Cystic Fibrosis, CFTR, and Colorectal Cancer

Department of Biomedical Sciences, University of Minnesota Medical School, Duluth, MN 55812, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(8), 2891; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082891
Submission received: 20 March 2020
/
Revised: 17 April 2020
/
Accepted: 19 April 2020
/
Published: 21 April 2020
(This article belongs to the Special Issue Therapeutic Approaches for Cystic Fibrosis)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cystic fibrosis (CF), caused by biallelic inactivating mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, has recently been categorized as a familial colorectal cancer (CRC) syndrome. CF patients are highly susceptible to early, aggressive colorectal tumor development. Endoscopic screening studies have revealed that by the age of forty 50% of CF patients will develop adenomas, with 25% developing aggressive advanced adenomas, some of which will have already advanced to adenocarcinomas. This enhanced risk has led to new CF colorectal cancer screening recommendations, lowering the initiation of endoscopic screening to age forty in CF patients, and to age thirty in organ transplant recipients. The enhanced risk for CRC also extends to the millions of people (more than 10 million in the US) who are heterozygous carriers of CFTR gene mutations. Further, lowered expression of CFTR is reported in sporadic CRC, where downregulation of CFTR is associated with poor survival. Mechanisms underlying the actions of CFTR as a tumor suppressor are not clearly understood. Dysregulation of Wnt/β-catenin signaling and disruption of intestinal stem cell homeostasis and intestinal barrier integrity, as well as intestinal dysbiosis, immune cell infiltration, stress responses, and intestinal inflammation have all been reported in human CF patients and in animal models. Notably, the development of new drug modalities to treat non-gastrointestinal pathologies in CF patients, especially pulmonary disease, offers hope that these drugs could be repurposed for gastrointestinal cancers.
1. Overview
In 2020, worldwide, it is estimated that there will be more than 1.8 million new cases of colorectal cancer (CRC) and close to 900,000 deaths, with more than 50,000 CRC deaths in the US [1]. Overall, CRC is the major site of cancer incidence and mortality in the digestive system. Genetic susceptibility to CRC is considered high. While only ~5% of CRC is inherited in a dominant fashion (e.g., Lynch syndrome and familial adenomatous polyposis), it has been proposed that between 25% and 50% of CRCs demonstrate some form of familial susceptibility [2]. Notably, the full constellation of driver genes for colorectal cancer, especially their contributions to genetic disease susceptibility and their underlying mechanisms of action remain to be fully elucidated. One of the most recently described familial CRC syndromes involves individuals with cystic fibrosis (CF) caused by biallelic inactivating mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, who are highly susceptible to early, aggressive colorectal tumor development [3]. The enhanced risk for CRC also extends to heterozygous carriers of CFTR mutations [4]. Further, low expression of CFTR is associated with poor survival in sporadic CRC [5]. Thus, CFTR deficiency may impact on CRC risk and mortality in large segments of the population. This review focuses on the important role of the CFTR ion channel in colorectal cancer in individuals with germline CFTR mutations (both CF patients and carriers) and in sporadic cases of CRC.
1.1. Cystic Fibrosis
Biallelic inactivating germline mutations in the CFTR gene found on chromosome 7 cause the inherited life-threatening disease cystic fibrosis, the most common autosomal recessive disease among people of European (primarily northern Europe) ancestry [6]. CFTR produces an mRNA transcript of 6128 nucleotides [7] encoding a protein of 1480 amino acids that functions as a chloride (Cl−) and bicarbonate (HCO3−) anion channel. CFTR is located on the apical surfaces of luminal epithelia. While the most severe clinical manifestations of CF are found in the lung [8], CFTR is expressed in a variety of extra-pulmonary tissues [9,10,11], where its deficiency is associated with CF disease pathologies. Intestinal CFTR-deficiency causes obstruction in the ileum (infantile meconium ileus) and proximal colon [11]. CF causes nutrient malabsorption linked to pancreatic enzyme deficiency and potentially, defective uptake of lipids [11]. CF patients are also susceptible to intestinal celiac disease, which results from aberrant TH1 immune and antibody responses caused by eating gluten, a protein found in wheat, barley, and rye [12].
1.2. CFTR: Its Role in Intestinal Tract Homeostasis
Chloride channels, including CFTR, play a key role in homeostasis of the gastrointestinal (GI) tract. Their functions involve osmoregulation, transport of major ions across epithelia, polarity of cells, cellular metabolism including glucose metabolism, cellular autophagy and protein turnover, migration of cells, mucus secretion, innate and adaptive immune responses, cell–cell interactions, membrane potential, mitochondrial function and related oxidative stress, tissue inflammation, microbiota composition, cellular pH, programmed cell death, and intestinal stem cell regulation [13]. Notably, all of these functions are linked to the well-characterized hallmarks of cancer. As the GI tract is constantly exposed to environmental insults dysregulation of these ion channel functions can readily contribute to carcinogenesis [13].
CFTR is expressed along the entire length of the intestinal tract, with a gradient of decreasing expression proximal (duodenum) to distal (ileum) in the small intestine. In both the small and large intestine CFTR expression is strongest at the base of the crypt, the location of the intestinal stem cell compartment [14,15]. Some CFTR expression is also located on the brush border of villus cells, and there are rare CFTR-high-expressing cells scattered along the small intestine outside of the crypt base [16]. CFTR expression in the colon follows an expression gradient of highest in the cecum and proximal colon to lower amounts in the distal colon. The membrane-spanning domains of CFTR form an aqueous channel that permits the passage of Cl− and HCO3− ions down their electrochemical gradients, which in the intestine is from the cytoplasm of epithelial cells to the intestinal lumen, especially the intestinal crypt lumen. This movement of ions out of the cell increases osmotic pressure for the passage of water in the same direction. Thus, indirectly, CFTR also determines water homeostasis [17,18]. CFTR also regulates Na+, K+, Ca2+, and other Cl− channels. For example, CFTR is implicated in inhibition of the activity of SCNN1 (sodium channel epithelial 1 alpha subunit), a Na+ channel, resulting in a further enhancement of water outflow in intestinal cells [19]. Further, CFTR is also implicated in maintenance of intestinal epithelial tight junctions, and in adjustments to the pH of cellular secretions. CFTR is also involved in sphingosine-1 phosphate (S1P) extracellular transport. S1P is a bioactive lipid mediator that is a critical regulator of inflammatory signaling and cell adhesion, among diverse roles [20]. In addition to its membrane-spanning domains, CFTR contains a cytoplasmic C-terminal PDZ (post-synaptic density protein 95 (PSD-95)-Drosophila discs large tumor suppressor (DLG1)-zona occludins 1 (ZO-1)-binding motif that interacts with PDZ-containing proteins that contribute to the regulation of intracellular signaling and the actin cytoskeleton [21,22]. Summarizing, in the GI tract, CFTR function is critical for ion and water homeostasis. Moreover, CFTR localization to the intestinal crypt stem cell compartment permits it to influence stem cell function and therefore, ultimately, CRC development as the intestinal stem cell is the likely cancer progenitor cell [23].
1.3. CF Patients Are at a High Risk for CRC
Development of more-effective therapeutic modalities for pulmonary dysfunction in people with CF has dramatically increased lifespan such that the expected average life expectancy of newborns with CF is ~44 years [24]. As people with CF have aged longer they are at increased risk for developing specific cancers. The first evidence for this was a 20-year epidemiological study using data from the US CF Registry comparing the incidence of cancer in people with CF to the predicted age-adjusted risk in the general population. It was reported that the risk of all types of GI cancers was increased and in particular the risk of CRC was increased 6-fold [25]. The same study also reported an enhanced risk for cancers of the esophago-gastric junction, biliary tract, small intestine, and testes, as well as for lymphoid leukemia. Further, meta-analysis of six population-based studies involving more than 99,000 patients confirmed the particular high risk for gastrointestinal cancers [26]. More directly, based on endoscopic screening studies, larger, more aggressive colon polyps were found in people with CF compared with non-CF individuals. These studies reported that by age forty 50% of individuals with CF developed adenomas. Moreover, half of these tumors were classified as aggressive advanced adenomas. Further, by age forty 3% of individuals with CF had already developed adenocarcinomas [3,27]. CF has now been declared a hereditary colon cancer syndrome by the Cystic Fibrosis Foundation [28]. It is now recommended that CRC endoscopic screening be initiated in CF patients by age forty, and in immunocompromised CF lung transplant patients, at particular risk for cancer, by age thirty [28].
1.4. CFTR Carriers Are Also Susceptible to GI Cancers
Germline CFTR mutations may be an enhanced risk factor for the estimated 3%–4% of the US population (>10 million people) that are heterozygote carriers for inactivating CFTR mutations [29] and therefore potentially at risk due to loss of heterozygosity (LOH) or haploinsufficiency. A recent population-based study using diagnostic codes to compare the prevalence of CF-related conditions in more than 19,000 CFTR heterozygous mutant carriers and 99,000 healthy controls, found that CF carriers were at an increased risk for 59 CF-related diagnostic conditions [4]. Specifically, carriers had a 44% increased prevalence of GI cancers (colon, stomach, and other GI organs). This increase was detected in spite of the fact that 96% of this cohort was under the age of 46 years [4] and thus this population was not representative of the age range in which most sporadic GI cancers occur, e.g., in the USA the average age for sporadic CRC is 68 years in males and 72 years in females, with a similar average age for gastric cancer [1].
1.5. CFTR Deficiency Causes Enhanced Risk in Sporadic CRC
CFTR deficiency is also linked to an increased risk for sporadic CRC. Than et al. reported that in a study of 90 Stage II CRC patients classified by tumor CFTR expression, 3-year disease-free survival (DFS) in the quartile of patients with lowest CFTR expression was 30% lower than in the remaining patients with higher CFTR expression [5]. A separate study in a different patient population by Sun et al. reported that CFTR mRNA and protein expression was lower in CRC tumors vs. normal tissue and CFTR mRNA expression was lower in metastatic CRC vs. non-metastatic CRC. Further, the same group found that CFTR-depleted CRC cell lines demonstrated more aggressive oncogenic characteristics including increased invasion, migration, and colony formation [30]. The factors that cause downregulation of CFTR expression in CRC are not known. However, intestinal-specific transcriptional regulatory elements have been identified. Enhancer elements in introns 1 and 11 have been shown to promote transcription [31] while the minor alleles of several exon 11 SNPs originally identified in CF patients caused decreased expression of CFTR in human Caco-2 CRC cell reporter assays [32]. Low CFTR expression in sporadic tumors may also be caused by silencing of the CFTR gene by promoter hypermethylation as was reported in other cancers such as lung, breast, head and neck, and bladder cancers [33,34,35,36,37,38].
1.6. Risk for GI Cancer Caused by CFTR Deficiency Was Confirmed in Mouse Genetic Studies
In Sleeping Beauty (SB) transposon-mediated genetic screens Cftr was identified as a common insertion site (CIS) gene in mouse GI tumors and likely candidate tumor suppressor gene. Cftr was ranked in the top 10% to 50% of CIS CRC cancer-driver genes in screens in adenomatous polyposis coli (Apc) wild-type [39], Apc-deficient [40], and transforming growth factor-beta (TGF beta)-deficient [41] genetic backgrounds. In a functional test of Cftr deficiency, Than et al. reported that in mice carrying an intestinal specific Cftr conditional knockout allele crossed to the tumor-sensitized ApcMin mouse, mice deficient for Cftr (both heterozygotes and homozygotes) developed significantly more tumors than ApcMin mice that were wildtype for Cftr. Furthermore, invasive lesions were only observed in the Cftr-deficient cohort. Notably, Cftr-deficiency alone was sufficient to cause tumors in >60% of Apc wildtype mice by 10–12 months of age [5].
