FGCaMP7, an Improved Version of Fungi-Based Ratiometric Calcium Indicator for In Vivo Visualization of Neuronal Activity

 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
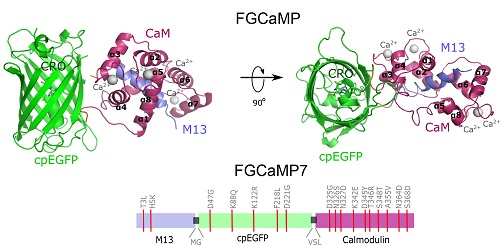
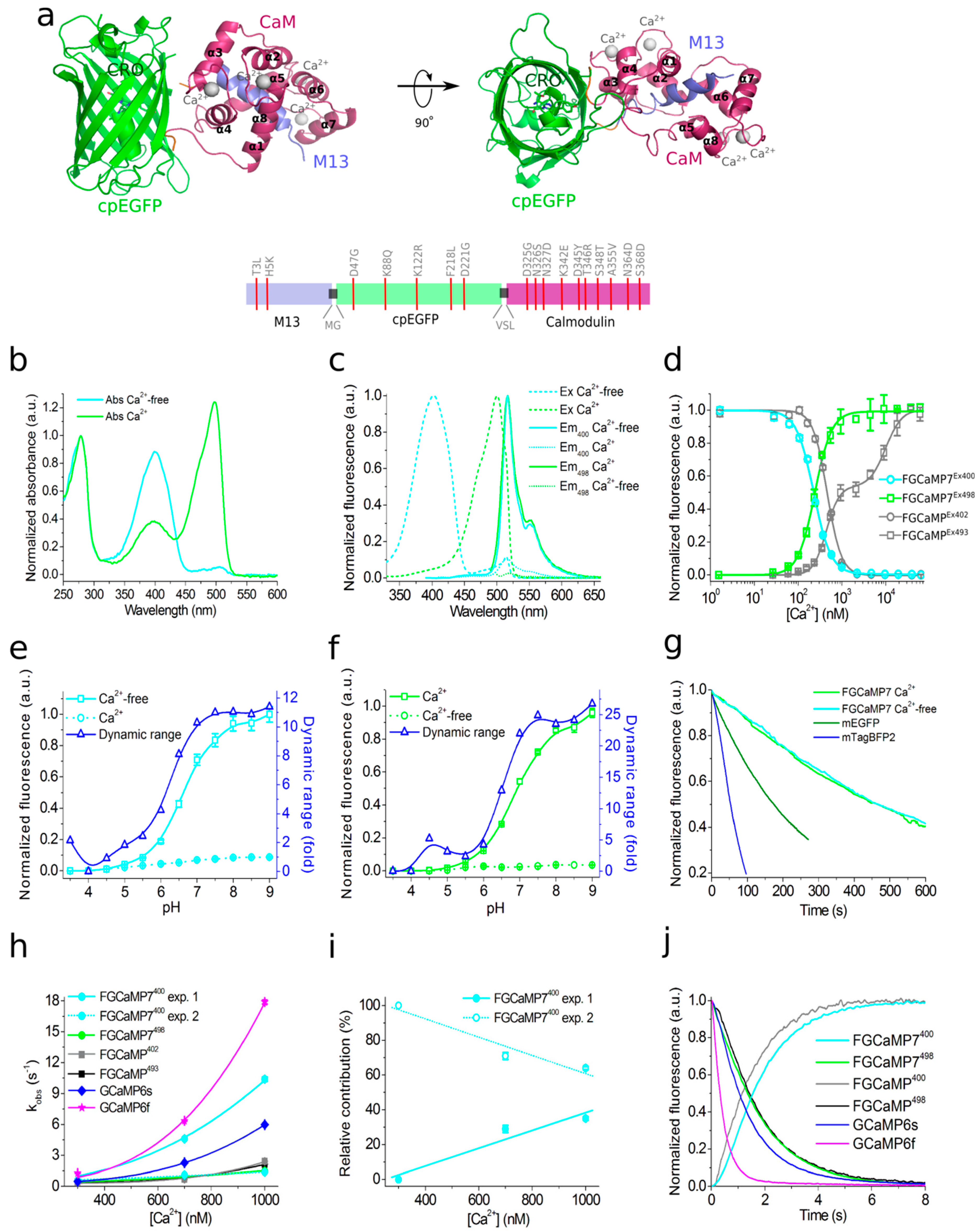
2.1. Structural Characterization of FGCaMP Calcium Indicator
2.2. Development of Improved Version of GECI Based on Fungi Calcium-Binding Parts
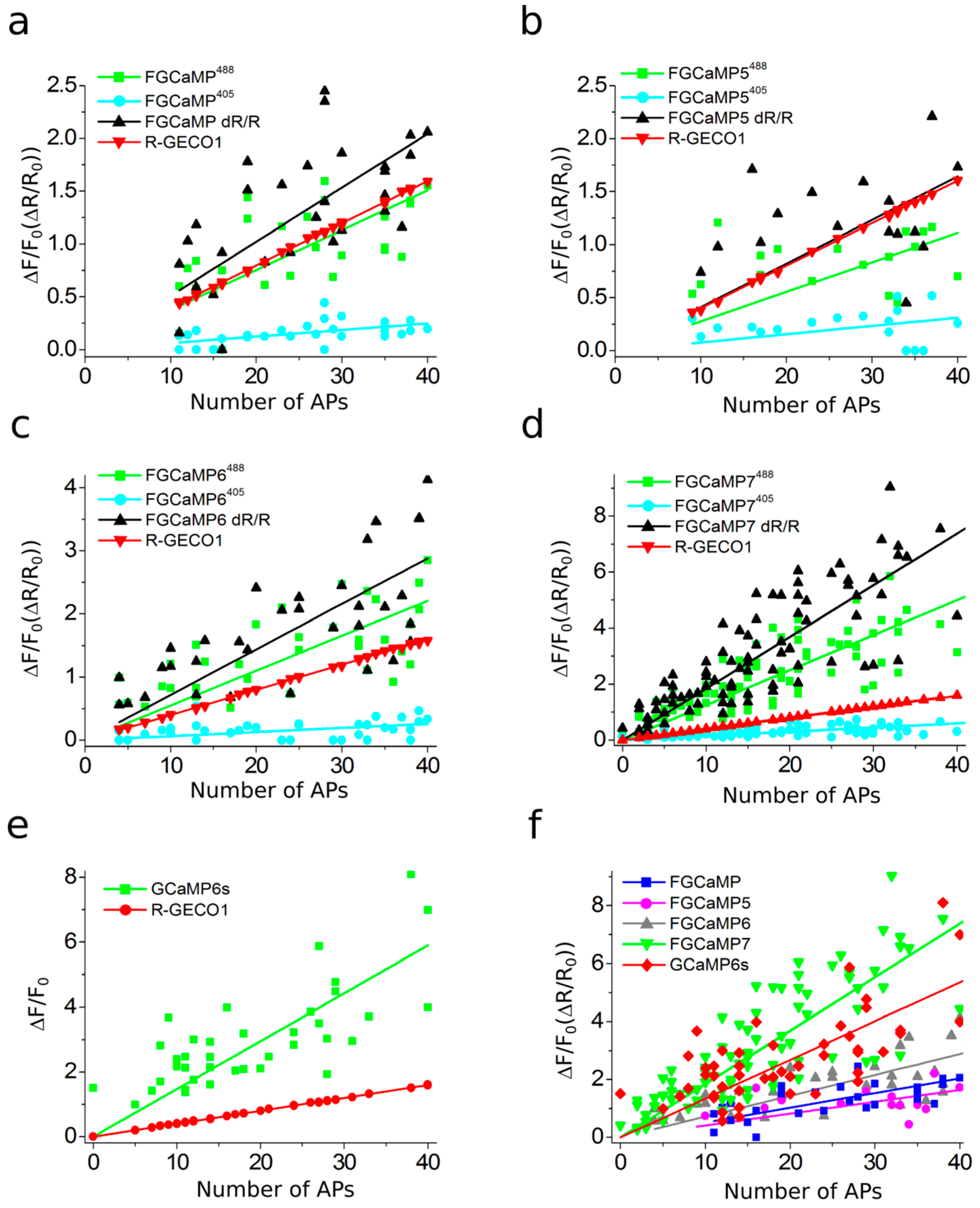
2.3. In Vitro Properties of Improved Purified FGCaMP Variants
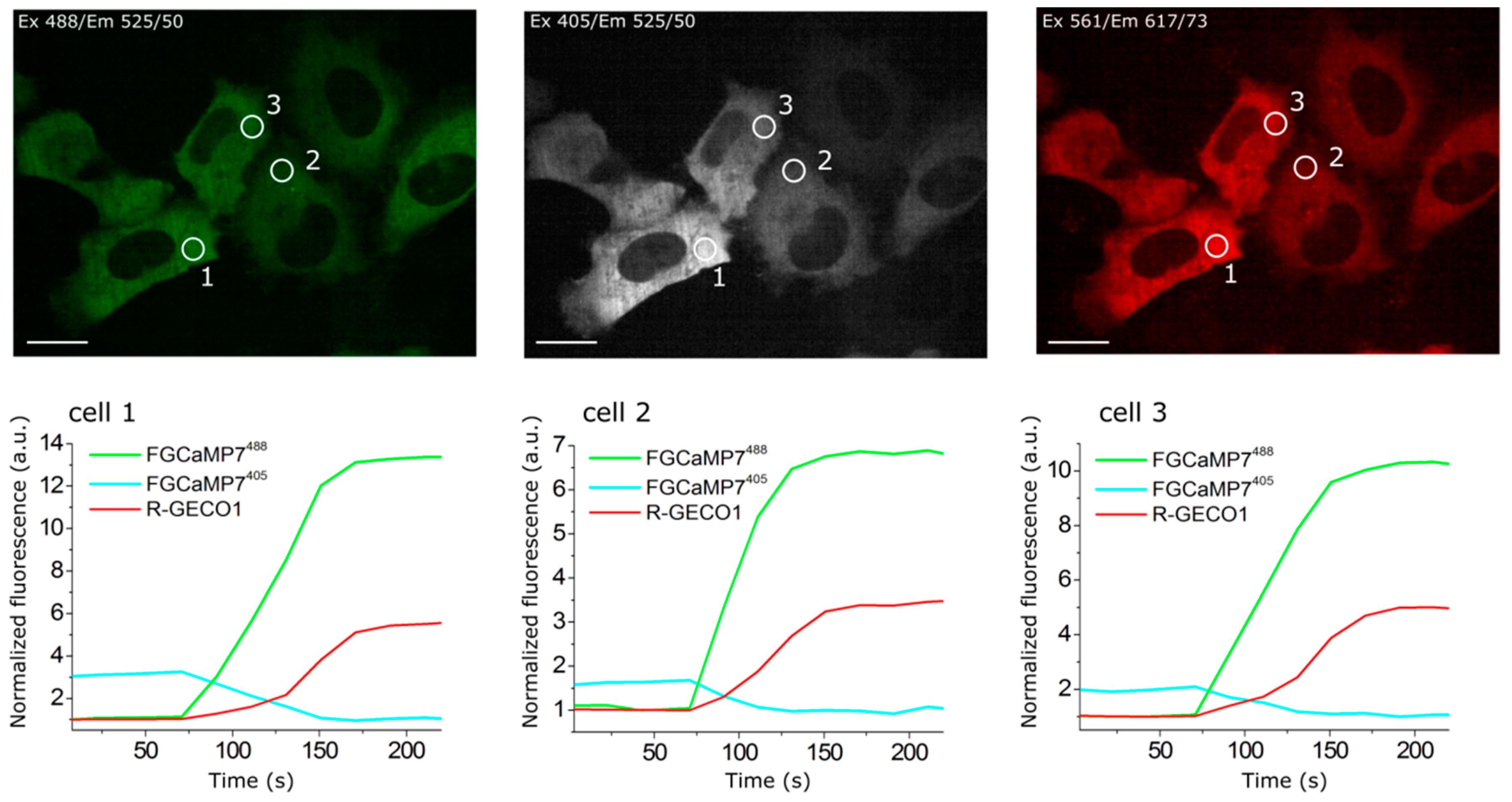
2.4. Calcium-Dependent Response of Improved FGCaMP Indicators in HeLa Cells
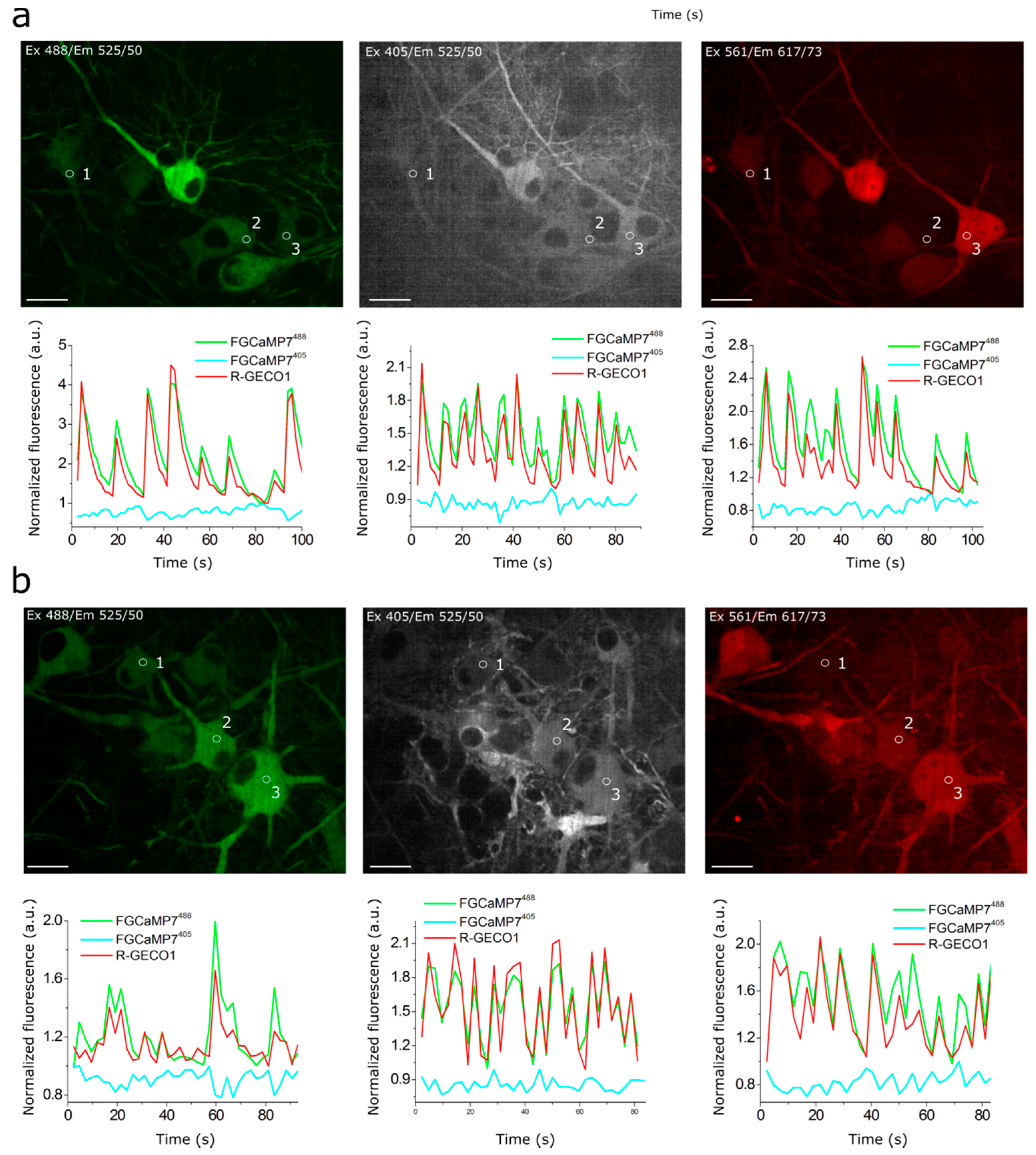
2.5. Visualization of Spontaneous Activity in Neuronal Cultures Using Enhanced Versions of FGCaMP Indicator
2.6. Responses of Improved FGCaMP Indicators to External Electric Stimulation of Neuronal Cultures
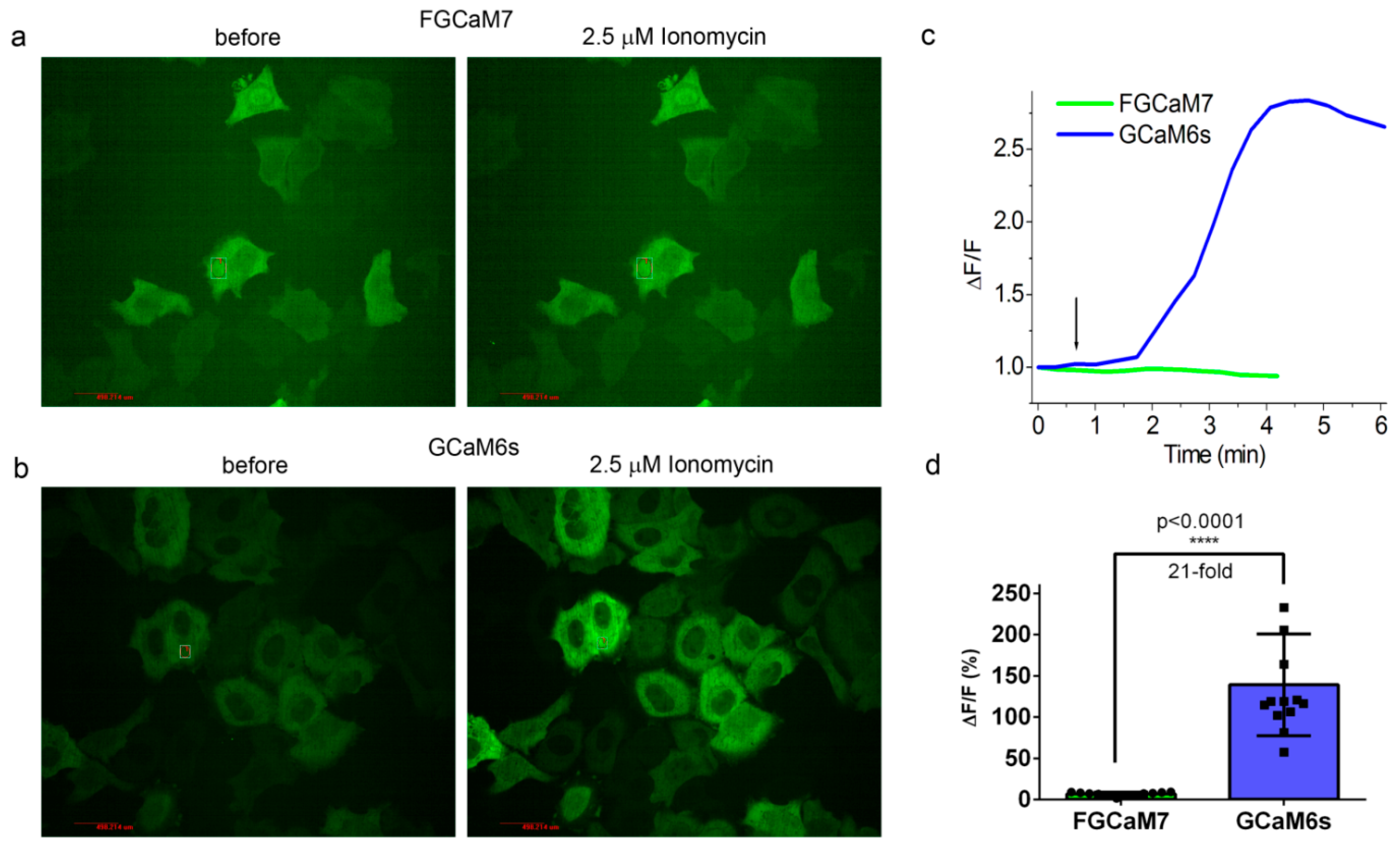
2.7. Characterization of Truncated Versions (with Deleted M13-Like Peptide) of FGCaMP7 and GCaMP6s Indicators In Vitro and in HeLa Cells
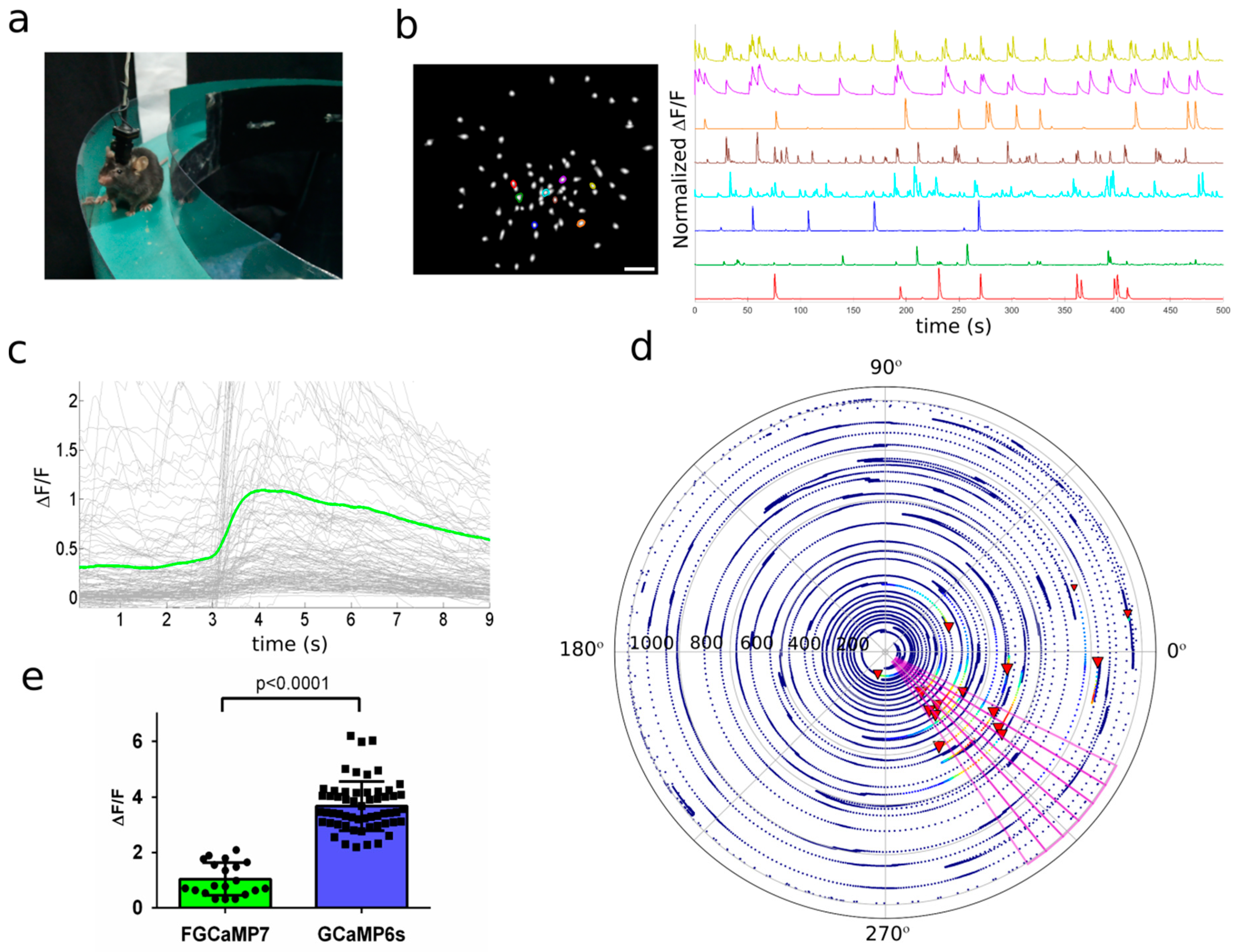
2.8. In Vivo Imaging of Hippocampal Neuronal Activity in Freely Moving Mice Using NVista Miniscope and FGCaMP7 Calcium Indicator
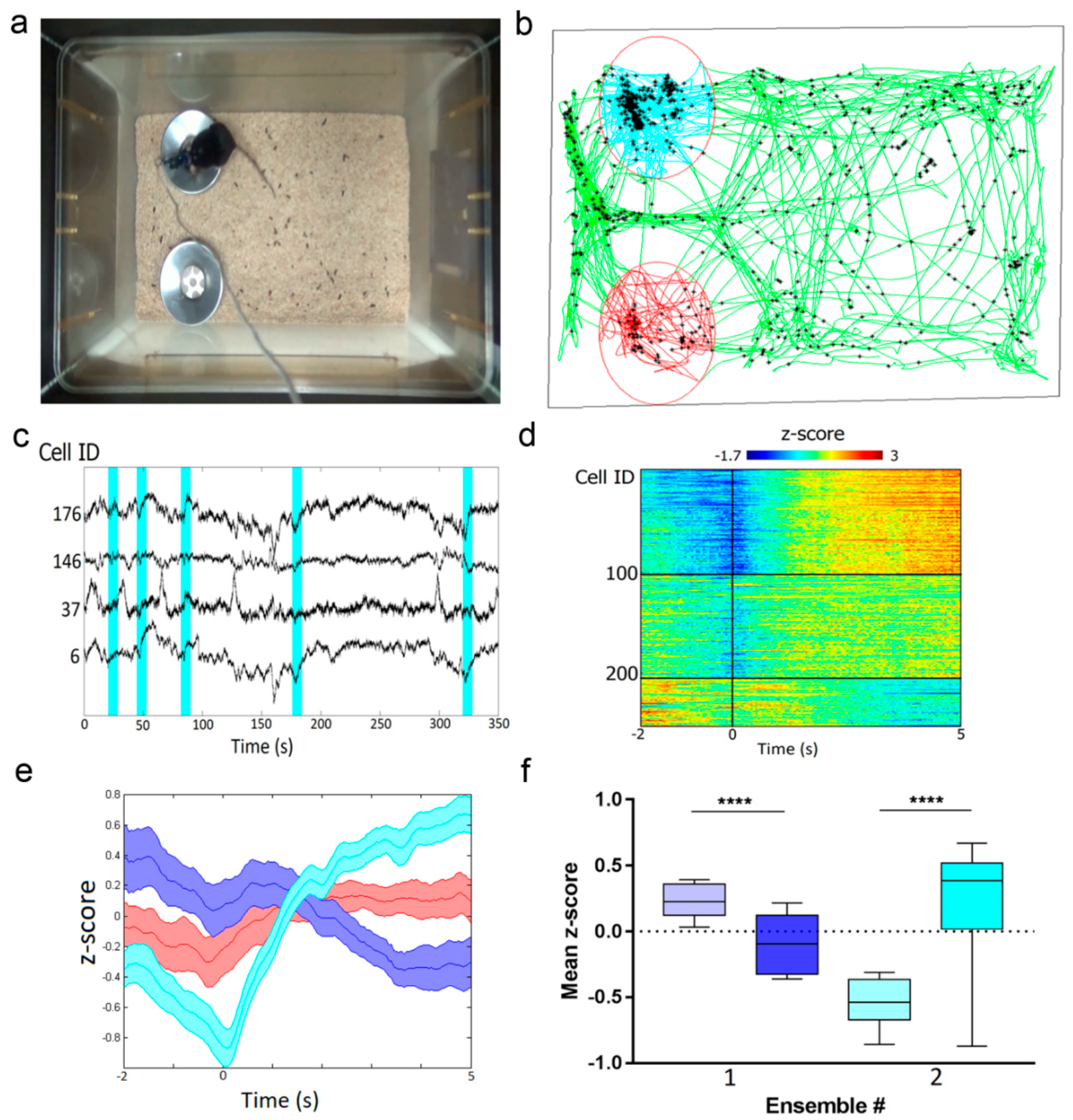
2.9. In Vivo Imaging of Hippocampal Neuronal Ensembles in Mice during Food Intake Using NVista Miniscope and FGCaMP7 Calcium Indicator
2.10. Two-Photon In Vivo Imaging of Visual Cortex in Awake Mice Expressing FGCaMP7 Calcium Indicator
2.11. In Vivo Ratiometric Two-Color Imaging of Neuronal Activity in Zebrafish Using FGCaMP7
3. Materials and Methods
3.1. Protein Purification for X-ray Crystallography
3.2. Crystallization of FGCaMP
3.3. Data Collection, Processing, Structure Solution and Refinement
3.4. Structure Analysis and Validation
3.5. Mutagenesis and Library Screening
3.6. Proteins Purification and Characterization
3.7. Stopped-Flow Fluorimetry
3.8. Mammalian Plasmid Construction
3.9. Cell Culture and Transfection
3.10. Mammalian Live-Cell Imaging
3.11. rAAV Particles Production and Isolation
3.12. Isolation, Transduction, and Imaging of Neuronal Cultures
