The Dichotomic Role of Macrophage Migration Inhibitory Factor in Neurodegeneration

 , , , and
, , , and 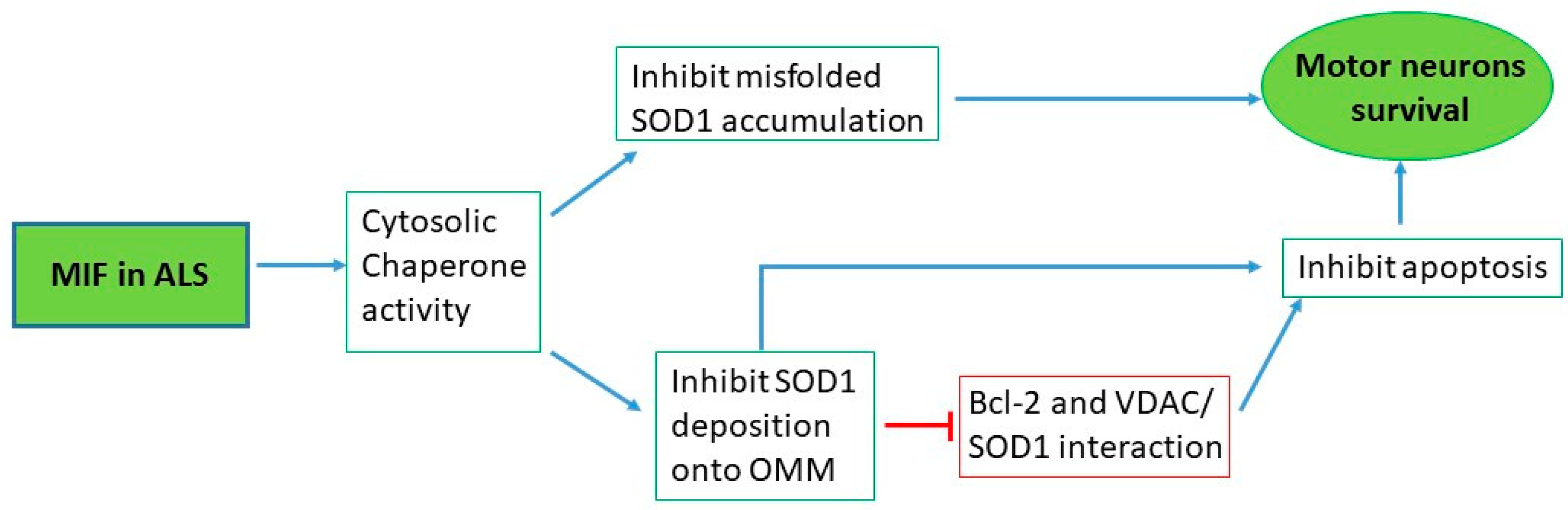{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Macrophage Migration Inhibitory Factor (MIF)
2. The Role of MIF in the Central Nervous System (CNS)
3. MIF in Neurodegenerative Diseases
4. ALS
5. MIF in ALS
5.1. Preclinical Studies
5.1.1. In Vitro Studies
5.1.2. In Vivo Studies
5.2. Clinical Studies
6. PD
7. MIF in PD
7.1. Preclinical Studies
7.1.1. In Vitro Studies
7.1.2. In Vivo Studies
7.2. Clinical Studies
8. HD
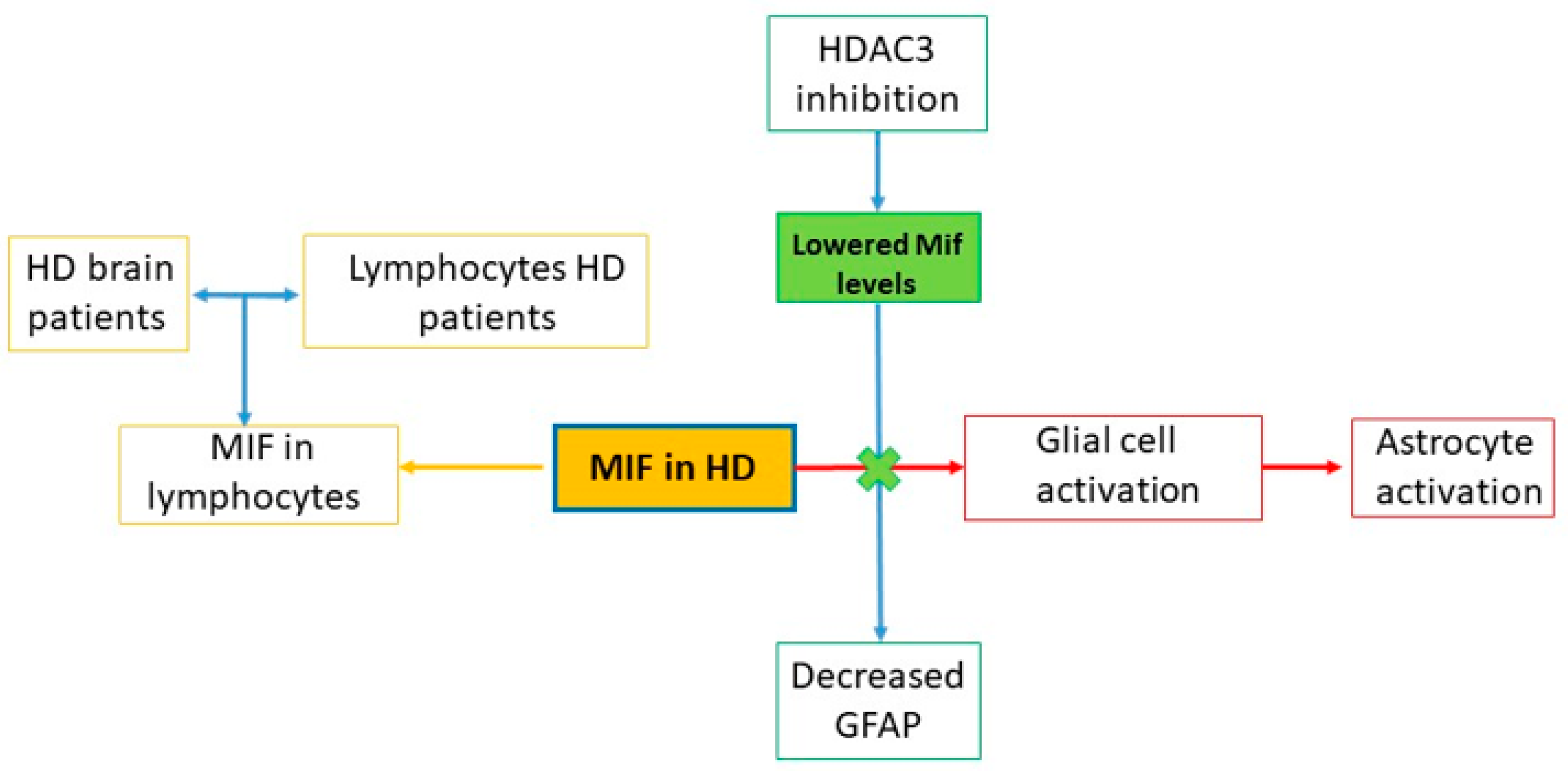
9. MIF in HD
9.1. Preclinical Studies
9.1.1. In Vitro Studies
9.1.2. In Vivo Studies
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bloom, B.R.; Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 1966, 153, 80–82. [Google Scholar] [CrossRef]
- Grieb, G.; Merk, M.; Bernhagen, J.; Bucala, R. Macrophage Migration Inhibitory Factor (MIF): A promising biomarker. Drug News Perspect. 2010, 23, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Kang, I.; Bucala, R. The immunobiology of MIF: Function, genetics and prospects for precision medicine. Nat. Rev. Rheumatol. 2019, 15, 427–437. [Google Scholar] [CrossRef]
- Bloom, J.; Sun, S.; Al-Abed, Y. MIF, a controversial cytokine: A review of structural features, challenges, and opportunities for drug development. Expert Opin. Ther. Targets 2016, 20, 1463–1475. [Google Scholar] [CrossRef]
- Günther, S.; Fagone, P.; Jalce, G.; Atanasov, A.G.; Guignabert, C.; Nicoletti, F. Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: From pathogenic factors to therapeutic targets. Drug Discov. Today 2019, 24, 428–439. [Google Scholar] [CrossRef]
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving complexity of MIF signaling. Cell. Signal. 2019, 57, 76–88. [Google Scholar] [CrossRef]
- Cavalli, E.; Ciurleo, R.; Petralia, M.C.; Fagone, P.; Bella, R.; Mangano, K.; Nicoletti, F.; Bramanti, P.; Basile, M.S. Emerging role of the macrophage migration inhibitory factor family of cytokines in neuroblastoma. Pathogenic effectors and novel therapeutic targets? Molecules 2020, 25, 1194. [Google Scholar] [CrossRef] [Green Version]
- Kleemann, R.; Hausser, A.; Geiger, G.; Mischke, R.; Burger-Kentischer, A.; Flieger, O.; Johannes, F.J.; Roger, T.; Calandra, T.; Kapurniotu, A.; et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 2000, 408, 211–216. [Google Scholar] [CrossRef]
- Sugimoto, H.; Taniguchi, M.; Nakagawa, A.; Tanaka, I.; Suzuki, M.; Nishihira, J. Crystallization and preliminary X-ray analysis of human D-dopachrome tautomerase. J. Struct. Biol. 1997, 120, 105–108. [Google Scholar] [CrossRef]
- Sugimoto, H.; Taniguchi, M.; Nakagawa, A.; Tanaka, I.; Suzuki, M.; Nishihira, J. Crystal Structure of Human D-Dopachrome Tautomerase, a Homologue of Macrophage Migration Inhibitory Factor, at 1.54 Å Resolution †,‡. Biochemistry 1999, 38, 3268–3279. [Google Scholar] [CrossRef]