2. Potential Mechanisms of CFTR Tumor Suppressor Action
2.1. CFTR Influences the Intestinal Stem Cell Compartment
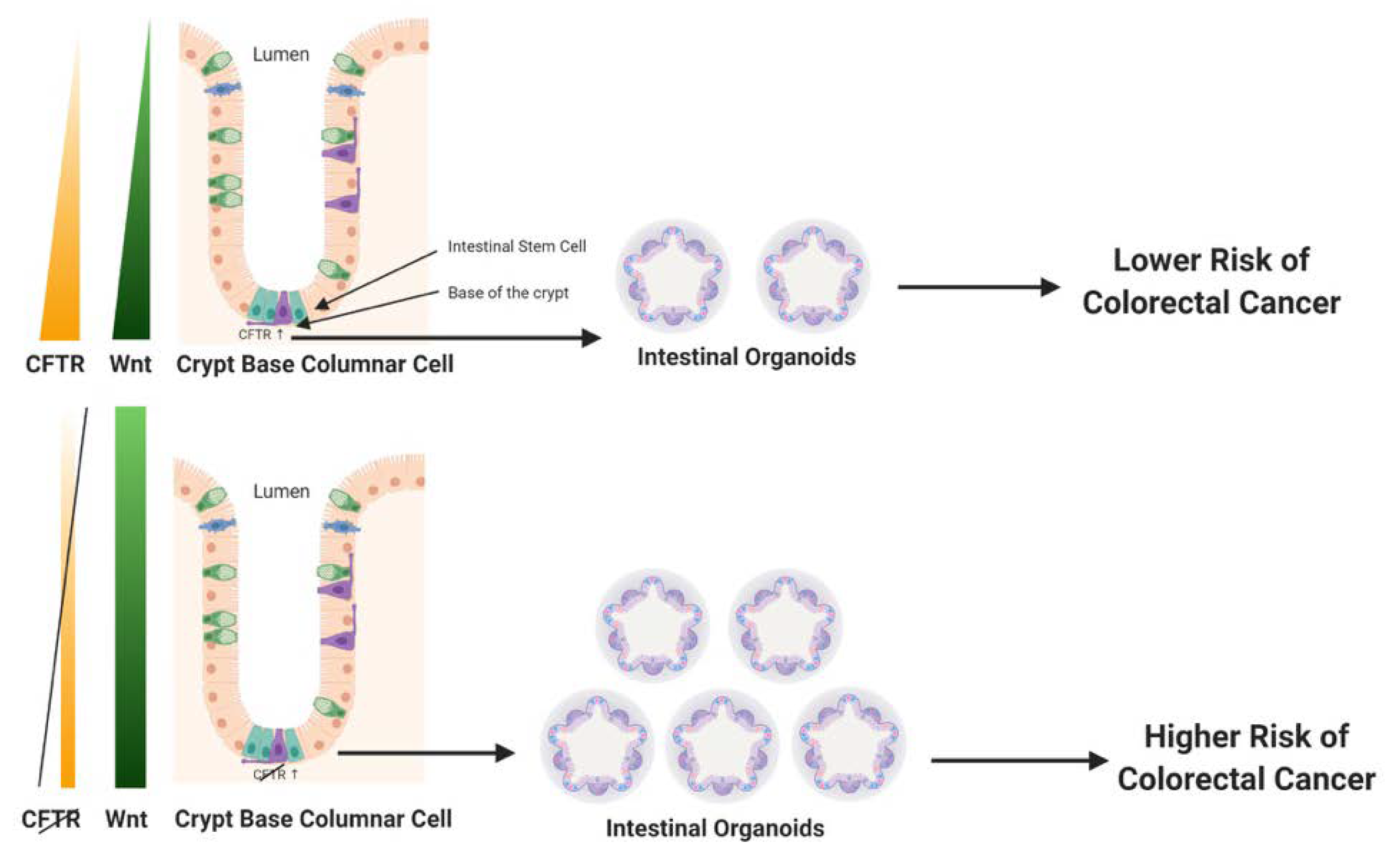
Long-lived multipotent intestinal stem cells located at the crypt base self-renew and repopulate crypts with specialized differentiated cells (e.g., enterocytes, secretory cells)—every 4–5 days [15]. There is substantial evidence that these intestinal stem cells are the primary source of CRC progenitor cells [23]. CFTR expression in the colon is strongest at the crypt base in the stem cell compartment [14], with some reports indicating that CFTR is expressed by the intestinal stem cells [42]. There is additional evidence that CFTR influences intestinal cell lineage differentiation of mouse embryonic stem (ES) cells. Further, Li et al. reported that CFTR F508del mutant mouse ES cells developed teratomas, and upregulated genes involved in proliferation, migration, and epithelial to mesenchymal transition (EMT) [43]. Accordingly, CFTR is well-placed to influence intestinal crypt epithelial cell renewal and the activity of CRC progenitor cells. Intestinal crypt structure and function can be modeled in vitro using three dimensional (3D) organoid cultures. Organoids can be created from single intestinal stem cells or isolated crypt bases containing stem cells. Organoids retain normal intestinal crypt structure, polarity, and physiological functions, including self-renewal and lineage differentiation into all of the differentiated cell types in the crypt [44]. Cftr expression and activity have been tested in organoids created from both humans and mice, and CFTR has been shown to retain its normal Cl− and HCO3− ion function in these organoids [45,46]. Importantly, in several studies, colon organoids have been shown to be valuable surrogates to measure the effectiveness of therapeutic drugs to treat CF patients with specific CFTR mutations [47,48,49]. Further, organoids created from CRC cells or pre-cancerous tissues recapitulate the oncogenic phenotypes of these tissues [50,51]. Organoid cultures have been used to test the effect of CFTR deficiency on oncogenicity. For example, it was reported that Cftr knockout (KO) mouse colon organoids developed a significant increase in clonogenicity [5]. Further, organoids created from mouse small intestine also showed enhanced proliferation along with the localization of Cftr to the leucine rich repeat containing G protein-coupled receptor 5-positive (LGR5+) intestinal stem cell [42]. Overall, these results confirm a likely role for Cftr in influencing the CRC progenitor cells and their microenvironment.
2.2. CFTR Is Linked to Regulation of Wnt/β-catenin Signaling
Wnt/β-catenin signaling is a critical mediator of intestinal tissue homeostasis, in particular as a regulator of intestinal cell survival, proliferation, and differentiation, and key characteristics of normal intestinal cell renewal and function. Wnt/β-catenin expression and target signaling is tightly controlled. Wnt expression forms a gradient in the intestinal crypt, where its expression is strongest at the base of the crypt in the stem cell compartment (depicted in Figure 1, left panel). Notably, and not surprisingly, dysregulation of the Wnt/β-catenin signaling pathway is implicated in up to 90% of human CRC, strongly contributing to both early tumor initiation as well as promoting progression of invasive CRC [52]. Deficiency of Cftr in the mouse intestine has been linked to increased Wnt/β-catenin signaling. A study of intestinal tumors isolated from mice carrying an intestinal-specific conditional knockout of Cftr alone (Apc wildtype) found enhanced nuclear localization of β-catenin, an indicator of activation, along with elevated expression of Wnt/β-catenin target genes such as Cyclin d1 (Ccnd1), Lgr5, and cluster of differentiation 44 (CD44) [5]. This finding was confirmed in crypts and organoids from the small intestine of constitutive Cftr knockout mice. Compared with wildtype control mice, Cftr KO mice demonstrated enhanced proliferation, including a 30% increase in intestinal stem cell proliferation and a significant increase in Wnt/β-catenin signaling [42]. In the same study, deficiency for Cftr or a transient inhibition of channel activity by CFTR-(inh)-172 caused higher levels of intracellular pH in LGR5+ intestinal stem cells. This elevation in intracellular pH was linked to enhanced physical association of dishevelled segment polarity protein 2 (Dvl2), a key part of the Wnt/β-catenin signaling pathway, with phospholipids at the plasma membrane, thus placing Dvl2 in an ideal location to promote Wnt/β-catenin signaling [42]. However, it is noted that not all reports of CFTR deficiency in the intestine have found an upregulation of Wnt/β-catenin signaling. Liu et al. reported that in both F508del Cftr mutant mice and in the human Caco-2 CRC cell line deficient for CFTR there was decreased Wnt/β-catenin signaling. The authors of this study proposed that deficiency for CFTR physically disrupted β-catenin localization to the plasma membrane thus promoting its degradation in the proteasome, with this event eventually causing NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) translocation to the nucleus leading to inflammation [53]. The significant differences between the Liu et al. study [53] and that of Than et al. [5] and Strubberg et al. [42] could be context dependent. For example, in the Liu et al. study total intestinal tissue was used for analysis, rather than intestinal epithelia, intestinal crypts, or organoids as was employed in Than et al. and Strubberg et al., thus the findings of Liu et al. could reflect an influence for CFTR independent of the stem cell compartment. Another contextual difference between the Liu et al. study and that of Than et al. and Strubberg et al. is in the specific mouse models of Cftr employed. Liu et al. employed the Cftrtm1Kth mouse that mimics the F508del mutation. It is considered a hypomorph that retains some protein expression and that demonstrates 40% survival to maturity, even when untreated with a polyethelene glycol (PEG) osmotic laxative to prevent intestinal obstruction. The Liu et al. paper does not mention treatment of these animals with a PEG osmotic laxative, thus presumably there was no treatment. In contrast, Strubberg et al. (Cftrtm1Unc) and Than et al. (Cftrfl10/fl10 × intestinal Cre-mediated excision) employed more severe mutant Cftr alleles (as low as 5% survival to maturity) that required treatment of the mice with a PEG osmotic laxative (Colyte) to prevent lethality due to intestinal obstruction. Treatment with Colyte significantly reduces bacterial load and inflammation [54]. Accordingly, it is likely that the mice described in Liu et al. (depicted in Fig. 1 in that paper [53]) were undergoing a level of intestinal inflammation not observed in the Strubberg et al. and Than et al. studies where mice were treated with Colyte, and this difference could in part explain differences in gene expression reported in the respective papers. It is also noted that the influence of deficiency for CFTR on Wnt/β-catenin signaling was context-/tissue-dependent in other published studies by the Liu et al. research group [55]. Further, the role of Wnt/β-catenin signaling in CRC could vary by CRC stage. For example, while dysregulation of canonical Wnt/β-catenin signaling is widely considered to be the primary driver for early CRC development and progression [52], there are some reports that decreased Wnt/β-catenin target gene expression is implicated in poor prognosis for some CRC patients [56]. In support of this idea, inhibition of Wnt/β-catenin signaling was reported to promote survival of latent metastatic CRC cells [57]. However, given that deficiency for CFTR has been reported in a wide range of cancers that arise independently of Wnt/β-catenin signaling, it is likely that CFTR’s function as a tumor suppressor in CRC extends beyond influencing the intestinal stem cell compartment and Wnt/β-catenin signaling. While these additional tumor suppressing mechanisms are not fully understood, from what is known observationally and experimentally about the functions of CFTR in normal intestinal tissue and the phenotypes of intestinal tissues that are CFTR-deficient, several potential mechanisms of action are suggested, possibly acting in concert. These mechanisms are described in the following sections. Figure 1 depicts a simple model of CFTR’s influence on the intestinal stem cell compartment and Wnt/β-catenin signaling.