3.13. Stimulation of Neuronal Cultures with Electric Field
3.14. Surgery and Imaging with an nVista HD Miniature Microscope
3.15. Viral Injection to the Neonatal Mouse Brain and Surgery for in Vivo Two-Photon Imaging
3.16. Two-Photon in Vivo Imaging in Mouse V1
3.17. In Vivo Imaging of Neuronal Activity in Zebrafish Larvae
3.18. Analysis of FGCaMP7 Dynamics in Zebrafish Larvae
3.19. Statistics
3.20. Ethical Approval and Animal Care
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| CaM | Calmodulin |
| H-bond | Hydrogen bond |
| GECI | Genetically Encoded Calcium Indicator |
| DPF | Days post-fertilization |
| FRET | Förster Resonance Energy Transfer |
| FP | Fluorescent Protein |
| EGFP | Enhanced Green Fluorescent Protein |
| PDB ID | Protein Data Bank IDentifier |
| BFP | Blue Fluorescent Protein |
| AP | Action Potential |
| FRAP | Fluorescence Recovery After Photobleaching |
| QY | Quantum Yield |
| MAD | Median Absolute Deviation |
| TEV | Tobacco Etch Virus |
| TLS | Translation Libration Screw-motion refinement |
| NCS | Non-Crystallographic Symmetry |
| SNR | Signal-to-Noise Ratio |
References
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882. [Google Scholar] [CrossRef]
- Thestrup, T.; Litzlbauer, J.; Bartholomaus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An expanded palette of genetically encoded Ca2+ indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Dana, H.; Sun, Y.; Mohar, B.; Hulse, B.K.; Kerlin, A.M.; Hasseman, J.P.; Tsegaye, G.; Tsang, A.; Wong, A.; Patel, R.; et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 2019, 16, 649–657. [Google Scholar] [CrossRef]
- Barykina, N.V.; Subach, O.M.; Doronin, D.A.; Sotskov, V.P.; Roshchina, M.A.; Kunitsyna, T.A.; Malyshev, A.Y.; Smirnov, I.V.; Azieva, A.M.; Sokolov, I.S.; et al. A new design for a green calcium indicator with a smaller size and a reduced number of calcium-binding sites. Sci. Rep. 2016, 6, 34447. [Google Scholar] [CrossRef]
