High Mobility Group AT-Hook 2 (HMGA2) Oncogenicity in Mesenchymal and Epithelial Neoplasia

1
Department of Anesthesiology, Columbia University Medical Center, 630 West 168th Street, P&S 12-402, New York, NY 10032, USA
2
Department of Biochemistry & Molecular Biology; Robert Wood Johnson Medical School, 675 Hoes Lane, Piscataway, NJ 08854, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(9), 3151; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093151
Submission received: 1 April 2020
/
Revised: 27 April 2020
/
Accepted: 28 April 2020
/
Published: 29 April 2020
(This article belongs to the Special Issue HMG Proteins in Development and Disease)
Abstract
:High mobility group AT-hook 2 (HMGA2) has been associated with increased cell proliferation and cell cycle dysregulation, leading to the ontogeny of varied tumor types and their metastatic potentials, a frequently used index of disease prognosis. In this review, we deepen our understanding of HMGA2 pathogenicity by exploring the mechanisms by which HMGA2 misexpression and ectopic expression induces mesenchymal and epithelial tumorigenesis respectively and distinguish the pathogenesis of benign from malignant mesenchymal tumors. Importantly, we highlight the regulatory role of let-7 microRNA family of tumor suppressors in determining HMGA2 misexpression events leading to tumor pathogenesis and focused on possible mechanisms by which HMGA2 could propagate lymphangioleiomyomatosis (LAM), benign mesenchymal tumors of the lungs. Lastly, we discuss potential therapeutic strategies for epithelial and mesenchymal tumorigenesis based on targeting the HMGA2 signaling pathway.
1. Introduction
High mobility group AT-hook 2 (HMGA2) belongs to the HMGA family of small, non-histone chromatin-associated proteins [1]. This protein is encoded by the HMGA2 gene localized to human chromosome 12 at band q14.3 [2], comprising five exons dispersed over a genomic region of ≥140 kb. Each of the first three exons contain conserved DNA binding domains called AT-hook motifs separated from an acidic C-terminal tail in the fifth exon by a spacer domain encoded by the fourth exon [1]. This structural feature determines HMGA2’s binding preference for AT-rich regions in the minor groove of DNA that causes ordered architectural changes which influence the conformation of bound DNA substrates, functional interactions between transcription factors, changes in chromatin structure, DNA replication, and gene transcription [3]. These physiological changes play fundamental roles in mammalian growth and development such that homozygous Hmga2−/− mice exhibit complete histological composition but yield a pygmy phenotype displaying dramatic reduction in adipose tissue accumulation and birth weight, and impairment of skeletal muscle development and myoblast proliferation [4,5]. Organ systems affected by HMGA2 mutations thus highlight its potential role in fate specification of mammalian tissues to a mesenchymal lineage during embryonic development. This tissue lineage specificity for Hmga2 during development is further supported by evidence describing mesodermal differentiation, self-renewal and proliferation of human embryonic stem cells (hESCs) induced by HMGA2 expression [6].
Indeed, Hmga2 is ubiquitously expressed in undifferentiated tissues at early mammalian embryogenetic stages, beginning at 9.5 days post-coitum (dpc) in the mouse embryo and with time, expression becomes increasingly restricted to mostly undifferentiated tissue regions of mesenchymal origin, and some parts of the central nervous system [7]. Between developmental stages 14.5 – 17.5 dpc, the pattern of Hmga2 expression declines and is observed to be akin to the distribution of connective tissues in the mouse tissue mesenchyme, and further restricted to proliferative tissue regions [7]. As tissue differentiation progresses in the maturing human fetus, HMGA2 expression is also restricted to specific regions of the lungs, kidneys, and synovia [7,8], and associated with the activation and renewal of endogenous tissue-resident stem cells in adult stages [5,9]. These HMGA2-positive adult stem cells are possible undifferentiated tissue remnants of ontogenetic development, although more characterization studies of tissue-specific stem cell populations still need to be performed. Given that HMGA2 expression dynamics are functionally superimposable to mammalian embryogenic differentiation paradigms, genetic anomalies at the HMGA2 locus during this tissue maturation period could account for anomalous cell fate specification, which could lead to multi-systemic neoplasms, determining tissues that would become tumorigenic, and the timing of tumor ontogeny.