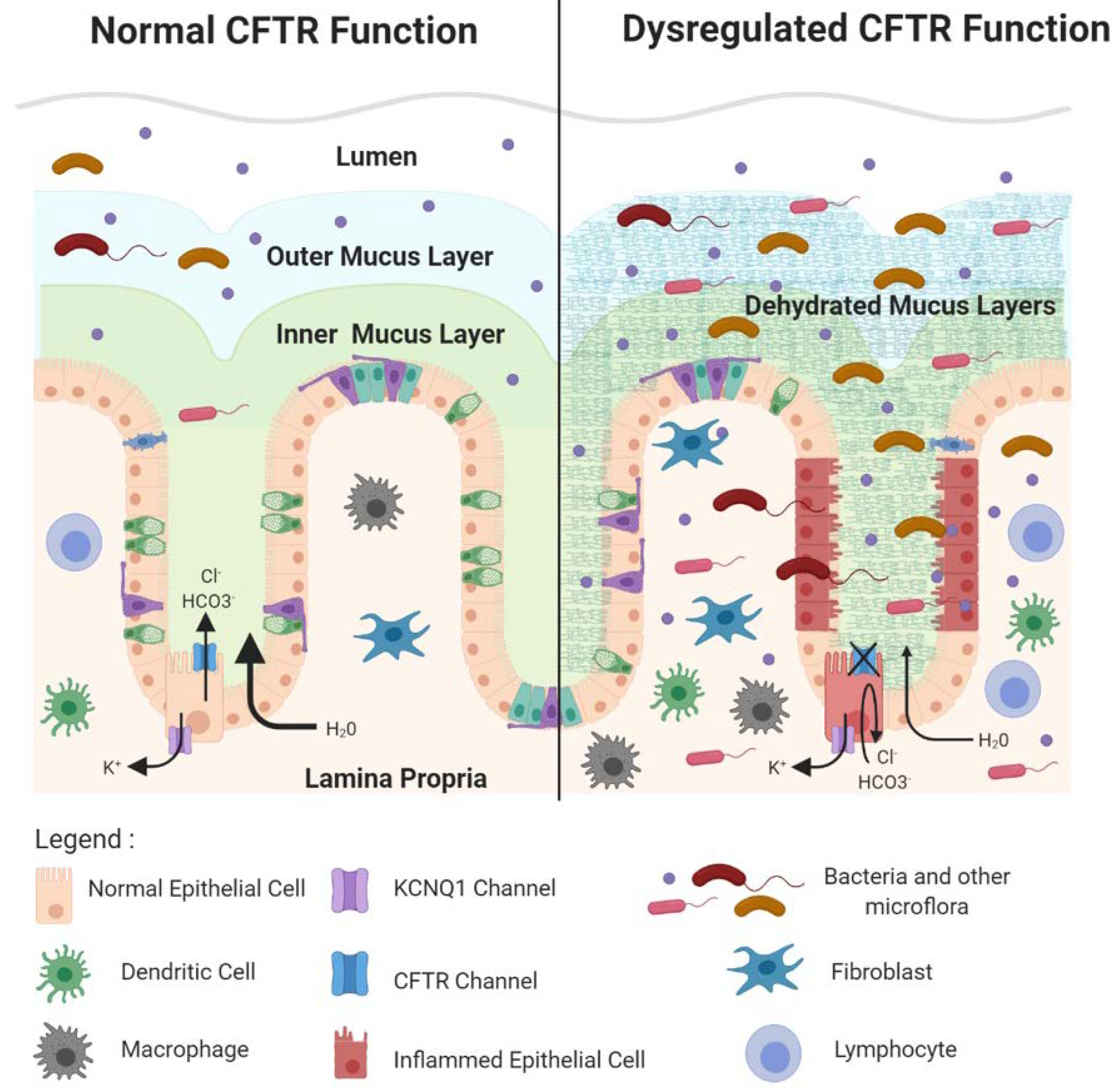
2.3. CFTR Deficiency Disrupts Protective Physical Barriers
CFTR influences several inter-related processes in the intestine that are critical for tissue homeostasis. These include the composition of the gut microflora, maintenance of essential barriers protecting the single cell epithelial layer, and homeostasis in the innate and adaptive immune response. Clinical manifestations of CF in the GI tract such as inflammation and obstruction are associated with the dysregulation of these processes, events that also create a favorable landscape for cancer development. In the lumen of the intestinal tract, primarily in the large intestine, trillions of commensal bacteria and other flora such as algae and fungi are found. The microflora, primarily bacteria, are important for nutrient production, such as the production of butyrate, a short chain fatty acid made from bacterial fermentation of dietary fibers. Butyrate is important for colonic health by providing a fuel source in the colon and by its anti-inflammatory action, among a variety of beneficial effects. Microflora also produce signaling molecules that regulate immune cell homeostasis such as lipopolysaccharide (LPS) molecules that can bind to pattern recognition receptors on epithelial cells to activate the intestinal cell immune response. However, it is critically important to prevent contact between microflora and the single cell layer of the intestinal epithelium, principally in the colon. Two primary physical barriers maintain the impenetrability of the colonic epithelium to bacteria. First, the apical surface of the colonic epithelium, facing the crypt and intestinal lumens, is covered by protective dense inner and looser outer mucus layers that are produced by constitutive secretion of mucin 2 (MUC2) mucins from luminal goblet cells. The dense inner mucus layer is largely sterile. The looser outer mucus layer results from partial enzymatic digestion of mucins in the inner layer that migrate out into the outer layer. The outer mucus layer contains an abundant population of commensal bacteria [58]. Secreted MUC2 initially exists in a very condensed form that expands to create the dense inner layer in a process that requires both bicarbonate ions (HCO3−) and water [59]. The ion channel function of CFTR is directly involved in HCO3- ion efflux into the lumen and indirectly involved in promotion of H2O efflux as well [60]. CFTR deficiency, consequently, causes the mucus layers to become dehydrated and dysfunctional. In the distal small intestine of CF infants this causes obstruction, a disorder called meconium ileus [11]. Obstruction also occurs in the proximal large intestine, leading to inflammation and related pathologies in CF patients, a phenotype also observed in Cftr KO mice. Further, markedly dilated CFTR-deficient crypts are found to contain an accumulation of unsecreted mucus in the goblet cell population, indicating that deficiency for CFTR may regulate goblet cell secretion as well [61,62]. In addition, the dehydrated dysfunctional mucus layer permits illicit bacterial contact with the epithelia as enlarged bacterial colonies were found in the inner mucus layer of CFTR KO ferrets [61]. Than et al. used gene set enrichment analysis (GSEA) [63] to compare gene expression profiles from Cftr KO mouse intestine with Muc2 KO mouse intestine and reported an enrichment in inflammatory gene expression [5], supporting the idea that the loss of Cftr, like loss of Muc2, permits dysregulated bacterial contact with the epithelial layer. A second protective physical barrier is the tight junctions between the basolateral surfaces of intestinal epithelial cells. CFTR is involved in maintenance of these tight junctions as loss of Cftr in the small intestine of KO mice resulted in enhanced epithelial permeability and disruption of epithelial tight junctions [64]. Several reports indicate that CFTR may mediate tight junction integrity via its C-terminal PDZ-binding domain [64]. This was demonstrated by in vitro studies in airway epithelial cells that reported physical interaction between the CFTR–PDZ binding domain (CFTR–PDZBD) and the PDZ domain of NHERF1 (Na+/H+ exchange regulatory cofactor 1)/ SLC9A3R1 (solute carrier family 9, subfamily A, member 3 regulator), a protein critical for the organization of the actin cytoskeleton and further important for tight junctions [65]. In addition, loss of CFTR could disrupt tight junctions by preventing its protein–protein interaction with TJP1 (tight junction protein 1)/ZO-1 (zona occludins 1), a key protein in both adherens and tight junction assembly and structure [66]. CFTR protein–protein interactions may also mediate tumor suppression by acting as a signaling hub with metastasis suppressor proteins of the NM23 (non-metastatic clone 23)/NDPK (nucleotide diphosphate kinase) family that are proposed to negatively regulate the Ras/PI3K(phosphoinositide 3 kinase) signaling pathway [67]. For example, it was reported that NM23-H1 (NM23-human gene 1) and NM23-H2 (NM23-human gene 2) are present in a complex of proteins near the plasma membrane that bind different domains of CFTR, including NBD1 (nucleotide binding domain 1) [68]. Mutations in CFTR disrupt this complex leading to dysfunction of NM23/NDPK [67].
2.4. CFTR Deficiency Causes Dysbiosis
Bacterial dysbiosis is commonly observed in the gut of CF patients and in CF animal models [11,69]. Loss of CFTR causes a dysregulated environment in the intestinal lumen that is associated with disruption of the mucus layer (as described above) and, specifically in CF patients is exacerbated by factors such as nutrient malabsorption, high fat diets, and antibiotic therapy. The degree of dysbiosis is associated with the severity of CF GI disease, GI inflammation, and nutrient uptake deficiency [11]. The first experimental evidence for dysbiosis was bacterial overgrowth observed in the usually-sterile small intestine of Cftr KO mice. qRT-PCR of bacterial 16S DNA in flushed mouse small intestine found a 40-fold increase in total bacteria with reduced species diversity [70]. Another report employed phylogenetic microarray analysis of flushed mouse small intestine and found a significant reduction in bacterial richness, evenness, and species diversity. Specifically, there was a reduction in the abundance of protective bacteria such as Acinetobacter Iwoffii and Lactobacilliales members, but a rise in Mycobacteria species and Bacteriodes fragilis bacteria implicated in infection and immunomodulation in the GI tract [71]. In following studies, the microbiota composition of CF vs. non-CF individuals was evaluated employing species-specific and metagenomic analysis of 16s rRNA DNA from patient feces. Compared with non-CF controls the microflora in CF patients showed markedly decreased diversity, evidence that CF-related changes in the gut lumen not only permitted enhanced illicit bacterial contact with the epithelia but in addition altered the composition of the microbiota [72,73,74]. Recently, changes in the microbiome of CF patients have been linked to potentially oncogenic functional consequences [75]. Meeker et al. [76] employed germ-free Cftr KO mice treated with specified pathogen-free (SPF) fecal transplants and reported that Cftr mutations alone were sufficient to alter the fecal GI microbiome, with enrichment in the proportion of Escherichia/Shigella (>250-fold) and a depletion of Lachnoclostridium and Parabacteriodes. This result is consistent with findings in the fecal microbiome of children with CF [77]. The Meeker et al. study further showed that colonized Cftr KO mice had an increase in mesenteric lymph node and spleen TH17+ cells compared with non-Cftr KO mice, evidence that CFTR deficiency altered the adaptive immune response [76]. In addition, CF patients with specific CFTR mutations and bacterial dysbiosis are vulnerable for the development of Crohn’s disease [78]. In another recent study in human CF patients Dayama et al. [79] described interactions between the gut microbiome and host gene regulation, resulting in enrichment of oncogenic pathways. They conducted RNA-Seq analysis of colonic mucosa samples from CF patients and healthy controls as well as 16S rRNA sequencing to characterize the colonic microbiome. They found more than 1500 genes differentially expressed in CF patients, with enrichment for genes related to CRC, including CRC metastasis, tumor suppression, P53 (tumor protein 53), and the mTOR (mammalian target of rapamycin) pathway. Further, CF patients showed decreased microbial diversity, decreased abundance of butyrate producing bacteria, such as Ruminococcaceae and Butyricimonas, and an increased abundance of taxa such as Actinobacteria and Clostridium. Butyrate, a product of bacterial fermentation by Ruminococcaceae and Butyricimonas (and other bacteria) of dietary fibers, is considered to prevent CRC via epigenetic regulation of gene expression [80,81] while Actinobacteria and Clostridium are among the taxa associated with CRC development and progression [82,83]. An integrative analysis by Dayama et al. identified CRC-related genes, including lipocalin 2 (LCN2) and dual oxidase 2 (DUOX2), which were correlated with the abundance of CRC-associated bacteria such as Veillonella [79]. These findings are consistent with more general findings showing increased evidence of an association between alterations in the intestinal microbiome with CRC [75,84]. For example, a recent study by Hans Clevers and co-workers reported that a colibactin-induced genotoxic mutational signature caused by pks+ E. coli was identified in a subset of more than 3000 metastatic CRC patients [85].