- Nishibori, M.; Nakaya, N.; Tahara, A.; Kawabata, M.; Mori, S.; Saeki, K. Presence of macrophage migration inhibitory factor (MIF) in ependyma, astrocytes and neurons in the bovine brain. Neurosci. Lett. 1996, 213, 193–196. [Google Scholar] [CrossRef]
- Ogata, A.; Nishihira, J.; Suzuki, T.; Nagashima, K.; Tashiro, K. Identification of macrophage migration inhibitory factor mRNA expression in neural cells of the rat brain by in situ hybridization. Neurosci. Lett. 1998, 246, 173–177. [Google Scholar] [CrossRef]
- Bacher, M.; Meinhardt, A.; Lan, H.Y.; Dhabhar, F.S.; Mu, W.; Metz, C.N.; Chesney, J.A.; Gemsa, D.; Donnelly, T.; Atkins, R.C.; et al. MIF expression in the rat brain: Implications for neuronal function. Mol. Med. 1998, 4, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Conboy, L.; Varea, E.; Castro, J.E.; Sakouhi-Ouertatani, H.; Calandra, T.; Lashuel, H.A.; Sandi, C. Macrophage migration inhibitory factor is critically involved in basal and fluoxetine-stimulated adult hippocampal cell proliferation and in anxiety, depression, and memory-related behaviors. Mol. Psychiatry 2011, 16, 533–547. [Google Scholar] [CrossRef]
- Matsunaga, J.; Sinha, D.; Pannell, L.; Santist, C.; Solano, F.; Wistow, G.J.; Hearing, V.J. Enzyme activity of macrophage migration inhibitory factor toward oxidized catecholamines. J. Biol. Chem. 1999, 274, 3268–3271. [Google Scholar] [CrossRef] [Green Version]
- Bloom, J.; Al-Abed, Y. MIF: Mood improving/inhibiting factor? J. Neuroinflamm. 2014, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Mangano, K.; Mazzon, E.; Basile, M.S.; Di Marco, R.; Bramanti, P.; Mammana, S.; Petralia, M.C.; Fagone, P.; Nicoletti, F. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 2018, 9, 17951–17970. [Google Scholar] [CrossRef] [Green Version]
- Leyton-Jaimes, M.F.; Kahn, J.; Israelson, A. Macrophage migration inhibitory factor: A multifaceted cytokine implicated in multiple neurological diseases. Exp. Neurol. 2018, 301, 83–91. [Google Scholar] [CrossRef]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Basile, M.S.; Lenzo, V.; Quattropani, M.C.; Bendtzen, K.; Nicoletti, F. Pathogenic contribution of the Macrophage migration inhibitory factor family to major depressive disorder and emerging tailored therapeutic approaches. J. Affect. Disord. 2020, 263, 15–24. [Google Scholar] [CrossRef]