The HMGA family of proteins was first isolated from cancerous HeLa S3 cells in 1983 [10]. However, the correlation between HMGA2 and neoplastic transformation was not established until two years later, when HMGA nuclear phosphoproteins were detected in a rat thyroid cell line (FRTL5) after viral transformation [11]. The isolated HMGA proteins were associated with a highly malignant phenotype irrespective of whether transformed cells were chemically, virally or spontaneously derived [12]. More direct evidence for the oncogenic role of HMGA proteins was reported when rat and human cell lines with ectopic expression of HMGA1 transcript variant (HMGA1a) and HMGA2 formed tumors and led to distant metastases when injected in athymic nude mice [13]. Since then, numerous postulates have been put forward and experiments conducted to explain the causative biological mechanisms employed by HMGA proteins to induce both benign and malignant neoplasms. These mechanisms of neoplastic transformation have been found to be tumor-type specific and to differ between epithelial and mesenchymal tumors. These mechanisms are discussed in subsequent sections of this review. The lack of HMGA2 expression in proliferating fibroblasts of some pulmonary interstitial diseases further suggests that the gene’s misexpression defines neoplastic transformation of a normal cell rather than a hyper-proliferative index [14].
Summarily, ectopic HMGA2 expression drives epithelial tumor metastasis and multiplicity in cell culture and in in vivo mouse models mainly by the activation of the TGFβ pathway and epithelial–mesenchymal transitions (EMT) [15,16]. In contrast, the misexpression of full length, chimeric or truncated HMGA2 mRNA transcripts in differentiated benign mesenchymal tumors derived from abnormal chromosomal breaks governs mesenchymal tumorigenesis irrespective of the nature of the HMGA2 gene product [17,18]. In this review, we will explore these mechanisms by which HMGA2 induces epithelial and mesenchymal tumorigenesis, and discuss the gene’s role in lymphangioleiomyomatosis, a rare pulmonary mesenchymal neoplasm of unknown etiology that is often a clinical manifestation of tuberous sclerosis. Lastly, we identify potential therapeutic strategies for epithelial and mesenchymal tumorigenesis based on targeting the HMGA2 signaling pathway.
2. Mechanisms of HMGA2-Induced Mesenchymal Tumorigenicity
From our review of cytogenetic studies (see Supplementary Table S1), causative mechanisms of HMGA2-induced mesenchymal tumorigenesis results from HMGA2 misexpression in well differentiated mesenchymal tissues contrary to its canonical expression in undifferentiated mesenchyme [7,8]. These genetic changes enhance or repress genes and transcription factors that play crucial roles in cell proliferation [19], cell cycle regulation [20,21], DNA damage response [22,23,24], apoptosis [25] and cellular senescence [26,27], all causatively leading to tumor phenotypes in mesenchymal tissues [28]. In this section, we explore evidence for HMGA2-induced pathogenesis in benign and malignant mesenchymal neoplasms.
2.1. HMGA2 Misexpression
HMGA2 is canonically expressed in the undifferentiated mesenchyme and is undetectable in the differentiated tissue forms [8]. However, in several types of differentiated mesenchymal tumor, including lipomas [29,30], leiomyomata [8,31,32,33], pulmonary chondroid hamartomas [34,35], endometrial polyps [36], and soft tissue chondromas [37], certain genetic mechanisms enable its untimely re-expression, biologically termed HMGA2 misexpression. A variety of HMGA2 transcripts have been isolated from these tumors and believed to cause mesenchymal tumorigenesis through a re-expression of the HMGA2 gene in differentiated tissues. Some of these transcripts derive from chromosomal translocations at 12q13–15 which truncates the human HMGA2 open reading frame (ORF) resulting in loss of its C-terminus and/or fusion to partner ectopic sequences [38,39], while in other mesenchymal tumors, intact ORF have been resolved but with truncated 3’ untranslated region (UTR) which encodes binding sites for let-7, a negative regulator of HMGA2 expression [40,41]. These chromosomal translocations have been reported as the genetic mechanisms necessitating HMGA2 misexpression in mesenchymal tumors [17,42,43]. Similar translocations involving the chromosomal locus 6p21–23 corresponding to HMGA1 (HMGI-Y) have also been described in benign mesenchymal tumors, implicating the HMGI family of DNA-binding proteins [44,45].
