem. Pharmacol. 2009, 77, 1348–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, A.; Caruso, D.; Strudel, M.; Tomao, S.; Tomao, F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J. Transl. Med. 2016, 14, 267. [Google Scholar] [CrossRef]
- Wang, D.; Wang, M.; Jiang, N.; Zhang, Y.; Bian, X.; Wang, X.; Roberts, T.M.; Zhao, J.J.; Liu, P.; Cheng, H. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget 2016, 7, 13153–13166. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, F.; Wilkinson, M.; Baxter, E.; Brennan, D.J. Mitogen-Activated Protein Kinase (MAPK) and obesity-related cancer. Int. J. Mol. Sci. 2020, 21, 1241. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Papaconstantinou, J. The role of signaling pathways of inflammation and oxidative stress in development of senescence and aging phenotypes in cardiovascular disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef] [Green Version]
- Assi, M. The differential role of reactive oxygen species in early and late stages of cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R646–R653. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Choy, W.C.; Abid, M.R. Direct sensing of endothelial oxidants by vascular endothelial growth factor receptor-2 and c-Src. PLoS ONE 2011, 6, e28454. [Google Scholar] [CrossRef] [Green Version]
- Palit, S.; Kar, S.; Sharma, G.; Das, P.K. hesperetin induces apoptosis in breast carcinoma by triggering accumulation of ROS and activation of ASK1/JNK pathway. J. Cell Physiol. 2015, 230, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Prinz, E.; Aviram, S.; Aronheim, A. WDR62 mediates TNFalpha-dependent JNK activation via TRAF2-MLK3 axis. Mol. Biol. Cell 2018, 29, 2470–2480. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Lin, L.; Tan, X.; Yang, J.; Bu, G.; Mars, W.M.; Liu, Y. tPA protects renal interstitial fibroblasts and myofibroblasts from apoptosis. J. Am. Soc. Nephrol. 2008, 19, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, J.-J.; Sung, P.-H.; Wallace, C.G.; Yang, C.-C.; Chen, K.-H.; Shao, P.-L.; Chu, Y.-C.; Huang, C.-R.; Chen, Y.-L.; Ko, S.-F.; et al. Intravenous administration of iPS-MSCSPIONs mobilized into CKD parenchyma and effectively preserved residual renal function in CKD rat. J. Cell Mol. Med. 2020, 24, 3593–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, M.; Steer, E.K.; Chu, C.T. ERKed by LRRK2: A cell biological perspective on hereditary and sporadic Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Wainstein, E.; Seger, R. The dynamic subcellular localization of ERK: Mechanisms of translocation and role in various organelles. Curr. Opin. Cell Biol. 2016, 39, 15–20. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Kaplowitz, N. The regulation of JNK signaling pathways in cell death through the interplay with mitochondrial SAB and upstream post-translational effects. Int. J. Mol. Sci. 2018, 19, 3657. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, X.; Tan, S.; Liu, X.; Zhao, X.; Yuan, Z.; Nie, C. Mcl-1 expression and JNK activation induces a threshold for apoptosis in Bcl-xL-overexpressing hematopoietic cells. Oncotarget 2017, 8, 11042–11052. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: Therapeutic perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Qin, X.; Cai, X.; Yang, L.; Xing, Y.; Li, J.; Zhang, L.; Tang, Y.; Liu, J.; Zhang, X.; et al. Mitochondrial JNK activation triggers autophagy and apoptosis and aggravates myocardial injury following ischemia/reperfusion. Biochim. Biophys. Acta 2015, 1852, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Min, R.W.M.; Le, K.; Zhou, S.; Aghajan, M.; Than, T.A.; Win, S.; Kaplowitz, N. The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis. 2017, 8, e2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdelyi, K.; Pacher, P.; Virag, L.; Szabo, C. Role of poly(ADP-ribosyl)ation in a ’two-hit‘ model of hypoxia and oxidative stress in human A549 epithelial cells in vitro. Int. J. Mol. Med. 2013, 32, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Halmosi, R.; Berente, Z.; Osz, E.; Toth, K.; Literati-Nagy, P.; Sumegi, B. Effect of poly(ADP-ribose) polymerase inhibitors on the ischemia-reperfusion-induced oxidative cell damage and mitochondrial metabolism in Langendorff heart perfusion system. Mol. Pharmacol. 