- Petralia, M.C.; Battaglia, G.; Bruno, V.; Pennisi, M.; Mangano, K.; Lombardo, S.D.; Fagone, P.; Cavalli, E.; Saraceno, A.; Nicoletti, F.; et al. The Role of Macrophage Migration Inhibitory Factor in Alzheimer′s Disease: Conventionally Pathogenetic or Unconventionally Protective? Molecules 2020, 25, 291. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, E.; Mazzon, E.; Basile, M.S.; Mangano, K.; Di Marco, R.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Upregulated Expression of Macrophage Migration Inhibitory Factor, Its Analogue D-Dopachrome Tautomerase, and the CD44 Receptor in Peripheral CD4 T Cells from Clinically Isolated Syndrome Patients with Rapid Conversion to Clinical Defined Multiple Sclerosis. Medicina (B Aires) 2019, 55, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, S.D.; Mazzon, E.; Mangano, K.; Basile, M.S.; Cavalli, E.; Mammana, S.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Transcriptomic analysis reveals involvement of the macrophage migration inhibitory factor gene network in duchenne muscular dystrophy. Genes 2019, 10, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedek, G.; Meza-Romero, R.; Jordan, K.; Zhang, Y.; Nguyen, H.; Kent, G.; Li, J.; Siu, E.; Frazer, J.; Piecychna, M.; et al. MIF and D-DT are potential disease severity modifiers in male MS subjects. Proc. Natl. Acad. Sci. USA 2017, 114, E8421–E8429. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Mazzon, E.; Cavalli, E.; Bramanti, A.; Petralia, M.C.; Mangano, K.; Al-Abed, Y.; Bramati, P.; Nicoletti, F. Contribution of the macrophage migration inhibitory factor superfamily of cytokines in the pathogenesis of preclinical and human multiple sclerosis: In silico and in vivo evidences. J. Neuroimmunol. 2018, 322, 46–56. [Google Scholar] [CrossRef]
- Cavalli, E.; Mazzon, E.; Mammana, S.; Basile, M.S.; Lombardo, S.D.; Mangano, K.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Overexpression of macrophage migration inhibitory factor and its homologue d-dopachrome tautomerase as negative prognostic factor in neuroblastoma. Brain Sci. 2019, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Nobre, C.C.G.; de Araújo, J.M.G.; de MedeirosFernandes, T.A.A.; Cobucci, R.N.O.; Lanza, D.C.F.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol. Oncol. Res. 2017, 23, 235–244. [Google Scholar] [CrossRef]
- Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.S.; Petralia, M.C.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The role of macrophages in neuroinflammatory and neurodegenerative pathways of alzheimer’s disease, amyotrophic lateral sclerosis, and multiple sclerosis: Pathogenetic cellular effectors and potential therapeutic targets. Int. J. Mol. Sci. 2018, 19, 831. [Google Scholar] [CrossRef] [Green Version]
- Cluskey, S.; Ramsden, D.B. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. J. Clin. Pathol. Mol. Pathol. 2001, 54, 386–392. [Google Scholar]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2016; Volume 138, pp. 225–238. [Google Scholar]