Chromosomal abnormalities are a hallmark of cancers, and their causes, although poorly understood, are commonly attributed to environmental and occupational exposures and certain therapies [46,47]. Chromosomal abnormalities can occur in the form of balanced chromosomal rearrangements such as reciprocal translocations or inversions that result in the formation of chimeric fusion genes, predicted to be early initiating events in tumorigenesis [47]. With non-balanced chromosomal rearrangements, some genetic material is lost or gained by deletions or duplications resulting in reduction or enhancement of genetic activity respectively [47]. We reviewed cytogenetic analyses of mesenchymal tumors and found that most chromosomal abnormalities occurred by balanced chromosomal rearrangements involving HMGA2 locus 12q13–15 [48,49]. A listing of these chromosomal rearrangements involving HMGA2 in human mesenchymal tumors is catalogued in Supplementary Table S1. Our data compilation reveals that the most common translocation partners of HMGA2 in mesenchymal tumors are chromosomes 1–3, with a preponderance of intragenic chromosome 12 aberrations. In addition, certain tumor types were synonymous with HMGA2 gene translocations involving specific chromosomes, as observed with chromosome 14 in uterine leiomyomata and pulmonary chondroid harmatomas (Supplementary Table S1).
The nature of the chromosomal rearrangements and resulting mRNA HMGA2 transcript also differ by mesenchymal tumor type [50]. In most lipomas and pulmonary chondroid harmatomas, chromosomal translocations have breakpoints that preferentially cluster in the third intron of the HMGA2 gene to yield either oncogenic truncated forms of HMGA2 mRNA transcripts containing exons 1–3 and lacking the 3’ untranslated region (3’ UTR), and/or chimeric fusion transcripts co-joining truncated HMGA2 DNA binding domains to up- or downstream transcriptional regulatory sequences [51,52]. Common HMGA2 fusion partners are tumor suppressors whose functions are typically lost following these gene rearrangements [53,54]. On the other hand, in most uterine leiomyomata, these chromosomal breakpoints mostly occur 10–100 kilobases upstream of the HMGA2 coding region such that full-length gene transcripts are expressed in these tumor types with or without chimeric forms [41,50,55,56].
Overall, these chromosomal translocations lead to the misexpression of three main types of HMGA2 transcript: 1) full-length HMGA2 transcripts with no apparent disruption of the coding sequence; 2) truncated HMGA2 transcripts lacking the 3’-UTR; and 3) chimeric HMGA2 transcripts fused to other ectopic sequences. Some mesenchymal tumor types were found to express only truncated, chimeric or full-length HMGA2 transcripts, while in other mesenchymal tumors, multiple HMGA2 transcript types could be found, indicating that the nature of HMGA2 disruption in these tumors did not affect the gene’s transformative capability [17].
2.1.1. Full Length HMGA2 Transcript with No Apparent Disruption of the Coding Sequence
Despite previous studies showing that truncation and/or addition of ectopic sequences after the third AT-hook were necessary for neoplastic transformation of murine fibroblasts [57], many human uterine leiomyomata, mammary fibroadenomas, and salivary gland adenomas were found to harbor full-length HMGA2 coding regions [8,40,41,58]. Full length HMGA2 transcripts can form in these tumor types when chromosomal rearrangements occur in an extragenic location usually upstream of the HMGA2 coding region [17,43,59]. In human uterine fibroids, such chromosomal rearrangements often involved the HMGA2 gene and chromosome 14, and balanced intragenic chromosome 12 aberrations were rare [41]. These findings suggest that full-length HMGA2 has oncogenic properties, and that disruptions in regulatory elements proximal to the HMGA2 coding region could lead to HMGA2 misexpression, inducing mesenchymal tumorigenesis [41]. Consistently, misexpression of full-length Hmga2 induces benign mesenchymal tumors in mice [17].
Another mesenchymal tumorigenic mechanism allowing for the expression of full length HMGA2 transcripts has been described in well differentiated liposarcomas (WDLPS) and atypical lipomas (ALP) [29,60]. In these mesenchymal tumors, supernumerary rings and giant rod marker chromosomes comprised various amplified subregions of different chromosomes associated with 12q14–15 [29,60]. In addition to ectopic expression of HMGA2, other genes co-amplified as a result of these supernumerary structures include MDM2, CDK4 and TSPAN31. However, supernumerary ring chromosomes are a rare find in ordinary lipomas [60].
2.1.2. Truncated HMGA2 Transcript Lacking the 3’-UTR
The HMGA2 3’ UTR has considerable potential for posttranscriptional regulation by RNA-binding proteins and miRNA-induced silencing complexes (miRISCs) [61,62]. Chromosomal rearrangements and intragenic chromosomal breakpoints within the region 12q14~15 leading to the formation of truncated HMGA2 transcripts lacking the 3’ untranslated region (UTR) of HMGA2 mRNA have been observed in many benign mesenchymal tumors, including some uterine leiomyomata and pulmonary chondroid hamartomas [62,63]. Human HMGA2 3’ UTR is 2.9kb long and harbors up to 35 discrete positive and negative cis-regulatory elements that act independently, or less commonly in synergy, altogether functioning to repress HMGA2 expression post-transcriptionally [64]. Although most regulatory elements in the HMGA2 3’ UTR have been found to induce HMGA2 expression such as AU-rich elements (AREs) that interact with HuR ARE-binding protein [65], repressive regulatory elements include characteristic binding sites for the tumor suppressor microRNA let-7 family [62,64,66]. The role of let-7 in HMGA2-induced mesenchymal tumor pathogenesis is further discussed below.