2.5. Loss of CFTR Promotes Immune Infiltration of the Epithelial Layer and Lamina Propria
Disruption of physical barriers in the intestine permits illicit access of bacteria to resident immune cells of the lamina propria as well as infiltration of the epithelia by immune cells. While immune cell infiltration and inflammation can lead to damage in the epithelia, only a single study thus far has observed this type of damage in CF patients, duodenal lesions detected by capsule endoscopy [86]. However, infiltration of immune cells has been observed in the intestine of both CF patients and in mouse CF models but this infiltration did not cause apparent morphological and histological damage [87]. Immune cell infiltration in CF patients was found following whole gut lavage [88], and assays to detect fecal calprotectin [86,89] and rectal nitric oxide [90]. In Cftr KO mice, gene expression analysis in mouse small intestine demonstrated upregulation of genes expressed by granulocytes. This finding was confirmed by microscopic analysis that detected elevated numbers of mast cells and neutrophils in the same tissue [87]. In support, Liu et al. found immunocytochemical evidence of elevated numbers of macrophages and neutrophils in the small intestine of Cftr F508del mice [53]. There is also emerging evidence that CF patients demonstrate dysregulation of CFTR expression in various immune cell populations that contribute to aberrant immune cell activity in several organs, in particular the lung, but also likely in the GI tract. CFTR expression and dysregulation has been detected in monocytes/macrophages and dendritic cells of the peripheral innate immune response. These cells can also influence adaptive T cell responses via antigen presentation. CFTR may also influence the immune response through its expression in lymphocytes and NK cells [91,92]. Furthermore, CFTR dysregulation of neutrophils in CF patients has been shown to cause neutrophil intrinsic impairment linked to degranulation [93].
2.6. Loss of CFTR Promotes Pro-Inflammatory Signaling
Loss of CFTR has been associated with activation of NF-κB signaling pathways, primarily in in vitro models. Xie et al. and Zhang et al. studied cell lines generated from several different types of cancer. They reported that loss of CFTR led to NF-κB activation, upregulation of UPA (urokinase-type plasminogen activator) expression, and increased cellular migration and invasion [94,95]. Studies in CFTR-deficient Caco-2, HT-29, and HEK293 human CRC cells reported elevated levels of pro-inflammatory cytokines. Specifically, there was an increase in the expression of NF-κB pathway members, including an elevation in TNF-α (tumor necrosis factor alpha), IL-6 (interleukin-6), and IL-1β (interleukin-1 beta)-induced secretion of IL-8 (interleukin-8), COX-2 (cyclooxygenase-2), and PGE2 (prostaglandin E2), along with enhanced activities of ERK1/2 (extracellular signal-related kinases 1/2), MAPK (mitogen-activated protein kinase), IκBα (NF-kappa-B inhibitor alpha), and NF-κB [96,97,98,99]. Massip-Copiz et al. reported that loss of CFTR caused NF-κB activation following autocrine signaling by IL-1β [100]. Furthermore, loss of CFTR was reported to cause downregulation of the hedgehog signaling pathway, an effect linked to heightened inflammatory signaling [101]. Finally, dysregulation of calcium homeostasis has been reported in CF bronchial epithelium both as a result of chronic inflammation [102], and as a direct result of mislocalized F508del-CFTR [103,104,105]. The increased TRPC6 (transient receptor potential cation subfamily C, member 6) Ca2+ entry, increased store-operated Ca2+ entry (SOCE), and aberrant mitochondrial buffering capacity seen in CF cells is likely altering the inflammatory response as well as various other pathways utilizing Ca2+ as a second messenger. While evidence for this influence of dysregulated Ca2+ signaling in CFTR-deficiency mediated inflammatory signaling in CRC is thus far weak, it remains a research area for heightened investigation.
Dysregulation of barrier integrity, dysbiosis, immune cell infiltration, and inflammation work together in disruption of colonic homeostasis, altering the landscape to favor CRC development.
Dysregulation of physical barrier integrity permits illicit bacterial contact with the intestinal epithelia, further attracting cells of the innate and adaptive immune system that can promote both direct and indirect pro-inflammatory responses [106,107,108,109]. For example, shedding of lipopolysaccharides that comprise the outer membrane of Gram-negative bacteria, can directly promote the production of pro-inflammatory cytokines via their interactions with toll-like receptors (TLR) on the intestinal epithelia [110], while, indirectly, illicit bacterial contact can activate cytokine signaling to recruit lymphocytes. There is evidence that CFTR is involved in mediating some of these direct and indirect effects. In studies employing primary biliary epithelial cells, Strazzabosco and colleagues reported binding of CFTR to inhibitors of Src (SRC proto-oncogene, non-receptor tyrosine kinase) that promoted their physical interactions with Src to inhibit its activity. Knockout of CFTR in these cells caused these Src inhibitors to be mislocalized, resulting in Src activation. Src activation then synergized with bacterial products such as LPS causing activation of NF-κB signaling and eventually disruption of both adherens and tight junctions [111]. Inflammatory responses are also associated with dysregulation of intestinal stem cell homeostasis, causing enhanced proliferation and expansion of the stem cell compartment via reversion of differentiated early transit amplifying cells back to stem cells [112]. Disruption of physical barriers caused by illicit bacterial contact can also lead to altered migration and invasion of cancer cells [113]. Dysbiosis resulting from loss of CFTR may promote the production of bacterial taxa that are prevalent in colorectal cancer. For example, studies in Cftr KO mice found an increase in Bacteroides fragilis [71], a species of bacteria associated with CRC because of its link with Stat3 (signal transducer and activator of transcription 3) signaling via a Th17+ (T helper cell 17) immune response [114]. See Figure 2.
2.7. CFTR Is Associated with Altered Stress Responses
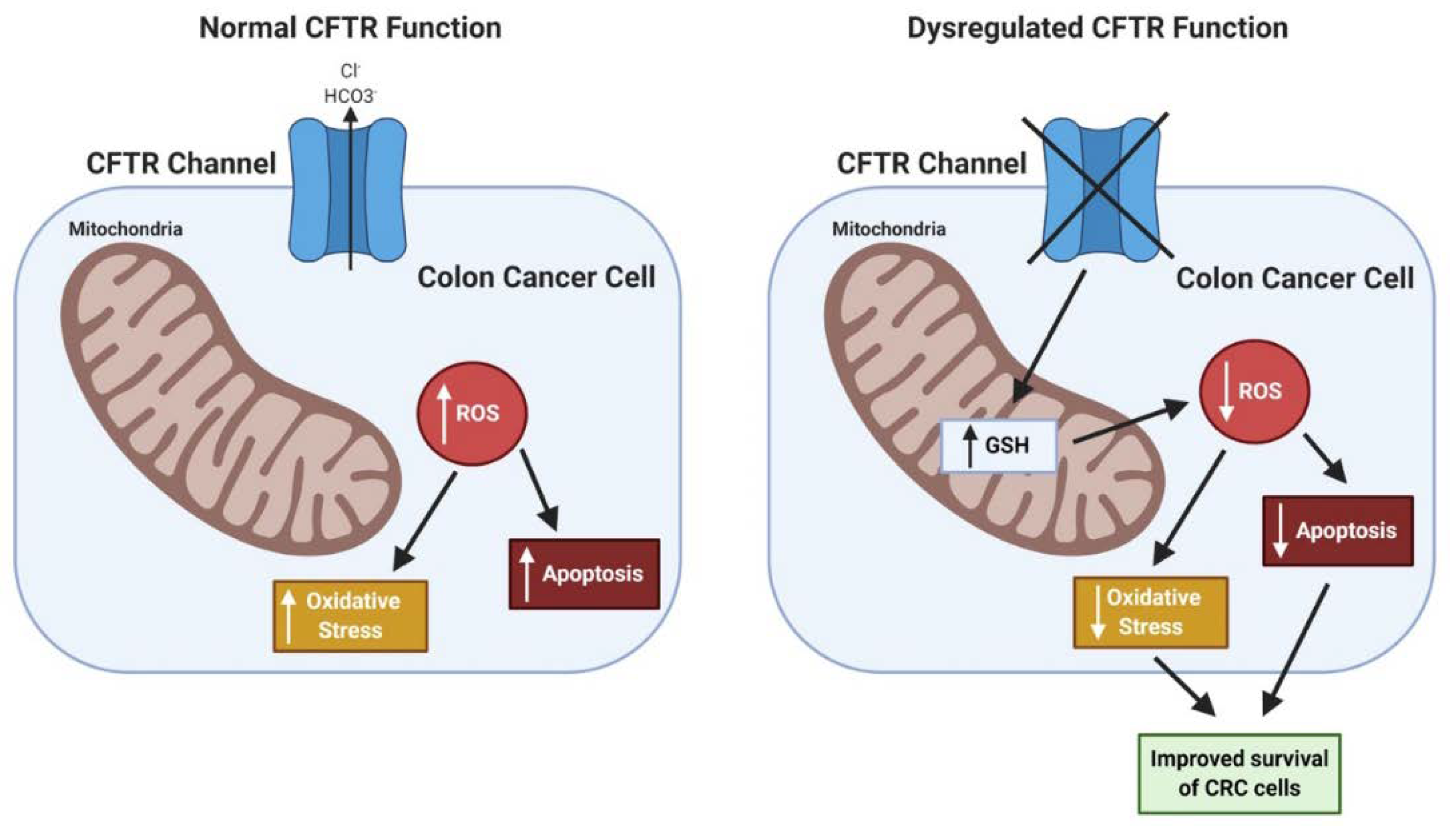
In the literature, loss of CFTR is associated with both a rise in cellular oxidative stress caused by dysfunction of mitochondria [97,115,116,117] and a reduction in cellular oxidative stress by retention in the cells of the antioxidant glutathione (GSH) [116]. Loss of CFTR in human Caco-2/15 CRC cells resulted in an elevation of lipid peroxidation along with a reduction in anti-oxidant factors such as the enzymes glutathione peroxidase and catalase [118]. Further, CFTR-deficiency has been linked to the downregulation of cellular autophagy caused by transglutaminase-2 (TG2) activation. TG2 cross-linking activity results in key autophagy proteins being bound up in aggresomes [119]. In addition, there is evidence that CFTR expression (mRNA, protein) and function can be repressed by HIF-1 in hypoxic epithelium, which is common in CRC, and most epithelial cancers [120]. How these various alterations in stress responses influence cancer development, particularly in the context of loss of CFTR, remains unclear. Overall, the potential impact of CFTR deficiency in CRC on oxidative stress may be context dependent, as may be true for cancers in general. Because of high rates of metabolism in cancer cells, oxidative stress and accompanying production of reactive oxygen species (ROS) is increased. While increased ROS can promote cancer development via mechanisms such as DNA damage and mutations, ROS is also harmful to both normal and cancer cells with excess DNA damage leading to apoptosis. One potential hypothesis is that CFTR deficiency protects CRC cells from ROS-induced cell death via retention of antioxidants such as GSH and other mechanisms. Preliminary studies in our research group employing CFTR-deficient human Caco-2 CRC cells indicate that CFTR-deficiency promotes the survival of CRC cells after treatment with oxidative-stress-inducing agents such as menadione [121]. Similar results have been observed in Cftr-deficient mouse colon organoids [122]. See Figure 3.