2001, 59, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Mester, L.; Szabo, A.; Atlasz, T.; Szabadfi, K.; Reglodi, D.; Kiss, P.; Racz, B.; Tamas, A.; Gallyas, F., Jr.; Sumegi, B.; et al. Protection against chronic hypoperfusion-induced retinal neurodegeneration by PARP inhibition via activation of PI-3-kinase Akt pathway and suppression of JNK and p38 MAP kinases. Neurotox. Res. 2009, 16, 68–76. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, S.; Liu, Z.G.; Han, J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J. Biol. Chem. 2006, 281, 8788–8795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alano, C.C.; Swanson, R.A. Players in the PARP-1 cell-death pathway: JNK1 joins the cast. Trends Biochem. Sci. 2006, 31, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Konta, T.; Nakayama, K.; Furusu, A.; Moreno-Manzano, V.; Lucio-Cazana, J.; Ishikawa, Y.; Fine, L.G.; Yao, J.; Kitamura, M. Cellular defense against H2O2-induced apoptosis via MAP kinase-MKP-1 pathway. Free Radic. Biol. Med. 2004, 36, 985–993. [Google Scholar] [CrossRef]
- Racz, B.; Hanto, K.; Tapodi, A.; Solti, I.; Kalman, N.; Jakus, P.; Kovacs, K.; Debreceni, B.; Gallyas, F., Jr.; Sumegi, B. Regulation of MKP-1 expression and MAPK activation by PARP-1 in oxidative stress: A new mechanism for the cytoplasmic effect of PARP-1 activation. Free Radic. Biol. Med. 2010, 49, 1978–1988. [Google Scholar] [CrossRef]
- Wong, H.R.; Dunsmore, K.E.; Page, K.; Shanley, T.P. Heat shock-mediated regulation of MKP-1. Am. J. Physiol. Cell Physiol. 2005, 289, C1152–C1158. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.-C.; Wang, Z.; Feng, X.; Chuang, P.; Fang, W.; Chen, Y.; Neves, S.; Maayan, A.; Xiong, H.; Liu, Y.; et al. Retinoic acid utilizes CREB and USF1 in a transcriptional feed-forward loop in order to stimulate MKP1 expression in human immunodeficiency virus-infected podocytes. Mol. Cell Biol. 2008, 28, 5785–5794. [Google Scholar] [CrossRef] [Green Version]
- Hocsak, E.; Szabo, V.; Kalman, N.; Antus, C.; Cseh, A.; Sumegi, K.; Eros, K.; Hegedus, Z.; Gallyas, F., Jr.; Sumegi, B.; et al. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic. Biol. Med. 2017, 108, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Mericskay, M. Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch. Cardiovasc. Dis. 2016, 109, 207–215. [Google Scholar] [CrossRef]
- Bai, P.; Nagy, L.; Fodor, T.; Liaudet, L.; Pacher, P. Poly(ADP-ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol. Metab. 2015, 26, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sileikyte, J.; Forte, M. The Mitochondrial Permeability Transition in Mitochondrial Disorders. Oxid. Med. Cell Longev. 2019, 2019, 3403075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Berger, N.A.; Sims, J.L.; Catino, D.M.; Berger, S.J. Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: Studies in normal and DNA-repair defective cells. Princess Takamatsu Symp. 1983, 13, 219–226. [Google Scholar]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Nicotinamide Adenine Dinucleotide Metabolism and Neurodegeneration. Antioxid Redox Signal 2018, 28, 1652–1668. [Google Scholar] [CrossRef]
- Di Lisa, F.; Menabo, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Omuro, H.; Liu, Y.-F.; Soya, M.; Shima, T.; McEwen, B.S.; Soya, H. Astrocytic glycogen-derived lactate fuels the brain during exhaustive exercise to maintain endurance capacity. Proc. Natl. Acad. Sci. USA 2017, 114, 6358–6363. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Vargas, J.M.; Rodriguez, M.I.; Majuelos-Melguizo, J.; Garcia-Diaz, A.; Gonzalez-Flores, A.; Lopez-Rivas, A.; Virag, L.; Illuzzi, G.; Schreiber, V.; Dantzer, F.; et al. Autophagy requires poly(adp-ribosyl)ation-dependent AMPK nuclear export. Cell Death Differ. 2016, 23, 2007–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alano, C.C.; Ying, W.; Swanson, R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 2004, 279, 18895–18902. [Google Scholar] [CrossRef] [Green Version]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.; Chen, Y.; Xu, Y.; Swanson, R.A. Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: A treatable cause of cell death in stroke. Transl. Stroke Res. 2014, 5, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C. Acute sources of mitochondrial NAD+ during respiratory chain dysfunction. Exp. Neurol. 2020, 327, 113218. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Seyfried, T.N. Mitochondrial Substrate-Level Phosphorylation as Energy Source for Glioblastoma: Review and Hypothesis. ASN Neuro. 2018, 10, 1759091418818261. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Xu, R.H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.M.; Ramdas, L.; Hu, L.M.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 2006, 175, 913–923. [Google Scholar] [CrossRef]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 30 disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrabi, S.A.; Umanah, G.K.E.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.-P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and quality control mechanisms inmitochondrial maintenance. Curr. Biol. CB 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322. [Google Scholar] [CrossRef]