2.1.3. Chimeric HMGA2 Transcript Fused to Ectopic Sequences
Translocation breakpoints in the chromosome 12 locus of the HMGA2 gene preferentially occurs in the large intronic space linking exons 3 and 4 resulting in a truncated gene with intact DNA and protein binding domains but lacking a carboxy terminus including the 3’ untranslated region (3’ UTR) [39,67]. These aberrant HMGA2 transcripts can fuse to ectopic sequences from translocation partners whose protein functions are frequently lost due to gene rearrangements [48] (Figure 1E). Table 1 lists the balanced chromosomal rearrangements in mesenchymal tumors that form chimeric HMGA2 fusion transcripts. Supporting previous cytogenetic reports, Table 1 shows that the lipoma preferred partner gene (LPP) was one of the most abundant fusion gene partners to HMGA2 in lipomas, in a case of pulmonary chondroid harmatomas, and in soft tissue chondromas, and involved translocation loci t(3;12)(q14~15;q12~21) [29,37,39,68,69,70,71,72,73,74]. The HMGA2-LPP gene codes for a transcription factor containing the AT-hook domains of HMGA2 fused to three LIM domains at the C-terminal [75]. The expression of the HMGA2-LPP fusion protein typically leads to neoplastic transformation although the expression of the fusion gene did not increase the transformative ability of the truncated HMGA2 [57,75]. Interestingly, another fusion gene, the SET binding protein 1 (SETBP1), encoding a protein that binds the SET nuclear oncogene involved in DNA replication, was also found to recur in lipomas and osteochondrolipomas (t(12;18)(q27~28;q13~15)) [42]. SETBP1 was fused to truncated HMGA2 containing exons 1–3, or to an intragenic sequence 18q12.3 that is 10kbp distal to SETBP1 [42]. In the HMGA2-SETBP1 chimeric transcripts, the translocation breakpoint in SETBP1 occurred at the 3’-UTR essentially deregulating HMGA2 and possibly influencing expression of SETBP1 [42]. Of note, intragenic chromosomal breakpoints predominated in sarcomas where in almost all cases investigated, chimeric fusion HMGA2 transcripts were formed [76]. On the contrary, in lipomas where rearrangements of the HMGA2 gene involve multiple chromosomal partners, chimeric fusion genes are rarely observed (Supplementary Table S1).
2.1.4. let-7 Regulation of HMGA2 Misexpression in Benign Mesenchymal Tumors
As all forms of HMGA2 transcript exhibit similar transformative potential, it can be hypothesized that a single regulatory regimen could chiefly govern the misexpression of these different transcript types and their propensity to drive the pathogenesis of benign mesenchymal tumors [18]. One such regulatory element is the let-7 microRNA family of tumor suppressors which have conserved complementary binding sites encoded in the 3’-UTR of the HMGA2 gene [61,62]. Let-7 expression is inversely related to HMGA2 expression, is undetectable during embryogenesis (Figure 1A), but increases after differentiation and in mature tissues [62]. In line with established models of miRNA action in mammalian cells [92], in the normal differentiated mesenchyme, let-7 post-transcriptionally recognizes its target sites in the full-length HMGA2 mRNA and recruits it to exonucleases leading to target mRNA decapping and degradation of the HMGA2 gene transcript (Figure 1B). This would account for the reciprocal relationship between HMGA2 and let-7 expression [62]. These let-7-HMGA2 molecular associations can further specify tissue-type differentiation of normal mesenchymal tissues, selectively enhancing human osteogenesis while repressing adipogenesis [93].