- Doronin, D.A.; Barykina, N.V.; Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Ivleva, O.A.; Isaakova, E.A.; Varizhuk, A.M.; Pozmogova, G.E.; Malyshev, A.Y.; et al. Genetically encoded calcium indicator with NTnC-like design and enhanced fluorescence contrast and kinetics. BMC Biotechnol. 2018, 18, 10. [Google Scholar] [CrossRef]
- Barykina, N.V.; Doronin, D.A.; Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Ivleva, O.A.; Gruzdeva, A.M.; Kunitsyna, T.A.; Ivashkina, O.I.; Lazutkin, A.A.; et al. NTnC-like genetically encoded calcium indicator with a positive and enhanced response and fast kinetics. Sci. Rep. 2018, 8, 15233. [Google Scholar] [CrossRef] [Green Version]
- Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Gruzdeva, A.M.; Barykina, N.V.; Ivashkina, O.I.; Anokhin, K.V.; Nikolaeva, A.Y.; Korzhenevskiy, D.A.; Vlaskina, A.V.; et al. Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein. Int. J. Mol. Sci. 2020, 21, 1644. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Rancic, V.; Wu, J.; Ballanyi, K.; Campbell, R.E. A Bioluminescent Ca2+ Indicator Based on a Topological Variant of GCaMP6s. ChemBioChem A Eur. J. Chem. Biol. 2018, 20, 516–520. [Google Scholar] [CrossRef]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef]
- Tian, L.; Hires, S.A.; Mao, T.; Huber, D.; Chiappe, M.E.; Chalasani, S.H.; Petreanu, L.; Akerboom, J.; McKinney, S.A.; Schreiter, E.R.; et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 2009, 6, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Tallini, Y.N.; Ohkura, M.; Choi, B.R.; Ji, G.; Imoto, K.; Doran, R.; Lee, J.; Plan, P.; Wilson, J.; Xin, H.B.; et al. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc. Natl. Acad. Sci. USA 2006, 103, 4753–4758. [Google Scholar] [CrossRef] [Green Version]
- Resendez, S.L.; Jennings, J.H.; Ung, R.L.; Namboodiri, V.M.; Zhou, Z.C.; Otis, J.M.; Nomura, H.; McHenry, J.A.; Kosyk, O.; Stuber, G.D. Visualization of cortical, subcortical and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat. Protoc. 2016, 11, 566–597. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, N.; He, Y.; Liu, Y.; Ge, L.; Zou, L.; Song, S.; Xiong, W.; Liu, X. Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat. Commun. 2018, 9, 1504. [Google Scholar] [CrossRef]