2.8. The Role of CFTR in Non-Colonic Cancers
CFTR is widely expressed in epithelial tissues, and as discussed above, CFTR influences epithelial cell homeostasis via a range of mechanisms, therefore it was not unexpected that CFTR dysregulation has been reported in a wide range of epithelial cancers. Upregulation of CFTR expression has been associated with several cancers such as gastric and ovarian cancers [123,124,125]. But for the great majority of cancer studies, loss of CFTR expression and/or activity is linked with cancer incidence, and in the majority of these reports, CFTR mutations or downregulation of CFTR expression in epithelial cancers is associated with rapid cancer growth, epithelial to mesenchymal transition (EMT), a reduction in cellular apoptosis, enhanced metabolic potential, and elevated patient morbidity and mortality. Down regulation of CFTR expression and/or mutations that disrupt CFTR function have been reported in non-small-cell lung cancer (NSCLC) [33,86,126,127], glioblastoma [128], bladder cancer [34,35,36], esophageal cancer [129,130], pancreatic cancer [131,132,133], nasopharyngeal cancer [134], prostate cancer [94], and breast cancer [37,95]. In many of these malignancies, loss of CFTR correlated with increased tumor stage and reduced disease-free or overall survival. Inherited heterozygous CFTR mutations have been associated with an increased risk of pancreatic cancer among both younger [131,132,133] and older patients [4]. Yet, a reduction in CFTR mRNA expression in most non-colonic cancers studied has not been linked to germline mutations, although germline mutations were not likely tested for in most cases. CF-related germline mutations are not the likely cause of reduced CFTR mRNA expression in these cancers as F508del, which represents ~ 70% of CF alleles, results in a 90% decrease in protein levels but a very modest or no reduction in mRNA [135]. Reasons for a loss of CFTR mRNA expression include promoter hypermethylation as has been reported in lung, breast, head and neck, and bladder cancer studies [34,35,36,37,38] and somatic CFTR mutations as seen in NSCLC [32]. Furthermore, cigarette smoke (CS) was reported to downregulate CFTR mRNA expression and downregulation of CFTR expression has been associated with the development of chronic obstructive pulmonary disease (COPD) [136]. A link between CS, downregulation of CFTR, and lung cancer was reported in a study that correlated CS and CFTR downregulation with enhanced oncogenesis in A549 lung cancer cells [137].
2.9. CFTR Therapies: Clinical Implications in CRC
CF therapies to correct the underlying defects caused by specific CFTR mutations are now in the clinic. For example, potentiator drugs can enhance anion efflux via CFTR channels already situated on the plasma membrane and corrector drugs can cause correct folding of mutant CFTR. Modulators such as ivacaftor, elexacaftor, tezacaftor, and lumacaftor are now in use to treat CF [138]. Ivacaftor is a potentiator employed in treatment of >25 CF-causing mutations that disrupt CFTR gating as well as residual function, and CFTR splice mutations. Tezacaftor, elexacaftor, and lumacaftor are correctors that can enhance the function of the F508delta mutation that represents ~ 70% of CF alleles. Most recently, a combination of three small molecules (TRIKAFTA) comprising two correctors (tezacaftor and elexacaftor) plus one potentiator (ivacaftor) have been approved by the FDA for patients who carry one or two copies of the F508del allele [139,140]. With the development of TRIKAFTA, modulator therapies are available to treat 90% of CF patients. Notably, these therapeutic drugs are now targeting CF-related gastrointestinal disorders, with the possibility of future application to cancer [141]. For example, CF patients with G155D mutations are now being treated with Ivacaftor, resulting in improvement for pancreatitis [142]; and of pH in the proximal small intestine, an improved clinical phenotype associated with increased bicarbonate secretion; increased cell motility; and overall improvement in clinical outcomes [143]. Lumacaftor was also demonstrated to improve bicarbonate permeability in CF patients with F508del mutations [144]. Ivacaftor treatment beneficially improved colonic microbiota, such as an increase in Akkermansia and a reduction in Enterobacteriaceae. Ivacaftor treatment also caused a major decrease in GI inflammation in CF patients [145]. Other drugs, either singly or in combination are being developed. For example, the combination of ivacaftor with 5-nitro-2-(3-phenylpropylamino) benzoate, has been shown to synergize in CF patients with G551D mutations [146]. Notably, these targeted therapies may be CRC preventative for the 3%–4% of Caucasians who carry germline CFTR-inactivating mutations. These individuals are at an enhanced risk for developing CRC [4], either via CFTR haploinsufficiency or LOH (loss of heterozygosity). Beyond US FDA-approved drugs, novel modulators known as amplifiers, such as PTI-428, are in current clinical trials. PTI-428 is proposed to increase the translation of CFTR mRNA and thus complement the action of corrector drugs [11]. PTI-428 may also be useful for augmenting CFTR activity in sporadic CRC patients with low CFTR protein expression. Testing protocols currently employed for screening and diagnosis of CF could be repurposed for adjusting the timing of early detection assays (such as colonoscopy screening), and even for the prevention of CRC. Genetic testing for the panel of >99% of CF-causing mutations is made available to all pregnant couples. This testing could also be adaptable for early detection of CRC in CF carriers. A major technical breakthrough to evaluate new CF therapies has been the development of patient-derived colorectal organoids [147]. Evidence for their applicability has been demonstrated in rectal organoids developed from CF patients that have been used to evaluate several CF drugs, including ivacaftor and lumacaftor [148].
3. Conclusions
Loss of CFTR causes disruption to a wide range of cellular processes that can promote carcinogenesis but it still remains unclear which of these dysregulated processes, alone or together, are most critical for CRC development and progression in the context of CFTR-deficiency. One area that cancer investigators might focus on is the influence of CFTR on the intestinal stem cell (ISC) compartment as ISCs are the source of putative CRC progenitor cells [23]. ISCs are located at the crypt base in the colon, the location of peak CFTR expression. To protect the vital long-lived ISCs from a hostile luminal environment the crypt contains several key protective mechanisms. These include a unique microflora, especially in the inner mucus layer, with a reduction in the quantity and diversity of bacteria compared with the outer intestinal lumen [149]. The dense inner mucus layer prevents illicit contact between bacteria and bacterial products and the single cell intestinal epithelial layer of the crypt. Sensors participate in this protective mechanism. When bacterial products such as LPS are detected by goblet cells at the crypt neck additional protective mucins are released [150]. Mitigation of harmful oxidative stress is another evolved protective mechanism in the crypt. For example, ISC survival is mediated by physical interactions between the cytosolic innate immune sensor Nod2 (nucleotide-binding oligomerization domain-containing protein 2) and the bacterial peptidoglycan motif MDP (muramyl dipeptide) [151]. These mechanisms are particularly important to block inflammation that can jeopardize crypt homeostasis and promote oncogenesis. For example, NF-κB has been demonstrated to synergize with Wnt/β-catenin signaling in crypt cells to cause reversion of early differentiated crypt cells to stem-like cells with the ability to cause tumorigenesis [112]. All of these observations emphasize the priority of investigating how loss of CFTR impacts on the crypt and ISCs, studies that can be especially informative by employing crypt-derived organoid models. Finally, repurposing of drugs to treat GI complications in CF patients have now reached the clinic. While these mutation-specific targeted CF drugs have not yet reached the cancer clinic, they could be used in the clinic in the near future, along with the re-purposing of other ion channel activators and blockers as more becomes known about the precise contribution of ion channels to cancer development.
Author Contributions
P.S., K.A., M.S., and R.C. All contributed to the writing of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
University of Minnesota Academic Health Center Faculty Development Grant (P.S.), University of Minnesota Masonic Cancer Center (R.C., P.S.), Mezin-Koats Colon Cancer Research Award (P.S., R.C.), Whiteside Institute for Clinical Research (P.S., R.C.), Essentia Health Systems (R.C.), and Annette Boman Cancer Research Fellowship (M.S.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- American Cancer Society. Cancer Facts & Figures 2020; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Burt, R. Inheritance of colorectal cancer. Drug Discov. Today Dis. Mech. 2007, 4, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Niccum, D.E.; Billings, J.L.; Dunitz, J.M.; Khoruts, A. Colonoscopic screening shows increased early incidence and progression of adenomas in cystic fibrosis. J. Cyst. Fibros. 2016, 15, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 2020, 117, 1621–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Than, B.L.; Linnekamp, J.F.; Starr, T.K.; Largaespada, D.A.; Rod, A.; Zhang, Y.; Bruner, V.; Abrahante, J.; Schumann, A.; Luczak, T.; et al. CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 2016, 35, 4179–4187. [Google Scholar] [CrossRef]
- Moskowitz, S.M.; Chmiel, J.F.; Sternen, D.L.; Cheng, E.; Gibson, R.L.; Marshall, S.G.; Cutting, G.R. Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders. Genet. Med. 2008, 10, 851–868. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-S.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J. Identification of the cystic fibrosis gene: Cloning and characterization of complementary cDNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Novak, I. The cystic fibrosis of exocrine pancreas. Cold Spring Harb. Perspect. Med. 2013, 3, a009746. [Google Scholar] [CrossRef] [PubMed]
- De Lisle, R.C.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb. Perspect. Med. 2013, 3, a009753. [Google Scholar] [CrossRef] [Green Version]
- Villela, V.R.; Venerando, A.; Cozza, G.; Esposito, S.; Ferrari, E.; Monzani, R.; Spinella, M.C.; Oikonomou, V.; Renga, G.; Tosco, A.; et al. A pathogenic role for cystic fibrosis transmembrane conductance regulator in celiac disease. EMBO J. 2019, 38, e100101. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef]
- Jakab, R.L.; Collaco, A.M.; Ameen, N.A. Physiological relevance of cell-specific distribution patterns of CFTR, NKCC1, NBCe1, and NHE3 along the crypt-villus axis in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G82–G98. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Jakab, R.L.; Collaco, A.M.; Ameen, N.A. Characterization of CFTR High Expresser cells in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G453–G465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitra, A. Cystic Fibrosis. In Robbins and Cotran Pathologic Basis of Disease, 9th ed.; Kumar, V., Abbas, A.K., Aster, J.C., Eds.; Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Sorscher, E.J. Cystic Fibrosis. In Harrison’s Principles of Internal Medicine; Kasper, D., Fauci, A., Hauser, S., Longo, D., Jameson, J.L., Loscalzo, J., Eds.; McGraw-Hill Education: New York, NY, USA, 2015. [Google Scholar]
- Tse, C.M.; Yin, J.; Singh, V.; Tse, C.M.; Yin, J.; Singh, V.; Sarker, R.; Lin, R.; Verkman, A.S.; Turner, J.R.; et al. cAMP Stimulates SLC26A3 Activity in Human Colon by a CFTR-Dependent Mechanism That Does Not Require CFTR Activity. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Liou, T.G. The clinical biology of cystic fibrosis transmembrane regulator protein: Its role and function in extrapulmonary disease. Chest 2019, 155, 605–616. [Google Scholar] [CrossRef]
- Li, C.; Naren, A.P. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 2005, 108, 208–223. [Google Scholar] [CrossRef]
- Guggino, W.B.; Stanton, B.A. New insights into cystic fibrosis: Molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 2006, 7, 426–436. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. Patient Registry. 2019. Available online: https://www.cff.org/ (accessed on 1 December 2019).