In uterine leiomyomas, the inverse physiological relationship between let-7 and HMGA2 also persists such that large leiomyomata were observed to express low levels of let-7 and high levels of HMGA2, while small leiomyomata expressed high levels of let-7 and low levels of HMGA2 [94]. These findings establish a direct correlation between endogenous HMGA2 and let-7 levels and suggest that additive binding of let-7 to 3’ UTR complementary sites in HMGA2 could progressively decrease protein translation and HMGA2 pathway activation, limiting tumor size. Given the multiple binding sites for let-7 in many chromosomal 3’ UTR loci, it is possible that HMGA2 is mis-expressed when the relative abundance of HMGA2 mRNA transcripts suffice to bind available let-7 and sustain HMGA2 re-expression in benign mesenchymal tumors. High levels of HMGA2 transcription in larger leiomyomata could encode more 3’ UTR binding sites to soak up available mature let-7 miRNA limiting the repressive and degradative function of let-7 on HMGA2 expression [40,41], and allowing for greater rates of tumorigenesis compared to smaller sized uterine fibroids. This potential tumorigenic mechanism is depicted in Figure 1C and could explain the expression of full length HMGA2 mRNA transcripts in these uterine leiomyomata. This mechanism could also account for slight differences in the tumor spectrum mediated by full length forms of Hmga2 versus truncated Hmga2 forms lacking the let-7 binding sites in mice [18,95]. Ectopic expression of truncated HMGA2 transgenes in an immortalized mesenchymal stem-like cell line stymied adipogenic differentiation and upregulated genes for transcription and intracellular protein transfer compared to wildtype HMGA2 indicative of higher tumorigenicity and lesser let-7 repressive activity in cells expressing truncated HMGA2 [96].
In many other mesenchymal tumors, activated LIN28A or LIN28B RNA-binding protein homologues directly interact with the terminal loop region of either pre-let-7 and/or primary let-7, preventing their biogenesis and tumor repressive function, and can induce their degradation [97,98,99]. Additionally, by repressing let-7, LIN28A/B indirectly upregulates cell cycle regulators targeted by let-7 such as cyclinD1/2, CDK6, CDC34, CDC25A, and TRIM71, and cell proliferation pathway targets PI3K/AKT, MAPK, MYC, RAS and BLIMP1 leading to aberrant proliferation of tumor cells [99]. Conversely, let-7 can also bind to complementary sites in the 3’ UTR of LIN28A/B mRNAs inhibiting their expression and function [100]. Other RNA-binding proteins like IMP3 exist in stable cytoplasmic granules which physically associate with and protect HMGA2 mRNA from let-7-dependent degradation [101].
Most frequently, chromosomal break points truncate the HMGA2 3’ UTR in mesenchymal tumors and can thus prevent the docking of let-7 to its binding sites and the canonical repression of HMGA2 expression in differentiated mesenchymal tumors [62] (Figure 1D). Truncated HMGA2 also occur as chimeras fused to ectopic sequences by chromosomal translocations (Figure 1E). It has been suggested that the loss of let-7 complementary sites in 3’UTR stabilizes HMGA2 mRNA in some of these tumors [63] leading to a deficiency in let-7-mediated regulation [61], increased cell proliferation and tumorigenesis [43,102]. These studies highlight the central role let-7 expression plays in multiple mechanisms of HMGA2 misexpression causing mesenchymal tumorigenesis. Given that multiple genes distributed throughout the human genome encode for let-7, loss of the 3’UTR might not suffice to explain all HMGA2-mediated tumorigenic events in the mesenchyme [62]. Indeed, in a recent study, let-7 accounted for only 15% of the total regulatory effects determining HMGA2 expression/misexpression [64]. This seemingly limited influence of let-7 in directly regulating HMGA2 misexpression could be more reflective of its cooperative roles with other translation regulatory networks in the ontogenesis of mesenchymal tumors. In mechanistic studies using adipogenic progenitor cells of lipomas and uterine leiomyomas, a p14Arf-MDM2-TP53-let-7 network stabilized HMGA2 expression, lowering the tendency for malignant transformation while maintaining stem-like proliferative characteristic of benign mesenchymal tumor cells [26]. p14Arf repressed HMGA2 via a TP53 mechanism that has been linked to increased expression of let-7, whereas FGF1-stimulated increase in HMGA2 increased p14Arf [26,103]. These study results are indicative of a bimodal mechanism of regulating cellular senescence and stem-like cell renewal forming the pathobiological basis for benign mesenchymal tumorigenesis [9].
Unlike the preponderance of studies performed on benign mesenchymal tumors, only a few preliminary studies have determined the role of HMGA2 misexpression in malignant mesenchymal tumors such as well differentiated liposarcomas and osteosarcomas [37,60,76,91]. This may suggest that HMGA2 misexpression is a relatively rare event in malignant mesenchymal tumors. In addition to canonical HMGA2 misexpression presently described, these tumors commonly possess supernumerary ring structures and giant rod chromosomes exhibiting gene amplifications at the 12q13–15 HMGA2 loci and in proximally located genes such as MDM2 [59,104,105].