- Garrabou, G.; Moren, C.; Lopez, S.; Tobias, E.; Cardellach, F.; Miro, O.; Casademont, J. The effects of sepsis on mitochondria. J. Infect. Dis. 2012, 205, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Arulkumaran, N.; Deutschman, C.S.; Pinsky, M.R.; Zuckerbraun, B.; Schumacker, P.T.; Gomez, H.; Gomez, A.; Murray, P.; Kellum, J.A.; Workgroup, A.X. Mitochondrial function in sepsis. Shock 2016, 45, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Hu, Q.; Huang, J.; Wu, X.; Ren, J. Mitochondria-derived damage-associated molecular patterns in sepsis: from bench to bedside. Oxid. Med. Cell Longev. 2019, 2019, 6914849. [Google Scholar] [CrossRef]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Kunze, F.A.; Hottiger, M.O. Regulating immunity via ADP-Ribosylation: Therapeutic implications and beyond. Trends Immunol. 2019, 40, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Atherosclerosis can be mitochondrial: A review. Cureus 2020, 12, e6987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.J.; Bourgeois, M.; Harbison, R.D. Poly(ADP-ribose) Polymerase (PARP) and PARP inhibitors: Mechanisms of action and role in cardiovascular disorders. Cardiovasc. Toxicol. 2018, 18, 493–506. [Google Scholar] [CrossRef]
- De Roeck, L.; Vandamme, S.; Everaert, B.R.; Hoymans, V.; Haine, S.; Vandendriessche, T.; Bosmans, J.; Ronsyn, M.W.; Miljoen, H.; Van Berendoncks, A.; et al. Adiponectin and ischemia-reperfusion injury in ST segment elevation myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2016, 5, 71–76. [Google Scholar] [CrossRef]
- Halmosi, R.; Deres, L.; Gal, R.; Eros, K.; Sumegi, B.; Toth, K. PARP inhibition and postinfarction myocardial remodeling. Int. J. Cardiol. 2016, 217, S52–S59. [Google Scholar] [CrossRef]
- Andreadou, I.; Iliodromitis, E.K.; Lazou, A.; Gorbe, A.; Giricz, Z.; Schulz, R.; Ferdinandy, P. Effect of hypercholesterolaemia on myocardial function, ischaemia-reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2017, 174, 1555–1569. [Google Scholar] [CrossRef]
- Medina de Chazal, H.; Del Buono, M.G.; Keyser-Marcus, L.; Ma, L.; Moeller, F.G.; Berrocal, D.; Abbate, A. Stress cardiomyopathy diagnosis and treatment: JACC State-of-The-Art review. J. Am. Coll. Cardiol. 2018, 72, 1955–1971. [Google Scholar] [CrossRef]
- Yamazaki, K.; Tanaka, S.; Sakata, R.; Miwa, S.; Oriyanhan, W.; Takaba, K.; Minakata, K.; Marui, A.; Ikeda, T.; Toyokuni, S.; et al. Protective effect of cardioplegia with poly (ADP-ribose) polymerase-1 inhibitor against myocardial ischemia-reperfusion injury: In vitro study of isolated rat heart model. J. Enzyme. Inhib. Med. Chem. 2013, 28, 143–147. [Google Scholar] [CrossRef]
- Faro, R.; Toyoda, Y.; McCully, J.D.; Jagtap, P.; Szabo, E.; Virag, L.; Bianchi, C.; Levitsky, S.; Szabo, C.; Sellke, F.W. Myocardial protection by PJ34, a novel potent poly (ADP-ribose) synthetase inhibitor. Ann. Thorac. Surg. 2002, 73, 575–581. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, J.; Jennings, L.K. Attenuation of neointima formation through the inhibition of DNA repair enzyme PARP-1 in balloon-injured rat carotid artery. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H659–H666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, D.A.; Brickman, C.M.; Murphy, S.A.; Baran, K.; Krakover, R.; Dauerman, H.; Kumar, S.; Slomowitz, N.; Grip, L.; McCabe, C.H.; et al. A randomized, placebo-controlled trial to evaluate the tolerability, safety, pharmacokinetics, and pharmacodynamics of a potent inhibitor of poly(ADP-ribose) polymerase (INO-1001) in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention: Results of the TIMI 37 trial. J. Thromb. Thrombolysis 2009, 27, 359–364. [Google Scholar] [PubMed]