- Salameh, J.S.; Brown, R.H.; Berry, J.D. Amyotrophic Lateral Sclerosis: Review. Semin. Neurol. 2015, 35, 469–476. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.C.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shvil, N.; Banerjee, V.; Zoltsman, G.; Shani, T.; Kahn, J.; Abu-Hamad, S.; Papo, N.; Engel, S.; Bernhagen, J.; Israelson, A. MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: Implications for familial ALS. Cell Death Dis. 2018, 9, 107. [Google Scholar] [CrossRef]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Leyton-Jaimes, M.F.; Benaim, C.; Abu-Hamad, S.; Kahn, J.; Guetta, A.; Bucala, R.; Israelson, A. Endogenous macrophage migration inhibitory factor reduces the accumulation and toxicity of misfolded SOD1 in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2016, 113, 10198–10203. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Ditsworth, D.; Sun, S.; Song, S.W.; Liang, J.; Hruska-Plochan, M.; McAlonis-Downes, M.; Abu-Hamad, S.; Zoltsman, G.; Shani, T.; et al. Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron 2015, 86, 218–232. [Google Scholar] [CrossRef] [Green Version]
- Leyton-Jaimes, M.F.; Kahn, J.; Israelson, A. AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc. Natl. Acad. Sci. USA 2019, 116, 14755–14760. [Google Scholar] [CrossRef] [Green Version]
- Maier, O.; Böhm, J.; Dahm, M.; Brück, S.; Beyer, C.; Johann, S. Differentiated NSC-34 motoneuron-like cells as experimental model for cholinergic neurodegeneration. Neurochem. Int. 2013, 62, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Iarkov, A.; Barreto, G.E.; Grizzell, J.A.; Echeverria, V. Strategies for the Treatment of Parkinson’s Disease: Beyond Dopamine. Front. Aging Neurosci. 2020, 12, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Nie, K.; Zhang, Q.; Guo, M.; Qiu, Y.; Li, Y.; Gao, Y.; Wang, L. Macrophage Migration Inhibitory Factor Mediates Neuroprotective Effects by Regulating Inflammation, Apoptosis and Autophagy in Parkinson’s Disease. Neuroscience 2019, 416, 50–62. [Google Scholar] [CrossRef]
- Jankovic, J.; Sherer, T. The future of research in Parkinson disease. JAMA Neurol. 2014, 71, 1351–1352. [Google Scholar] [CrossRef]
- Schwarz, S.C.; Schwarz, J.; Sautter, J.; Oertel, W.H. Effects of macrophage migration inhibitory factor and macrophage migration stimulatory factor on function and survival of foetal dopaminergic grafts in the 6-hydroxydopamine rat model of Parkinson’s disease. Exp. Brain Res. 1998, 120, 95–103. [Google Scholar] [CrossRef]
- Nakahara, K.; Fujikawa, K.; Hiraoka, H.; Miyazaki, I.; Asanuma, M.; Ito, A.; Takasugi, N.; Uehara, T. Attenuation of Macrophage Migration Inhibitory Factor-Stimulated Signaling via S-Nitrosylation. Biol. Pharm. Bull. 2019, 42, 1044–1047. [Google Scholar] [CrossRef] [Green Version]
- Weingarten, P.; Zhou, Q.Y. Protection of intracellular dopamine cytotoxicity by dopamine disposition and metabolism factors. J. Neurochem. 2001, 77, 776–785. [Google Scholar] [CrossRef]