2.2. Effects of HMGA2 Misexpression on Chromatin Structure
Accessibility to DNA within chromatin remains central to the epigenetic regulation of eukaryotic DNA-dependent nuclear processes such as transcription, replication, recombination, and repair [106]. Nucleosomes are the structural and functional units of chromatin, comprising DNA surrounding histone octamers to form nucleosome cores that are joined by linker DNA [107]. Two major epigenetic regulatory processes—post-translational modification of histones and chromosome remodeling—yield chromosomal conformations that allow transcriptional access to DNA in chromatin [108]. Some of the post-translational modifications necessitating architectural changes in chromatin include DNA methylation and histone acetylation [108,109,110]. As an example, in gliomas, HMGA2 was shown to form a complex with histone lysine acetyltransferase GCN5 and bind to AT-rich promoter region of matrix metalloproteinase 2 (MMP2), catalyzing the histone acetylation and chromatin conformational remodeling of the promoter that induced gene transcription and invasive phenotype of glioblastoma cells [111].
In addition, H1 histones can bind to linker DNA and increase the compactness of chromatin, providing a barrier to sequence-specific recognition sites on DNA [112,113]. In turn, the HMGA2 and HMG families of proteins also bind to nucleosomes and functional AT-rich motifs in DNA minor groove, competing with H1 histones for binding to linker DNA [114,115]. In doing so, HMGA2 modulates H1 histone binding to chromatin, replacing post-translationally modified H1 histones in some cases [106,116]. This induces loosening and conformational changes in the chromatin structure, that affects nucleosome accessibility in both genetically active euchromatin and inactive heterochromatin [106,116]. This ability of HMGA2 to constrain chromatin supercoiling has been shown to confer protection and genome stability in human fibrosarcoma, embryonic stem cells (HESCs), lung epithelial cells, and adenocarcinomas during DNA replication [22,117,118]. HMGA2 inhibits replication fork regression and cytotoxic double stranded breaks (DSBs) generated by chemotherapeutic agents (anti-topoisomerases) and/or during excessive DNA supercoiling [22,117,118]. As such, at high levels of expression, HMGA2 is a critical determinant of tumor response to chemotherapy and tumor cell survival and invasiveness [117,119].
For complete access to transcriptional regulatory elements on target genes, chromatin remodelers such as the FACT complex [120] and the anti-silencing function 1 histone chaperone (ASF1) [121] will be necessary participants. These protein complexes have been shown to facilitate eviction/deposition of histones from nucleosome cores, destabilizing/stabilizing nucleosome structure for transcription elongation processes [120,121]. Given that HMGA2 also binds to these chromatin remodeling protein complexes, a role of HMGA proteins in the dynamics of nucleosome core architectures during transcriptional regulation has been suggested [106]. These HMGA2–nucleosome associations help establish cell identity, and initiate tissue differentiation programs, such as the EMT-driven epithelial tumorigenesis [106] discussed in the subsequent section. However, causative mechanisms of mesenchymal tumorigenesis by HMGA2-induced alteration of chromatin structure is yet to be delineated for most mesenchymal tumors.
2.3. HMGA2-Induced Tumorigenesis in Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is the major pulmonary manifestation of the tuberous sclerosis complex disorder (TS) presenting as benign mesenchymal tumors and lesions that cause recurrent pneumothorax, lung cysts, chylous pleural effusions and renal and abdominal angiomyolipomas (AMLs) [122,123,124,125,126]. This disorder occurs either sporadically (S-LAM), or as an autosomal dominant inheritance of mutations in the Tsc1 or, more frequently, Tsc2 tumor suppressor genes (TSC-LAM) [126,127,128,129,130,131,132]. Heterozygous TSC1/2 mutations have been causatively linked to hyperactivation of the mammalian target of rapamycin (mTOR) pathway, leading to the aberrant cell growth and proliferation characterizing benign tumors in LAM [133,134,135,136,137,138]. However, as many LAM patient tumors do not exhibit TSC mutations especially in S-LAM cases [139], biochemical signaling due to activation of the mTOR pathway does not solely account for tumorigenesis in LAM [138,140,141]. This explains why pharmacological inhibition of mTOR pathway targets is not curative [142,143,144], and in most treatment cohorts, disease symptoms tend to recur upon discontinuation of therapy [145]. It is thus imperative that alternative etiologies to LAM pathogenesis in addition to genetic mechanisms at the TSC1/2 loci be considered.