- Steinmetz, N.A.; Buetfering, C.; Lecoq, J.; Lee, C.R.; Peters, A.J.; Jacobs, E.A.K.; Coen, P.; Ollerenshaw, D.R.; Valley, M.T.; de Vries, S.E.J.; et al. Aberrant Cortical Activity in Multiple GCaMP6-Expressing Transgenic Mouse Lines. eNeuro 2017. [Google Scholar] [CrossRef] [Green Version]
- Mank, M.; Santos, A.F.; Direnberger, S.; Mrsic-Flogel, T.D.; Hofer, S.B.; Stein, V.; Hendel, T.; Reiff, D.F.; Levelt, C.; Borst, A.; et al. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat. Methods 2008, 5, 805–811. [Google Scholar] [CrossRef]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef] [Green Version]
- Barykina, N.V.; Subach, O.M.; Piatkevich, K.D.; Jung, E.E.; Malyshev, A.Y.; Smirnov, I.V.; Bogorodskiy, A.O.; Borshchevskiy, V.I.; Varizhuk, A.M.; Pozmogova, G.E.; et al. Green fluorescent genetically encoded calcium indicator based on calmodulin/M13-peptide from fungi. PLoS ONE 2017, 12, e0183757. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Abdelfattah, A.S.; Ruangkittisakul, A.; Ballanyi, K.; Campbell, R.E.; Harrison, D.J. Microfluidic cell sorter-aided directed evolution of a protein-based calcium ion indicator with an inverted fluorescent response. Integr. Biol. (Camb.) 2014, 6, 714–725. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Bushey, D.; Schreiter, E.R.; Harrison, D.J.; Wong, A.M.; Campbell, R.E. Inverse-response Ca2+ indicators for optogenetic visualization of neuronal inhibition. Sci. Rep. 2018, 8, 11758. [Google Scholar] [CrossRef] [Green Version]
- Gifford, J.L.; Walsh, M.P.; Vogel, H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007, 405, 199–221. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Niwa, H.; Inouye, S.; Hirano, T.; Matsuno, T.; Kojima, S.; Kubota, M.; Ohashi, M.; Tsuji, F.I. Chemical nature of the light emitter of the Aequorea green fluorescent protein. Proc. Natl. Acad. Sci. USA 1996, 93, 13617–13622. [Google Scholar] [CrossRef] [Green Version]
- Brejc, K.; Sixma, T.K.; Kitts, P.A.; Kain, S.R.; Tsien, R.Y.; Ormo, M.; Remington, S.J. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA 1997, 94, 2306–2311. [Google Scholar] [CrossRef] [Green Version]