- Cheng, J.; Liao, Y.; Dong, Y.; Hu, H.; Yang, N.; Kong, X.; Li, S.; Li, X.; Guo, J.; Qin, L.; et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 2020. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Fagone, P.; Donzuso, G.; Mangano, K.; Dibilio, V.; Caponnetto, S.; Bendtzen, K.; Zappia, M.; Nicoletti, F. Parkinson’s disease is associated with increased serum levels of macrophage migration inhibitory factor. Cytokine 2011, 55, 165–167. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.A. Genetics of Huntington disease. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 144, pp. 3–14. [Google Scholar]
- Ghosh, R.; Tabrizi, S.J. Clinical features of huntington’s disease. In Advances in Experimental Medicine and Biology; Springer New York LLC: Cham, Switzerland, 2018; Volume 1049, pp. 1–28. [Google Scholar]
- Rodrigues, F.B.; Quinn, L.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: January 2019. J. Huntington’s. Dis. 2019, 8, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Barkley, D.S.; Hardiwidjaja, S.I.; Tourtellotte, W.W.; Menkes, J.H. Cellular immune responses in huntington disease: Specificity of brain antigenicity detected with huntington disease lymphocytes. Neurology 1978, 28, 32–35. [Google Scholar] [CrossRef]
- Jia, H.; Wang, Y.; Morris, C.D.; Jacques, V.; Gottesfeld, J.M.; Rusche, J.R.; Thomas, E.A. The effects of pharmacological inhibition of histone deacetylase 3 (HDAC3) in Huntington’s disease mice. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Barkley, D.; Hardiwidjaja, S.; Menkes, J. Huntington’s disease: Delayed hypersensitivity in vitro to human central nervous system antigens. Science 1977, 195, 314–316. [Google Scholar] [CrossRef]
- Barkley, D.S.; Hardiwidjaja, S.I.; Menkes, J.H.; Ellison, G.W.; Myers, L.W. Cellular immune responses in Huntington’s disease (H.D.). Detection of H.D. and multiple sclerosis (M.S.) brain antigenicity by H.D. but not M.S. lymphocytes. Cell. Immunol. 1977, 32, 385–390. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basile, M.S.; Battaglia, G.; Bruno, V.; Mangano, K.; Fagone, P.; Petralia, M.C.; Nicoletti, F.; Cavalli, E. The Dichotomic Role of Macrophage Migration Inhibitory Factor in Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083023
Basile MS, Battaglia G, Bruno V, Mangano K, Fagone P, Petralia MC, Nicoletti F, Cavalli E. The Dichotomic Role of Macrophage Migration Inhibitory Factor in Neurodegeneration. International Journal of Molecular Sciences. 2020; 21(8):3023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083023
Chicago/Turabian StyleBasile, Maria Sofia, Giuseppe Battaglia, Valeria Bruno, Katia Mangano, Paolo Fagone, Maria Cristina Petralia, Ferdinando Nicoletti, and Eugenio Cavalli. 2020. "The Dichotomic Role of Macrophage Migration Inhibitory Factor in Neurodegeneration" International Journal of Molecular Sciences 21, no. 8: 3023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083023