In our laboratory, we focused on the mechanisms of HMGA2 in LAM pathogenesis, and established that although HMGA2 was mis-expressed in 100% of tumors resected from LAM patients in our studies and from Tsc2+/− mice, an established animal model for LAM [14,146], Hmga2−/−Tsc2+/− mice exhibited minimal renal (epithelial) tumors and no extra-renal (mesenchymal) tumors) [146]. These results indicate that Hmga2 expression is absolutely required for mesenchymal tumorigenesis in the Tsc2+/− mice. We further observed analogous expression of IGF2BP2, an oncofetal protein and downstream target of HMGA2 in 100% of human LAM and TSC lesions [146], similar to the gene’s pattern of co-expression with HMGA2 in rhabdomyosarcoma and during myoblast proliferation [5,147]. Importantly, the tuberin protein product of Tsc2 expression was present in all Tsc2+/− mouse and human mesenchymal tumors, and about 80% of Tsc2+/− mouse renal carcinoma [146]. Adding that only 50% of human mesenchymal tumors, and 31% of Tsc2+/− mouse renal tumors exhibit altered mTOR pathway activation, supports the existence of tumorigenic mTOR-independent mechanisms causing LAM pathogenesis [146]. The lack of HMGA2 expression in similar proliferative interstitial lung diseases to LAM, like interstitial pneumonitis and pulmonary fibrosis, also suggests that HMGA2 misexpression transforms tumor cells in LAM, and is not solely due to an abnormal increase in cell proliferation rates [14].
However, there has been a dearth in studies investigating the mechanistic potential of HMGA2-induced neoplasms in LAM. As LAM pathogenesis is characterized by benign tumors with differentiating characteristics towards a mesenchymal phenotype, we postulated that the oncogenic triangle involving LIN28-let-7-HMGA2 observed in most mesenchymal tumors might account for the tumorigenic pathway employed by HMGA2 in LAM. Using an established Tsc2+/- mouse model of LAM [148], we have assessed the expression of Hmga2 pathway targets in both mesenchymal and renal (epithelial) tumors. Compared to matched normal tissues, our results reveal significant downregulation of let-7a expression and upregulation of Lin28a, Lin28b and Igf2bp2 in all mesenchymal tumors assessed including hepatic hemangiosarcomas, foot lymphomas, and hemangiomas, and in pulmonary adenomas by RT-PCR (unpublished data). In these mice, the absolute necessity for the repression of let-7a was observed for both epithelial and mesenchymal tumors supporting previous findings of the key role of the let-7–Hmga2 axis in these carcinogenic processes [63,99]. Cytogenetic studies in the Tsc2+/− mouse mesenchymal tumors will determine whether chromosomal rearrangements play a pathogenic role in these LAM mouse models.
3. Mechanisms of HMGA2-Induced Epithelial Tumorigenicity
Nascent epithelial tumors and early adenomas typically exhibit undetectable or low levels of HMGA2 mRNA [149] and low to moderate levels of the HMGA2 protein product [15]. However, an increasing expression of HMGA2 was observed to be distributed from non-neoplastic, well-differentiated centers of human colorectal tumors, human squamous carcinoma, and MMTV-Wnt1 transgenic mice mammary tumors towards the extremity invasive front where the overwhelming majority of tumor cells were HMGA2-positive [15,150]. In mammary and colorectal tumors, these invasive HMGA2-positive tumor cells exhibited membrane-to-nucleus re-localization of β-catenin, loss of E-cadherin, increase in levels of vimentin, and an analogous expression of HMGA2 downstream target IGF2BP2 [15,151]. This observed cytological transition in phenotype known as epithelial-mesenchymal transition (EMT) is a highly conserved gradual process that also occurs during embryonic development, histogenesis, and wound repair, and is precursory to tumor invasion and malignancy [15,152,153]. Indeed, HMGA2 expression increased proportionally with anchorage-independent growth and metastasis of many epithelial tumors including colon, gastric, and breast cancer cells [15,154,155]. In mammary tumor cell lines stably expressing HMGA2 by vector transfection, primary tumors formed at a faster rate and exhibited higher metastatic potential to the liver parenchyma and lungs than tumor cells with empty vectors after inoculation in mice [15]. In many studies, EMT has been reported to be the key mechanism for HMGA2-induced tumorigenesis in epithelial tissues [16,156,157].