- Needham, A.E. The Uniqueness of Biological Materials: International Series of Monographs in Pure and Applied Biology: Zoology; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Subach, O.M.; Cranfill, P.J.; Davidson, M.W.; Verkhusha, V.V. An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS ONE 2011, 6, e28674. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.I.; Barondeau, D.P.; Hitomi, C.; Kassmann, C.J.; Tainer, J.A.; Getzoff, E.D. Defining the role of arginine 96 in green fluorescent protein fluorophore biosynthesis. Biochemistry 2005, 44, 16211–16220. [Google Scholar] [CrossRef]
- Barondeau, D.P.; Putnam, C.D.; Kassmann, C.J.; Tainer, J.A.; Getzoff, E.D. Mechanism and energetics of green fluorescent protein chromophore synthesis revealed by trapped intermediate structures. Proc. Natl. Acad. Sci. USA 2003, 100, 12111–12116. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Schenkelberg, C.D.; Jordan, T.B.; Reimertz, J.M.; Crone, E.E.; Crone, D.E.; Bystroff, C. Mispacking and the Fitness Landscape of the Green Fluorescent Protein Chromophore Milieu. Biochemistry 2017, 56, 736–747. [Google Scholar] [CrossRef]
- Ormo, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [Green Version]
- Jung, G.; Wiehler, J.; Zumbusch, A. The photophysics of green fluorescent protein: Influence of the key amino acids at positions 65, 203, and 222. Biophys. J. 2005, 88, 1932–1947. [Google Scholar] [CrossRef] [Green Version]
- Brocard, J.B.; Rajdev, S.; Reynolds, I.J. Glutamate-induced increases in intracellular free Mg2+ in cultured cortical neurons. Neuron 1993, 11, 751–757. [Google Scholar] [CrossRef]
- Perez Koldenkova, V.; Nagai, T. Genetically encoded Ca2+ indicators: Properties and evaluation. Biochim. Biophys. Acta 2013, 1833, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Maravall, M.; Mainen, Z.F.; Sabatini, B.L.; Svoboda, K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 2000, 78, 2655–2667. [Google Scholar] [CrossRef] [Green Version]