- Gerace, E.; Pellegrini-Giampietro, D.E.; Moroni, F.; Mannaioni, G. Poly(ADP-Ribose)Polymerase 1 (PARP-1) Activation and Ca(2+) Permeable alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid (AMPA) channels in post-ischemic brain damage: New therapeutic opportunities? CNS Neurol. Disord Drug Targets 2015, 14, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Magi, S.; Piccirillo, S.; Amoroso, S.; Lariccia, V. Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. Int. J. Mol. Sci. 2019, 20, 5674. [Google Scholar] [CrossRef] [Green Version]
- Owens, K.; Park, J.H.; Schuh, R.; Kristian, T. Mitochondrial dysfunction and NAD(+) metabolism alterations in the pathophysiology of acute brain injury. Transl. Stroke Res. 2013, 4, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Shalbuyeva, N.; Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calcium-dependent spontaneously reversible remodeling of brain mitochondria. J. Biol. Chem. 2006, 281, 37547–37558. [Google Scholar] [CrossRef] [Green Version]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [Green Version]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- Gotz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. 2019, 14, 239–261. [Google Scholar] [CrossRef]
- Huang, M.L.H.; Chiang, S.; Kalinowski, D.S.; Bae, D.-H.; Sahni, S.; Richardson, D.R. The role of the antioxidant response in mitochondrial dysfunction in degenerative diseases: Cross-talk between antioxidant defense, autophagy, and apoptosis. Oxid. Med. Cell Longev. 2019, 2019, 6392763. [Google Scholar] [CrossRef] [Green Version]
- Oliver, D.; Reddy, P.H. Dynamics of Dynamin-related protein 1 in Alzheimer’s disease and other neurodegenerative diseases. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.-H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 ameliorates synaptic depression, Abeta deposition, and cognitive impairment in an Alzheimer’s disease model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects against amyloid-beta induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. (Vienna) 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Abdel-Aleem, G.A.; Khaleel, E.F.; Mostafa, D.G.; Elberier, L.K. Neuroprotective effect of resveratrol against brain ischemia reperfusion injury in rats entails reduction of DJ-1 protein expression and activation of PI3K/Akt/GSK3b survival pathway. Arch. Physiol. Biochem. 2016, 122, 200–213. [Google Scholar] [CrossRef]
- Santos, D.; Esteves, A.R.; Silva, D.F.; Januario, C.; Cardoso, S.M. The impact of mitochondrial fusion and fission modulation in sporadic Parkinson’s Disease. Mol. Neurobiol. 2015, 52, 573–586. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Milakovic, T.; Johnson, G.V.W. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lei, Y.-H.; Zhou, J.-P.; Hou, Y.-Y.; Wan, Z.; Wang, H.-L.; Meng, H. Role of PGC-1alpha in mitochondrial quality control in neurodegenerative diseases. Neurochem. Res. 2019, 44, 2031–2043. [Google Scholar] [CrossRef]
- Cardinale, A.; Paldino, E.; Giampa, C.; Bernardi, G.; Fusco, F.R. PARP-1 inhibition is neuroprotective in the R6/2 mouse model of Huntington’s Disease. PLoS ONE 2015, 10, e0134482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paldino, E.; Cardinale, A.; D’Angelo, V.; Sauve, I.; Giampa, C.; Fusco, F.R. Selective Sparing of Striatal Interneurons after Poly (ADP-Ribose) Polymerase 1 Inhibition in the R6/2 Mouse Model of Huntington’s Disease. Front. Neuroanat. 2017, 11, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Interplay of poly(ADP-ribose) polymerase (PARP) with Akt-mediated mitochondrial protection in oxidative stress. (a) Effects of oxidative stress in the absence of PARP inhibition. ROS-induced DNA strand breaks induce excess PARP activation leading to NAD+ then ATP depletion. PARylated ATM–NEMO are retained in the nucleus. ROS-induced mitochondrial dysfunction contributes to cell death caused by energy failure. (b) Effects of oxidative stress in the presence of PARP inhibition. The PARP inhibitor blocks excess PARP activation and NAD+ consumption. Activated ATM–NEMO complex translocates to the cytoplasm and attaches to the outer mitochondrial membrane. Akt and mTOR are complexed with ATM–NEMO to form an ATM–NEMO–Akt–mTOR signallosome. Akt becomes activated and protect the mitochondria against the ROS-induced damages. Pointed arrows denote activation while arrows with flat end represent inhibition. P indicates phosphorylation. Red and yellow spots designate ADP-ribose and nicotinamide, respectively.