- Maisonneuve, P.; Marshall, B.C.; Knapp, E.A.; Lowenfels, A.B. Cancer risk in cystic fibrosis: A 20-year nationwide study from the United States. J. Natl. Cancer Inst. 2013, 105, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, A.; Komaki, Y.; Komaki, F.; Micic, D.; Zullow, S.; Sakuraba, A. Risk of gastrointestinal cancers in patients with cystic fibrosis: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 758–767. [Google Scholar] [CrossRef]
- Billings, J.L.; Dunitz, J.M.; McAllister, S.; Herzog, T.; Bobr, A.; Khoruts, A. Early colon screening of adult patients with cystic fibrosis reveals high incidence of adenomatous colon polyps. J. Clin. Gastroenterol. 2014, 48, e85–e88. [Google Scholar] [CrossRef] [PubMed]
- Hadjiliadis, D.; Khoruts, A.; Zauber, A.G.; Hempstead, S.E.; Maisonneuve, P.; Lowenfels, A.B. Cystic Fibrosis Colorectal Cancer Screening Task Force. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology 2018, 154, 736–745.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cystic Fibrosis Foundation. Carrier Testing for Cystic Fibrosis. 2019. Available online: https://www.cff.org/ (accessed on 1 December 2019).
- Sun, T.T.; Wang, Y.; Cheng, H.; Xiao, H.Z.; Xiang, J.J.; Zhang, J.T.; Yu, S.B.; Martin, T.A.; Ye, L.; Tsang, L.L.; et al. Disrupted interaction between CFTR and AF-6/afadin aggravates malignant phenotypes of colon cancer. Biochim. Biophys. Acta 2014, 1843, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Swahn, H.; Harris, A. Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes (Basel) 2019, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Kerschner, J.L.; Ghosh, S.; Paranjapye, A.; Cosme, W.R.; Audrézet, M.P.; Nakakuki, M.; Ishiguro, H.; Férec, C.; Rommens, J.; Harris, A. Screening for Regulatory Variants in 460 kb Encompassing the CFTR Locus in Cystic Fibrosis Patients. J. Mol. Diagn. 2019, 21, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Son, J.W.; Kim, Y.J.; Cho, H.M.; Lee, S.Y.; Lee, S.M.; Kang, J.K.; Lee, J.U.; Lee, Y.M.; Kwon, S.J.; Choi, E.; et al. Promoter hypermethylation of the CFTR gene and clinical/pathological features associated with non-small cell lung cancer. Respirology 2011, 16, 1203–1209. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, T.; Wang, Z.; Zhang, H.; Qian, Z.; Xu, H.; Gao, B.; Wang, W.; Gu, L.; Meng, J.; et al. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin. Cancer Res. 2007, 13, 7296–7304. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Guo, S.; Sun, J.; Huang, Z.; Zhu, T.; Zhang, H.; Gu, J.; He, Y.; Wang, W.; Ma, K.; et al. Methylcap-seq reveals novel DNA methylation markers for the diagnosis and recurrence prediction of bladder cancer in a Chinese population. PLoS ONE 2012, 7, e35175. [Google Scholar] [CrossRef]
- Van der Heijden, A.G.; Mengual, L.; Ingelmo-Torres, M.; Lozano, J.J.; van Rijt-van de Westerlo, C.C.M.; Baixauli, M.; Geavlete, B.; Moldoveanud, C.; Ene, C.; Dinney, C.P.; et al. Urine cell-based DNA methylation classifier for monitoring bladder cancer. Clin. Epigenetics 2018, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Dong, F.; Gao, H.; Guo, Y.; Li, H.; Yang, F.; Zhao, P.; Dai, Y.; Wang, J.; Zhou, W.; et al. Promoter hypermethylation of the CFTR gene as a novel diagnostic and prognostic marker of breast cancer. Cell Biol. Int. 2020, 44, 603–609. [Google Scholar] [CrossRef]
- Shin, Y.; Kim, M.; Won, J.; Kim, J.; Oh, S.B.; Lee, J.-H.; Park, K. Epigenetic modification of CFTR in head and neck cancer. J. Clin. Med. 2020, 9, 734. [Google Scholar] [CrossRef] [Green Version]
- Starr, T.K.; Allaei, R.; Silverstein, K.A.; Staggs, R.A.; Sarver, A.L.; Bergemann, T.L.; Gupta, M.; O’Sullivan, M.G.; Matise, I.; Dupuy, A.J.; et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009, 323, 1747–1750. [Google Scholar] [CrossRef] [Green Version]
- March, H.N.; Rust, A.G.; Wright, N.A.; ten Hoeve, J.; de Ridder, J.; Eldridge, M.; van der Weyden, L.; Berns, A.; Gadiot, J.; Uren, A.; et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat. Genet. 2011, 43, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M.; Davison, J.; Carter, K.T.; O’Leary, R.M.; Trobridge, P.; Knoblaugh, S.E.; Myeroff, L.L.; Markowitz, S.D.; Brett, B.T.; Scheetz, T.E.; et al. Transposon mutagenesis identifies candidate genes that cooperate with loss of transforming growth factor-beta signaling in mouse intestinal neoplasms. Int. J. Cancer 2017, 140, 853–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strubberg, A.M.; Liu, J.; Walker, N.M.; Stefanski, C.D.; MacLeod, R.J.; Magness, S.T.; Clarke, L.L. Cftr Modulates Wnt/beta-Catenin Signaling and Stem Cell Proliferation in Murine Intestine. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 253–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Singh, J.; Sun, Y.; Ma, X.; Yuan, P. CFTR constrains the differentiation from mouse embryonic stem cells to intestine lineage cells. Biochem. Biophys. Res. Commun. 2019, 510, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Liu, J.; Walker, N.M.; Cook, M.T.; Ootani, A.; Clarke, L.L. Functional Cftr in crypt epithelium of organotypic enteroid cultures from murine small intestine. Am. J. Physiol. Cell Physiol. 2012, 302, C1492–C1503. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholtem, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [Green Version]
- Beekman, J.M. Individualized medicine using intestinal responses to CFTR potentiators and correctors. Pediatr. Pulmonol. 2016, 51, S23–S34. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.R.; Steele, M.S.; Valerio, D.M.; Miron, A.; Mann, R.J.; LePage, D.F.; Conlon, R.A.; Cotton, C.U.; Drumm, M.L.; Hodges, C.A. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS ONE 2018, 13, e0199573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhang, X.; Zhang, J.T.; Tsang, L.L.; Jiang, X.; Chan, H.C. Defective CFTR- B-catenin interaction promotes NF-KB nuclear translocation and intestinal inflammation in cystic fibrosis. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- De Lisle, R.C.; Roach, E.; Jansson, K. Effects of laxative and N-acetylcysteine on mucus accumulation, bacterial load, transit, and inflammation in the cystic fibrosis mouse small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G577–G584. [Google Scholar] [CrossRef]
- Zhang, J.T.; Wang, Y.; Chen, J.J.; Zhang, X.H.; Dong, J.D.; Tsang, L.L.; Huang, X.R.; Cai, Z.; Lan, H.Y.; Jiang, X.H.; et al. Defective CFTR leads to aberrant β-catenin activation and kidney fibrosis. Sci. Rep. 2017, 7, 5233. [Google Scholar] [CrossRef] [Green Version]
- De Sousa, E.M.F.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; van den Bergh, S.P.; et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011, 9, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massagué, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.E.; Larsson, J.M.; Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4659–4665. [Google Scholar] [CrossRef] [Green Version]
- Morrison, C.B.; Markovetz, M.R.; Ehre, C. Mucus, mucins, and cystic fibrosis. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S84–S96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjövall, H.; Hansson, G.C. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 2012, 209, 1263–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Olivier, A.K.; Yi, Y.; Hayden, H.S.; Liang, B.; Sui, H.; Zhou, W.; Hager, K.R.; Zhang, Y.; Liu, X.; et al. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am. J. Pathol. 2014, 184, 1309–1322. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Walker, N.M.; Ootani, A.; Strubberg, A.M.; Clarke, L.L. Defective goblet cell exocytosis contributes to murine cystic fibrosis-associated intestinal disease. J. Clin. Investig. 2015, 125, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- De Lisle, R.C. Disrupted tight junctions in the small intestine of cystic fibrosis mice. Cell Tissue Res. 2014, 355, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Broadbent, D.; Ahmadzai, M.M.; Kammala, A.K.; Yang, C.; Occhiuto, C.; Das, R.; Subramanian, H. Roles of NHERF Family of PDZ-Binding Proteins in Regulating GPCR Functions. Adv. Immunol. 2017, 136, 353–385. [Google Scholar] [CrossRef]
- Ruan, Y.C.; Wang, Y.; Da Silva, N.; Kim, B.; Diao, R.Y.; Hill, E.; Brown, D.; Chan, H.C.; Breton, S. CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J. Cell Sci. 2014, 127, 4396–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mátyási, B.; Farkas, Z.; Kopper, L.; Sebestyén, A.; Boissan, M.; Mehta, A.; Takács-Vellai, K. The Function of NM23-H1/NME1 and Its Homologs in Major Processes Linked to Metastasis. Pathol. Oncol. Res. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muimo, R.; Alothaid, H.M.; Mehta, A. NM23 proteins: Innocent bystanders or local energy boosters for CFTR? Lab. Investig. 2018, 98, 272–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munck, A. Cystic fibrosis: Evidence for gut inflammation. Int. J. Biochem. Cell Biol. 2014, 52, 180–183. [Google Scholar] [CrossRef]
- Norkina, O.; Burnett, T.G.; De Lisle, R.C. Bacterial overgrowth in the cystic fibrosis transmembrane conductance regulator null mouse small intestine. Infect. Immun. 2004, 72, 6040–6049. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Goldfarb, K.C.; Wild, Y.K.; Kong, W.; De Lisle, R.C.; Brodie, E.L. Cystic fibrosis transmembrane conductance regulator knockout mice exhibit aberrant gastrointestinal microbiota. Gut Microbes 2013, 4, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Vernocchi, P.; Del Chierico, F.; Russo, A.; Majo, F.; Rossitto, M.; Valerio, M.; Casadei, L.; La Storia, A.; De Filippis, F.; Rizzo, C.; et al. Gut microbiota signatures in cystic fibrosis: Loss of host CFTR function drives the microbiota enterophenotype. PLoS ONE 2018, 13, e0208171. [Google Scholar] [CrossRef]