- Rose, T.; Goltstein, P.M.; Portugues, R.; Griesbeck, O. Putting a finishing touch on GECIs. Front. Mol. Neurosci. 2014, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar]
- Yao, H.; Ma, E.; Gu, X.Q.; Haddad, G.G. Intracellular pH regulation of CA1 neurons in Na+/H+ isoform 1 mutant mice. J. Clin. Investig. 1999, 104, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Shen, Y.; Dana, H.; Abdelfattah, A.S.; Patel, R.; Shea, J.; Molina, R.S.; Rawal, B.; Rancic, V.; Chang, Y.F.; Wu, L.; et al. A genetically encoded Ca2+ indicator based on circularly permutated sea anemone red fluorescent protein eqFP578. BMC Biol. 2018, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 2016, 5, e12727. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, C.; Singh-Alvarado, J.; Zhou, Z.C.; Frohlich, F.; Mooney, R.; Wang, F. MIN1PIPE: A Miniscope 1-Photon-Based Calcium Imaging Signal Extraction Pipeline. Cell Rep. 2018, 23, 3673–3684. [Google Scholar] [CrossRef] [PubMed]
- Grundemann, J.; Bitterman, Y.; Lu, T.; Krabbe, S.; Grewe, B.F.; Schnitzer, M.J.; Luthi, A. Amygdala ensembles encode behavioral states. Science 2019, 364, eaav8736. [Google Scholar] [CrossRef]
- Zhang, X.; Li, B. Population coding of valence in the basolateral amygdala. Nat. Commun. 2018, 9, 5195. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Piatkevich, K.D.; Mc Larney, B.; Abdelfattah, A.S.; Mehta, S.; Murdock, M.H.; Gottschalk, S.; Molina, R.S.; Zhang, W.; Chen, Y.; et al. A genetically encoded near-infrared fluorescent calcium ion indicator. Nat. Methods 2019, 16, 171–174. [Google Scholar] [CrossRef]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. DBiol. Crystallogr. 2011, 67 Pt 4, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Padilla, J.E.; Yeates, T.O. A statistic for local intensity differences: Robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr. Sect. DBiol. Crystallogr. 2003, 59 Pt 7, 1124–1130. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. DBiol. Crystallogr. 2006, 62 Pt 1, 72–82. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Cryst. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. DBiol. Crystallogr. 1994, 50 Pt 5, 760–763.