Figure 1.
Interplay of poly(ADP-ribose) polymerase (PARP) with Akt-mediated mitochondrial protection in oxidative stress. (a) Effects of oxidative stress in the absence of PARP inhibition. ROS-induced DNA strand breaks induce excess PARP activation leading to NAD+ then ATP depletion. PARylated ATM–NEMO are retained in the nucleus. ROS-induced mitochondrial dysfunction contributes to cell death caused by energy failure. (b) Effects of oxidative stress in the presence of PARP inhibition. The PARP inhibitor blocks excess PARP activation and NAD+ consumption. Activated ATM–NEMO complex translocates to the cytoplasm and attaches to the outer mitochondrial membrane. Akt and mTOR are complexed with ATM–NEMO to form an ATM–NEMO–Akt–mTOR signallosome. Akt becomes activated and protect the mitochondria against the ROS-induced damages. Pointed arrows denote activation while arrows with flat end represent inhibition. P indicates phosphorylation. Red and yellow spots designate ADP-ribose and nicotinamide, respectively.

Figure 2.
Interplay of PARP with MAPK-mediated mitochondrial damage in oxidative stress. (a) Effects of oxidative stress in the absence of PARP inhibition. ROS-induced DNA strand breaks induce excess PARP activation leading to NAD+, then ATP depletion. PARP PARylates both itself and ATF4, and replaces PARylated ATM4 at the promoter of MKP-1 coding DNA region. ROS activates JNK and p38 MAPK, which exaggerate ROS-induced mitochondrial dysfunction, and contributes to cell death caused by energy failure. (b) Effects of oxidative stress in the presence of PARP inhibition. The PARP inhibitor blocks excess PARP activation and NAD+ consumption. ATF4 is not PARylated, therefore is able to bind to the promoter of MKP-1 coding DNA region. MKP-1 mRNA is transcribed, MKP-1 protein is translated and inactivates JNK and p38 MAPK. The antioxidant defences compensate for the ROS-induced mitochondrial damages. Pointed arrows denote activation while arrows with flat end represent inhibition. P indicates phosphorylation. Red and yellow spots designate ADP-ribose and nicotinamide, respectively.
Figure 2.
Interplay of PARP with MAPK-mediated mitochondrial damage in oxidative stress. (a) Effects of oxidative stress in the absence of PARP inhibition. ROS-induced DNA strand breaks induce excess PARP activation leading to NAD+, then ATP depletion. PARP PARylates both itself and ATF4, and replaces PARylated ATM4 at the promoter of MKP-1 coding DNA region. ROS activates JNK and p38 MAPK, which exaggerate ROS-induced mitochondrial dysfunction, and contributes to cell death caused by energy failure. (b) Effects of oxidative stress in the presence of PARP inhibition. The PARP inhibitor blocks excess PARP activation and NAD+ consumption. ATF4 is not PARylated, therefore is able to bind to the promoter of MKP-1 coding DNA region. MKP-1 mRNA is transcribed, MKP-1 protein is translated and inactivates JNK and p38 MAPK. The antioxidant defences compensate for the ROS-induced mitochondrial damages. Pointed arrows denote activation while arrows with flat end represent inhibition. P indicates phosphorylation. Red and yellow spots designate ADP-ribose and nicotinamide, respectively.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gallyas Jr, F.; Sumegi, B. Mitochondrial Protection by PARP Inhibition. Int. J. Mol. Sci. 2020, 21, 2767. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082767
AMA Style
Gallyas Jr F, Sumegi B. Mitochondrial Protection by PARP Inhibition. International Journal of Molecular Sciences. 2020; 21(8):2767. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082767
Chicago/Turabian StyleGallyas Jr, Ferenc, and Balazs Sumegi. 2020. "Mitochondrial Protection by PARP Inhibition" International Journal of Molecular Sciences 21, no. 8: 2767. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082767
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.