- Burke, D.G.; Fouhy, F.; Harrison, M.J.; Rea, M.C.; Cotter, P.D.; O’Sullivan, O.; Stanton, C.; Hill, C.; Shanahan, F.; Plant, B.J.; et al. The altered gut microbiota in adults with cystic fibrosis. BMC Microbiol. 2017, 17, 58. [Google Scholar] [CrossRef] [Green Version]
- Bruzzese, E.; Callegari, M.L.; Raia, V.; Viscovo, S.; Scotto, R.; Ferrari, S.; Morelli, L.; Buccigrossi, V.; Lo Vecchio, A.; Ruberto, E.; et al. Disrupted intestinal microbiota and intestinal inflammation in children with cystic fibrosis and its restoration with Lactobacillus GG: A randomised clinical trial. PLoS ONE 2014, 9, e87796. [Google Scholar] [CrossRef] [Green Version]
- Garg, M.; Ooi, C.Y. The enigmatic gut in cystic fibrosis: Linking inflammation, dysbiosis, and the increased risk of malignancy. Curr. Gastroenterol. Rep. 2017, 19, 6. [Google Scholar] [CrossRef]
- Meeker, S.M.; Mears, K.S.; Sangwan, N.; Brittnacher, M.J.; Weiss, E.J.; Treuting, P.M.; Tolley, N.; Pope, C.E.; Hager, K.R.; Vo, A.T.; et al. CFTR dysregulation drives active selection of the gut microbiome. PLoS Pathog. 2020, 16, e1008251. [Google Scholar] [CrossRef] [PubMed]
- Manor, O.; Levy, R.; Pope, C.E.; Hayden, H.S.; Brittnacher, M.J.; Carr, R.; Radey, M.C.; Hager, K.R.; Heltshe, S.L.; Ramsey, B.W.; et al. Metagenomic evidence for taxonomic dysbiosis and functional imbalance in the gastrointestinal tracts of children with cystic fibrosis. Sci. Rep. 2016, 6, 22493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigo Salado, C.; Leo Carnerero, E.; de la Cruz Ramírez, M.D. Crohn’s disease and cystic fibrosis: There is still a lot to learn. Rev. Esp. Enferm Dig. 2018, 110, 835–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayama, G.; Priya, S.; Niccum, D.E.; Khoruts, A.; Blekhman, R. Interactions between the gut microbiome and host gene regulation in cystic fibrosis. Genome Med. 2020, 12, 12. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Luo, Y.; Lv, Y.; Huang, C.; Sheng, Q.; Zhao, P.; Ye, J.; Jiang, W.; Liu, L. Clostridium difficile colonization in preoperative colorectal cancer patients. Oncotarget 2017, 8, 11877–11886. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Mori, G.; Rampelli, S.; Orena, B.S.; Rengucci, C.; De Maio, G.; Barbieri, G.; Passardi, A.; Casadei Gardini, A.; Frassineti, G.L.; Gaiarsa, S.; et al. Shifts of Faecal Microbiota During Sporadic Colorectal Carcinogenesis. Sci. Rep. 2018, 8, 10329. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Huber, A.R.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020. [Google Scholar] [CrossRef]
- Werlin, S.L.; Benuri-Silbiger, I.; Kerem, E.; Adler, S.N.; Goldin, E.; Zimmerman, J.; Malka, N.; Cohen, L.; Armoni, S.; Yatzkan-Israelit, Y.; et al. Evidence of intestinal inflammation in patients with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Norkina, O.; Kaur, S.; Ziemer, D.; De Lisle, R.C. Inflammation of the cystic fibrosis mouse small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G1032–G1041. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.L.; Croft, N.M.; O’Hea, U.; Marshall, T.G.; Ferguson, A. Intestinal inflammation in cystic fibrosis. Arch. Dis. Child. 2000, 82, 394–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhaliwal, J.; Leach, S.; Katz, T.; Nahidi, L.; Pang, T.; Lee, J.M.; Strachan, R.; Day, A.S.; Jaffe, A.; Ooi, C.Y. Intestinal inflammation and impact on growth in children with cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Bruzzese, E.; Raia, V.; Gaudiello, G.; Polito, G.; Buccigrossi, V.; Formicola, V.; Guarino, A. Intestinal inflammation is a frequent feature of cystic fibrosis and is reduced by probiotic administration. Aliment. Pharmacol. Ther. 2004, 20, 813–819. [Google Scholar] [CrossRef]
- Mulcahy, E.M.; Cooley, M.A.; McGuire, H.; Asad, S.; Fazekas de St Groth, B.; Beggs, S.A.; Roddam, L.F. Widespread alterations in the peripheral blood innate immune cell profile in cystic fibrosis reflect lung pathology. Immunol. Cell Biol. 2019, 97, 416–426. [Google Scholar] [CrossRef]
- Johansson, J.; Vezzalini, M.; Verze, G.; Caldrer, S.; Bolognin, S.; Buffelli, M.; Bellisola, G.; Tridello, G.; Assael, B.M.; Melotti, P.; et al. Detection of CFTR protein in human leukocytes by flow cytometry. Cytometry 2014, 85, 611–620. [Google Scholar] [CrossRef]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef]
- Xie, C.; Jiang, X.H.; Zhang, J.T.; Sun, T.T.; Dong, J.D.; Sanders, A.J.; Diao, R.Y.; Wang, Y.; Fok, K.L.; Tsang, L.L.; et al. CFTR suppresses tumor progression through miR-193b targeting urokinase plasminogen activator (uPA) in prostate cancer. Oncogene 2013, 32, 2282–2291. [Google Scholar] [CrossRef]
- Zhang, J.T.; Jiang, X.H.; Xie, C.; Cheng, H.; Da Dong, J.; Wang, Y.; Fok, K.L.; Zhang, X.H.; Sun, T.T.; Tsang, L.L.; et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim. Biophys. Acta 2013, 1833, 2961–2969. [Google Scholar] [CrossRef] [Green Version]
- Crites, K.S.; Morin, G.; Orlando, V.; Patey, N.; Cantin, C.; Martel, J.; Brochiero, E.; Mailhot, G. CFTR Knockdown induces proinflammatory changes in intestinal epithelial cells. J. Inflamm. (Lond.) 2015, 12, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleme, M.L.; Sané, A.T.; Garofalo, C.; Levy, E. Targeted CFTR gene disruption with zinc-finger nucleases in human intestinal epithelial cells induces oxidative stress and inflammation. Int. J. Biochem. Cell Biol. 2016, 74, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Mazur, S.; Zeitlin, P.L. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS ONE 2009, 4, e4664. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Roesch, E.A.; Perez, A.; Weiner, R.L.; Henderson, L.C.; Cummings, L.; Consiglio, P.; Pajka, J.; Eisenberg, A.; Yeh, L.; et al. Inactivation of CFTR by CRISPR/Cas9 alters transcriptional regulation of inflammatory pathways and other networks. J. Cyst. Fibros. 2020, 19, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Massip-Copiz, M.M.; Clauzure, M.; Valdivieso, A.G.; Santa-Coloma, T.A. CFTR impairment upregulates c-Src activity through IL-1beta autocrine signaling. Arch. Biochem. Biophys. 2017, 616, 1–12. [Google Scholar] [CrossRef]
- Liu, K.; Wang, X.; Zou, C.; Chen, H.; Tsang, L.; Yu, M.K.; Chung, Y.W.; Wang, J.; Dai, Y.; Liu, Y.; et al. Defective CFTR promotes intestinal proliferation via inhibition of the hedgehog pathway during cystic fibrosis. Cancer Lett. 2019, 446, 15–24. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Paradiso, A.M.; Carew, M.A.; Shears, S.B.; Boucher, R.C. Cystic fibrosis airway epithelial Ca2+ i signaling: The mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia. J. Biol. Chem. 2005, 280, 10202–10209. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Norez, C.; Becq, F.; Vandebrouck, C. CFTR and Ca Signaling in Cystic Fibrosis. Front. Pharmacol. 2011, 2, 67. [Google Scholar] [CrossRef] [Green Version]
- Vachel, L.; Norez, C.; Becq, F.; Vandebrouck, C. Effect of VX-770 (ivacaftor) and OAG on Ca2+ influx and CFTR activity in G551D and F508del-CFTR expressing cells. J. Cyst. Fibros. 2013, 12, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Philippe, R.; Antigny, F.; Buscaglia, P.; Norez, C.; Becq, F.; Frieden, M.; Mignen, O. SERCA and PMCA pumps contribute to the deregulation of Ca2+ homeostasis in human CF epithelial cells. Biochim. Biophys. Acta 2015, 1853, 892–903. [Google Scholar] [CrossRef]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef]
- Su, L.; Shen, L.; Clayburgh, D.R.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted Epithelial Tight Junction Dysfunction Causes Immune Activation and Contributes to Development of Experimental Colitis. Gastroenterology 2009, 136, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorotto, R.; Villani, A.; Kourtidis, A.; Scirpo, R.; Amenduni, M.; Geibel, P.J.; Cadamuro, M.; Spirli, C.; Anastasiadis, P.Z.; Strazzabosco, M. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity. Hepatology 2016, 64, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Jiang, X.; Zhang, X.; Liu, K.S.; Zhang, J.; Chen, J.; Yu, M.K.; Tsang, L.L.; Chung, Y.W.; Wang, Y.; et al. Dynamically Regulated CFTR Expression and Its Functional Role in Cutaneous Wound Healing. J. Cell. Physiol. 2015, 230, 2049–2058. [Google Scholar] [CrossRef]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Kleme, M.L.; Sane, A.; Garofalo, C.; Seidman, E.; Brochiero, E.; Berthiaume, Y.; Levy, E. CFTR deletion confers mitochondrial dysfunction and disrupts lipid homeostasis in intestinal epithelial cells. Nutrients 2018, 10, 836. [Google Scholar] [CrossRef] [Green Version]
- Favia, M.; de Bari, L.; Bobba, A.; Atlante, A. An Intriguing Involvement of Mitochondria in Cystic Fibrosis. J. Clin. Med. 2019, 8, 1890. [Google Scholar] [CrossRef] [Green Version]
- Valdivieso, A.G.; Santa-Coloma, T.A. CFTR activity and mitochondrial function. Redox Biol. 2013, 5, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.; Sun, L.; Yuan, C.; Jiang, M.; Lou, Q.; Xu, Y. CFTR ameliorates high glucose-induced oxidative stress and inflammation by mediating the NF-κB and MAPK signaling pathways in endothelial cells. Int. J. Mol. Med. 2018, 41, 3501–3508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell. Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Kuhlicke, J.; Jäckel, K.; Eltzschig, H.K.; Singh, A.; Sjöblom, M.; Riederer, B.; Weinhold, C.; Seidler, U.; Colgan, S.P.; et al. Hypoxia inducible factor-1 (HIF-1)-mediated repression of cystic fibrosis transmembrane conductance regulator (CFTR) in the intestinal epithelium. FASEB J. 2009, 23, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhania, M.; University of Minnesota Medical School, Duluth, Minnesota, USA. Unpublished data. Personal communication, 2020.