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 Pt 12 Pt 1, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 Pt 12 Pt 1, 2256–2268. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Pnevmatikakis, E.A.; Giovannucci, A. NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 2017, 291, 83–94. [Google Scholar] [CrossRef]
- Lopes, G.; Bonacchi, N.; Frazao, J.; Neto, J.P.; Atallah, B.V.; Soares, S.; Moreira, L.; Matias, S.; Itskov, P.M.; Correia, P.A.; et al. Bonsai: An event-based framework for processing and controlling data streams. Front. Neuroinform. 2015, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Grunke, S.D.; Levites, Y.; Golde, T.E.; Jankowsky, J.L. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J. Vis. Exp. 2014, 91, e51863. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Ash, R.T.; Ceballos-Diaz, C.; Levites, Y.; Golde, T.E.; Smirnakis, S.M.; Jankowsky, J.L. Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits in vivo. Eur. J. Neurosci. 2013, 37, 1203–1220. [Google Scholar] [CrossRef] [Green Version]
- Gunaydin, L.A.; Grosenick, L.; Finkelstein, J.C.; Kauvar, I.V.; Fenno, L.E.; Adhikari, A.; Lammel, S.; Mirzabekov, J.J.; Airan, R.D.; Zalocusky, K.A.; et al. Natural neural projection dynamics underlying social behavior. Cell 2014, 157, 1535–1551. [Google Scholar] [CrossRef] [Green Version]
- Muir, J.; Lorsch, Z.S.; Ramakrishnan, C.; Deisseroth, K.; Nestler, E.J.; Calipari, E.S.; Bagot, R.C. In Vivo Fiber Photometry Reveals Signature of Future Stress Susceptibility in Nucleus Accumbens. Neuropsychopharmacology 2018, 43, 255–263. [Google Scholar] [CrossRef]
- McMahon, S.M.; Jackson, M.B. An Inconvenient Truth: Calcium Sensors Are Calcium Buffers. Trends Neurosci. 2018, 41, 880–884. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
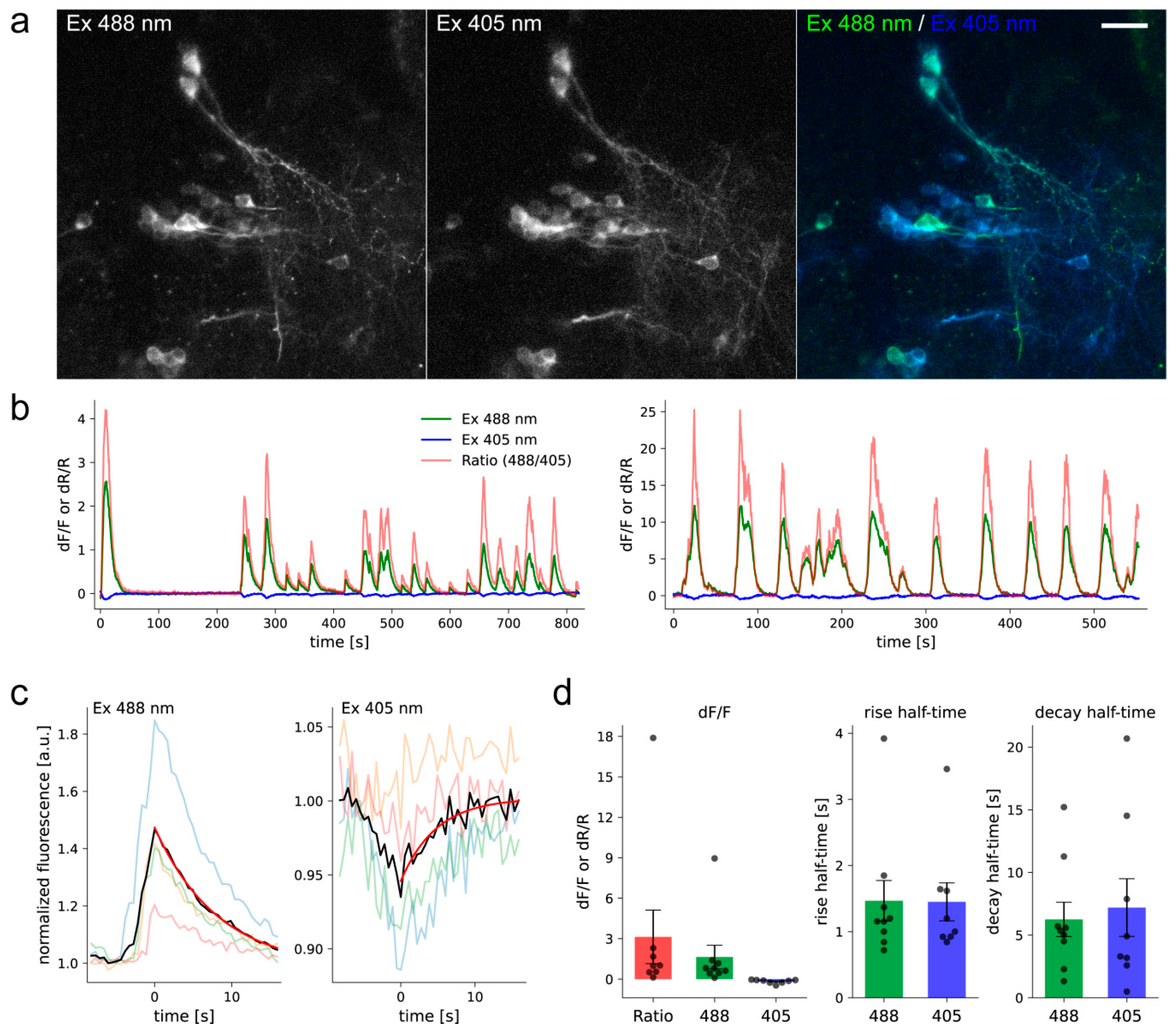{kind=link}
| Properties | Proteins | ||||
|---|---|---|---|---|---|
| FGCaMP | FGCaMP7 | ||||
| Apo | Sat | Apo | Sat | ||
| Absorbance maximum (nm) | 402 | 493 | 400 | 498 | |
| Emission maximum (nm) | 516 | 516 | |||
| Quantum yield a | 0.48 ± 0.02 | 0.46 ± 0.01 | 0.40 ± 0.02 | 0.55 ± 0.04 | |
| ε (mM−1cm−1) b | 55 ± 5 | 106 ± 20 | 82 ± 11 | 103 ± 14 | |
| Brightness (%) c | 100 | 100 | 123 | 116 | |
| Fluorescence contrast (fold) | Ex 400/402 | 6.9 ± 0.5 | 9.3 ± 0.3 | ||
| Ex 498/493 | 14.7 ± 0.6 | 38.4 ± 0.9 | |||
| Fluorescence contrast with 1 mM MgCl2 (fold) | Ex 400/402 | 7.1 ± 0.2 | 10.1 ± 1.0 | ||
| Ex 498/493 | 15.3 ± 0.5 | 32.7 ± 1.5 | |||
| pKa (Ex 400) d | 6.56 ± 0.03 | 7.0 ± 0.6 | 6.63 ± 0.20 | 6.00 ± 0.20 | |
| pKa (Ex 498) d | 6.2 ± 0.2 | 7.33 ± 0.07 | 5.28 ± 0.10; 7.81 ± 0.10 | 6.87 ± 0.01 | |
| Kd (nM) e | Ex 400 | 276 ± 9 (n = 2.8 ± 0.3) | 130 ± 10 (n = 2.7 ± 0.1) | ||
| Ex 498 | 273 ± 7 (n = 3.5 ± 0.3) 4700 ± 200 (n = 1.9 ± 0.2) | 160 ± 10 (n = 2.6 ± 0.2) | |||
| Kd (nM) with 1 mM MgCl2 e | Ex 400 | 460 ± 60 (n = 2.3 ± 0.6) | 230 ± 5 (n = 2.2 ± 0.1) | ||
| Ex 498 | 460 ± 40 (n = 2.8 ± 0.4) 4400 ± 800 (n = 1.9 ± 0.2) | 240 ± 6 (n = 2.3 ± 0.2) | |||
| kobs (s−1) (300 nM Ca2+) f | Ex 400 | 0.37 ± 0.01 | 0.60 ± 0.01 | ||
| Ex 498 | 0.35 ± 0.01 | 0.42 ± 0.01 | |||
| t1/2off (s) g | Ex 400 | 1.2 ± 0.1 | 1.5 ± 0.1 | ||
| Ex 498 | 1.4 ± 0.1 | 1.34 ± 0.02 | |||
| Protein state | monomer | monomer | |||
| Maturation half-time (min) h | ND | 27 ± 4 | ND | ND | |
| Photobleaching half-time (sec) i | 54 ± 9 | 260 ± 40 | 462 ± 126 | 464 ± 97 | |
| Protein | ||||||
|---|---|---|---|---|---|---|
| FGCaMP | FGCaMP7 | FGCaMP5 | FGCaMP6 | GCaMP6s | ||
| ΔF/F0 in HeLa (% vs. R-GECO1) | Ex405 | 62 ± 22 (n = 11) | 42 ± 19 p = 0.0493 a (n = 10) | 44 ± 16 p = 0.0243 a (n = 11) | 40 ± 22 p = 0.0230 a (n = 10) | NA |
| Ex488 | 162 ± 20 p = 0.0007 b (n = 11) | 237 ± 81 p < 0.0001b (n = 10) | 87 ± 25 p = 0.0305 b (n = 11) | 160 ± 55 p = 0.0297 b (n = 10) | 113 ± 35 (n = 15) | |
| ΔF/F0 in neurons (% vs. R-GECO1) | Ex405 | 50 ± 20 (n = 12) | 34 ± 13 p = 0.0267 a (n = 11) | 19 ± 10 p = 0.0014 a (n = 11) | 46 ± 37 p = 0.2412 a (n = 13) | NA |
| Ex488 | 150 ± 53 p = 0.3299 b (n = 12) | 142 ± 35 p = 0.4966 b (n = 10) | 58 ± 25 p < 0.0001 b (n = 16) | 143 ± 40 p = 0.3932 b (n = 14) | 133 ± 58 (n = 17) | |
| ΔR/R0 c in neurons (% vs. R-GECO1) | 245 ± 110 p = 0.0030 b (n = 11) | 219 ± 78 p = 0.0014 b (n = 10) | 103 ± 42 p = 0.1436 b (n = 11) | 231 ± 70 p = 0.0003 b (n = 13) | NA | |
| rise half-time (s) d | Ex405 | 1.93 ± 0.62 | 1.36 ± 0.47 | 2.45 ± 1.09 | 2.01 ± 0.93 | NA |
| Ex488 | 2.08 ± 0.57 | 1.36 ± 0.47 | 2.01 ± 1.23 | 1.65 ± 0.71 | 1.14 ± 0.61 | |
| decay half-time (s) e | Ex405 | 3.56 ± 1.37 | 3.40 ± 0.72 | 5.78 ± 2.56 | 4.41 ± 2.18 | NA |
| Ex488 | 4.01 ± 1.65 | 3.84 ± 0.88 | 6.51 ± 2.50 | 4.89 ± 1.92 | 3.77 ± 1.27 | |
| ΔF/F0 per 1 AP in stimulated neurons (%) | Ex405 | 0.6 ± 0.4 f (n = 29) | 2.1 ± 1.5 g p < 0.0001 a (n = 87) | 1.1 ± 0.8 f (p = 0.0148) a (n = 17) | 0.9 ± 0.9 h (p = 0.2678) a (n = 34) | NA |
| Ex488 | 4.1 ± 1.7 f p < 0.0001 b (n = 29) | 15.0 ± 7.5 g (p = 0.8805) b (n = 90) | 3.5 ± 1.6 f p < 0.0001 b (n = 17) | 7.3 ± 4.2 h p < 0.0001 b (n = 34) | 15.0 ± 7.0 g (n = 50) | |
| ΔR/R0 c per 1 AP in stimulated neurons (%) | 5.4 ± 2.3 f p < 0.0001 b (n = 29) | 20.3 ± 9.2 g (p = 0.0002) b (n = 86) | 5.1 ± 2.4 f p < 0.0001 b (n = 17) | 9.2 ± 4.7 h p < 0.0001 b (n = 34) | NA | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barykina, N.V.; Sotskov, V.P.; Gruzdeva, A.M.; Wu, Y.K.; Portugues, R.; Subach, O.M.; Chefanova, E.S.; Plusnin, V.V.; Ivashkina, O.I.; Anokhin, K.V.; et al. FGCaMP7, an Improved Version of Fungi-Based Ratiometric Calcium Indicator for In Vivo Visualization of Neuronal Activity. Int. J. Mol. Sci. 2020, 21, 3012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083012
Barykina NV, Sotskov VP, Gruzdeva AM, Wu YK, Portugues R, Subach OM, Chefanova ES, Plusnin VV, Ivashkina OI, Anokhin KV, et al. FGCaMP7, an Improved Version of Fungi-Based Ratiometric Calcium Indicator for In Vivo Visualization of Neuronal Activity. International Journal of Molecular Sciences. 2020; 21(8):3012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083012
Chicago/Turabian StyleBarykina, Natalia V., Vladimir P. Sotskov, Anna M. Gruzdeva, You Kure Wu, Ruben Portugues, Oksana M. Subach, Elizaveta S. Chefanova, Viktor V. Plusnin, Olga I. Ivashkina, Konstantin V. Anokhin, and et al. 2020. "FGCaMP7, an Improved Version of Fungi-Based Ratiometric Calcium Indicator for In Vivo Visualization of Neuronal Activity" International Journal of Molecular Sciences 21, no. 8: 3012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083012