- Anderson, K.; University of Minnesota Medical School, Duluth, Minnesota, USA. Unpublished data. Personal communication, 2020.
- Zhang, J.; Wang, Y.; Jiang, X.; Chan, H.C. Cystic fibrosis transmembrane conductance regulator-emerging regulator of cancer. Cell Mol. Life Sci. 2018, 75, 1737–1756. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Z.; Wu, Y.; Babovic-Vuksanovic, D.; Li, Y.; Cunningham, J.M.; Pankratz, V.S.; Yang, P. Cystic fibrosis transmembrane conductance regulator gene mutation and lung cancer risk. Lung Cancer 2010, 70, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yan, T.; Gong, Y.; Liu, X.; Sun, H.; Xu, W.; Wang, C.; Naren, D.; Zheng, Y. High CFTR expression in Philadelphia chromosome-positive acute leukemia protects and maintains continuous activation of BCR-ABL and related signaling pathways in combination with PP2A. Oncotarget 2017, 8, 24437–24448. [Google Scholar] [CrossRef] [Green Version]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, J.T.; Jiang, X.; Shi, X.; Shen, J.; Feng, F.; Chen, J.; Liu, G.; He, P.; Jiang, J.; et al. The cystic fibrosis transmembrane conductance regulator as a biomarker in non-small cell lung cancer. Int. J. Oncol. 2015, 46, 2107–2115. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Chen, H.Q.; Yang, X.L.; Wang, Q.; Chen, W.; Li, C. CFTR activation suppresses glioblastoma cell proliferation, migration and invasion. Biochem. Biophys. Res. Commun. 2019, 508, 1279–1285. [Google Scholar] [CrossRef]
- Li, W.; Wang, C.; Peng, X.; Zhang, H.; Huang, H.; Liu, H. CFTR inhibits the invasion and growth of esophageal cancer cells by inhibiting the expression of NF-kappaB. Cell Biol. Int. 2018, 42, 1680–1687. [Google Scholar] [CrossRef]
- Gharahkhani, P.; Fitzgerald, R.C.; Vaughan, T.L.; Palles, C.; Gockel, I.; Tomlinson, I.; Buas, M.F.; May, A.; Gerges, C.; Anders, M.; et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: A large-scale meta-analysis. Lancet Oncol. 2016, 17, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Cazacu, I.M.; Farkas, N.; Garami, A.; Balaskó, M.; Mosdósi, B.; Alizadeh, H.; Gyöngyi, Z.; Rakonczay, Z.J., Jr.; Vigh, É.; Habon, T.; et al. Pancreatitis-Associated Genes and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. Pancreas 2018, 47, 1078–1086. [Google Scholar] [CrossRef]
- McWilliams, R.R.; Petersen, G.M.; Rabe, K.G.; Holtegaard, L.M.; Lynch, P.J.; Bishop, M.D.; Highsmith, W.E., Jr. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations and risk for pancreatic adenocarcinoma. Cancer 2010, 116, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Earl, J.; Galindo-Pumariño, C.; Encinas, J.; Barreto, E.; Castillo, M.E.; Pachón, V.; Ferreiro, R.; Rodríguez-Garrote, M.; González-Martínez, S.; Ramon, Y.; et al. A comprehensive analysis of candidate genes in familial pancreatic cancer families reveals a high frequency of potentially pathogenic germline variants. EBioMedicine 2020, 53, 102675. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Chen, Q.; Zhang, J.T.; Jiang, X.; Xia, Y.; Chan, H.C. CFTR is a potential marker for nasopharyngeal carcinoma prognosis and metastasis. Oncotarget 2016, 7, 76965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshani, T.; Kerem, E.; Szeinberg, A.; Augarten, A.; Yahav, Y.; Cohen, D.; Rivlin, J.; Tal, A.; Kerem, B. Similar levels of mRNA from the W1282X and the delta F508 cystic fibrosis alleles, in nasal epithelial cells. J. Clin. Investig. 1994, 93, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, H.; Yuan, C.; Luo, M.; Wei, J.; Liu, X. Cigarette Smoke-Induced Acquired Dysfunction of Cystic Fibrosis Transmembrane Conductance Regulator in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Oxid. Med. Cell Longev. 2018, 2018, 6567578. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, N.; Wang, J.; Wang, Y.; Yuan, C.; Wu, J.; Luo, M.; Yang, J.; Chen, J.; Shi, J.; et al. Nicotine Induces Progressive Properties of Lung Adenocarcinoma A549 Cells by Inhibiting Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Expression and Plasma Membrane Localization. Technol. Cancer Res. Treat. 2018, 17, 1533033818809984. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. CFTR Modulator Therapies. 2019. Available online: https://www.cff.org (accessed on 1 December 2019).
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, F.; Wilschanski, M. CFTR Protein Function Modulation Therapy Is Finally Targeting Cystic Fibrosis-related Gastrointestinal Disease. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 372–373. [Google Scholar] [CrossRef]
- Carrion, A.; Borowitz, D.S.; Freedman, S.D.; Siracusa, C.M.; Goralski, J.L.; Hadjiliadis, D.; Srinivasan, S.; Stokes, D.C. Reduction of Recurrence Risk of Pancreatitis in Cystic Fibrosis With Ivacaftor: Case Series. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Gelfond, D.; Heltshe, S.; Ma, C.; Rowe, S.M.; Frederick, C.; Uluer, A.; Sicilian, L.; Konstan, M.; Tullis, E.; Roach, R.N.; et al. Impact of CFTR modulation on intestinal pH, motility, and clinical outcomes in patients with cystic fibrosis and the G551D mutation. Clin. Transl. Gastroenterol. 2017, 8, e81. [Google Scholar] [CrossRef]
- Ferrera, L.; Baroni, D.; Moran, O. Lumacaftor-rescued F508del-CFTR has a modified bicarbonate permeability. J. Cyst. Fibros. 2019. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Syed, S.A.; Rossi, L.; Garg, M.; Needham, B.; Avolio, J.; Young, K.; Surette, M.G.; Gonska, T. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci. Rep. 2018, 8, 17834. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.Y.; Sohma, Y.; Hwang, T.C. Synergistic Potentiation of Cystic Fibrosis Transmembrane Conductance Regulator Gating by Two Chemically Distinct Potentiators, Ivacaftor (VX-770) and 5-Nitro-2-(3-Phenylpropylamino) Benzoate. Mol. Pharmacol. 2016, 90, 275–285. [Google Scholar] [CrossRef] [Green Version]
- De Poel, E.; Lefferts, J.W.; Beekman, J.M. Intestinal organoids for Cystic Fibrosis research. J. Cyst. Fibros. 2020, 19 (Suppl. 1), S60–S64. [Google Scholar] [CrossRef] [Green Version]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.; Houwen, R.H.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Pedron, T.; Mulet, C.; Dauga, C.; Frangeul, L.; Chervaux, C.; Grompone, G.; Sansonetti, P.J. A crypt-specific core microbiota resides in the mouse colon. MBio 2012, 3, e00116-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, G.; Rossi, R.; Commere, P.H.; Jay, P.; Sansonetti, P.J. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 2014, 15, 792–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
↓Cystic fibrosis transmembrane conductance regulator (CFTR) → ↑ Wnt/β-catenin signaling → ↑ proliferation. CFTR deficiency leads to increased cellular response to Wnt/β-catenin signaling and cellular proliferation. For example, CFTR deficiency enhances survival of colon organoids from CFTR knockout (KO) mice [5]. Increases in Wnt/β-catenin signaling is a hallmark of colorectal cancer (CRC) development. Figure created using BioRender.com.
Figure 1.
↓Cystic fibrosis transmembrane conductance regulator (CFTR) → ↑ Wnt/β-catenin signaling → ↑ proliferation. CFTR deficiency leads to increased cellular response to Wnt/β-catenin signaling and cellular proliferation. For example, CFTR deficiency enhances survival of colon organoids from CFTR knockout (KO) mice [5]. Increases in Wnt/β-catenin signaling is a hallmark of colorectal cancer (CRC) development. Figure created using BioRender.com.

Figure 2.
CFTR deficiency disrupts protective physical barriers and leads to dysbiosis. CFTR deficiency causes a failure of intestinal cell chloride and bicarbonate ion efflux and accompanying water efflux. This results in dehydration of the mucus layer, making it permissive to bacterial passage, and also causing intestinal obstruction. Disruption of the epithelial barrier leads to infiltration of commensal and pathogenic bacteria, inflammation, epithelial tissue damage, and immune cell infiltration. These alterations in the intestinal landscape (mutations, inflammatory signaling) create favorable conditions for CRC initiation and progression. Figure created using BioRender.com.
Figure 2.
CFTR deficiency disrupts protective physical barriers and leads to dysbiosis. CFTR deficiency causes a failure of intestinal cell chloride and bicarbonate ion efflux and accompanying water efflux. This results in dehydration of the mucus layer, making it permissive to bacterial passage, and also causing intestinal obstruction. Disruption of the epithelial barrier leads to infiltration of commensal and pathogenic bacteria, inflammation, epithelial tissue damage, and immune cell infiltration. These alterations in the intestinal landscape (mutations, inflammatory signaling) create favorable conditions for CRC initiation and progression. Figure created using BioRender.com.

Figure 3.
CFTR deficiency improves survival of CRC cells under oxidative stress. Cancer cells are subject to oxidative stress via increased production of reactive oxygen species (ROS) and are therefore susceptible to oxidative-stress-induced cell death. A potentially protective mechanism for CRC cells involves CFTR-deficiency leading to increased retention of the antioxidant glutathione (GSH) in mitochondria. Enhanced GSH could cause decreased ROS levels, decreased oxidative stress and decreased apoptosis. Through this mechanism CFTR-deficiency may promote survival of CRC cells. Figure created using BioRender.com.
Figure 3.
CFTR deficiency improves survival of CRC cells under oxidative stress. Cancer cells are subject to oxidative stress via increased production of reactive oxygen species (ROS) and are therefore susceptible to oxidative-stress-induced cell death. A potentially protective mechanism for CRC cells involves CFTR-deficiency leading to increased retention of the antioxidant glutathione (GSH) in mitochondria. Enhanced GSH could cause decreased ROS levels, decreased oxidative stress and decreased apoptosis. Through this mechanism CFTR-deficiency may promote survival of CRC cells. Figure created using BioRender.com.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Scott, P.; Anderson, K.; Singhania, M.; Cormier, R. Cystic Fibrosis, CFTR, and Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 2891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082891
AMA Style
Scott P, Anderson K, Singhania M, Cormier R. Cystic Fibrosis, CFTR, and Colorectal Cancer. International Journal of Molecular Sciences. 2020; 21(8):2891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082891
Chicago/Turabian StyleScott, Patricia, Kyle Anderson, Mekhla Singhania, and Robert Cormier. 2020. "Cystic Fibrosis, CFTR, and Colorectal Cancer" International Journal of Molecular Sciences 21, no. 8: 2891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082891
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.