How does ectopic expression of HMGA2 induce EMT for epithelial tumorigenesis, metastasis and invasion? In both in vitro and in vivo models of epithelial tumorigenesis, HMGA2 has been observed to activate the TGFβ pathway via TGFβRII leading to the phosphorylation and translocation of Smad3 from the cytoplasm to the nucleus [15], a mechanism necessary for EMT initiation [157,158]. For HMGA2 to induce epithelial tumorigenesis and invasiveness via EMT, it is necessary that TGFβ pathway is activated [15,157]. It is suggested that epithelial tumor cells, upon HMGA2 induction, might become more responsive to the TGFβ ligand typically present in tumor microenvironments, signaling for increased tumor metastasis [15]. Indeed, downstream of SMAD proteins, HMGA2 was resolved to directly activate zinc-finger transcription families Snail, Slug, and Twist and downregulate Inhibitor of differentiation 2 (Id2) in mammary tumor cells which altogether are known to repress E-cadherin (CDH1) expression [157,159,160]. In addition, ectopic HMGA2 expression also remodels chromatin in mammary tumor and breast cancer to a closed conformation at the Cdh1 locus by hypermethylation governed by HMGA2 interaction with DNA methyltransferases (DNMT3A) [161]. This biochemical mechanism represents a second layer of epigenetic control of E-cadherin expression during EMT-induced epithelial tumorigenesis.
Other small molecule regulators including microRNA and RNA-binding proteins also govern epithelial tumorigenesis induced by HMGA2. The let-7 miRNA family have been shown to exert repressive control over HMGA2-induced epithelial tumorigenesis by binding to the 3’-UTR of human HMGA2 gene [162]. In the course of EMT advancing carcinogenesis, ovarian cancer cells expressing an epithelial gene signature exhibited significantly higher levels of seven of the twelve members of the let-7 family compared to cells defined by mesenchymal genes [162]. Individual let-7 family members have also been implicated in a variety of tumor pathogenic mechanisms involving HMGA2. For instance, let-7c was shown to suppress EMT and proliferation of head and neck squamous cell carcinoma by targeting HMGA2 and IGF1R [163], while let-7a knockdown exhibited an analogous inhibitory function in nasopharyngeal carcinomas induced by HMGA2 expression, decreasing HMGA2 and the expression of EMT marker genes Snail, Slug, and vimentin [164]. However, LIN28 RNA-binding protein, serves as a competitive inhibitor of let-7 binding preventing its maturation by inducing terminal uridylation and degradation of let-7 precursors, and can thus de-repress HMGA2 expression, restoring tumorigenicity and invasiveness of epithelial tumors [165]. The LIN28-let-7-HMGA2 signaling axis is further modulated by Raf-1-kinase inhibitory protein (RKIP), a metastasis suppressor which induces expression of epithelial miR-200b that directly inhibits Lysyl Oxidase (LOX) expression leading to decreased transcription of LIN28, elevated let-7 expression and inhibition of HMGA2 [166,167]. In another study of prostate cancer metastasis, overexpression of BTB and CNC Homology 1 (BACH1) transcription factor led to a significant decrease in let-7A expression and subsequent increase in HMGA2 which facilitated metastasis by promoting EMT [168]. Given the genomic ubiquity of let-7 in regulating HMGA2 expression [62], further studies are required to clearly distinguish the role of let-7 in mesenchymal versus epithelial tumorigenesis.
Ectopic expression of HMGA2 can also promote epithelial tumor cell proliferation and metastasis by influencing the cell cycle in a tumor cell-type dependent manner, where, for instance, its knockdown arrests ovarian cancer cells at G1 [169] and G2/M arrest for leukemia cells [19]. In some instances, HMGA2 can exert tumorigenic effects on cell cycle by directly inducing cyclin A2 [20], activating protein-1 (AP1) expression [170], and MDM2-mediated p53 ubiquitination [171] facilitating cell proliferation. Indirectly, HMGA2 can activate phosphatidylinositide 3-kinase (PI3K)/AKT/mTOR/p70S6k signaling which inhibits tumor suppressors p16INK4A, p21CIP1/WAF1 [172], retinoblastoma protein (pRB) [173] and p14Arf [9] to enable transitions through cell cycle checkpoints, facilitate cell proliferation and restrain cellular senescence. During these replicative cycles, HMGA2 has also been postulated to either inhibit DNA damage response (DDR) mechanisms that ensure genomic stability at replication forks, which leads to increased DNA mutational rates at onset of tumorigenesis, or augment these DDR mechanisms to reduce replication recovery times after replication fork arrest in stem and cancer cells [22,174]. HMGA2 can also mediate epithelial tumorigenesis by modulating apoptosis in cancer cells. Breast adenocarcinoma cells overexpressing HMGA2 exhibit fewer apoptotic events compared to cells with low HMGA2 expression by a mechanism involving HMGA2-mediated inhibition of miR-34a and subsequent de-repression of Bcl-2 [25]. HMGA2 also protects cancer cells from apoptosis by hyperactivating the PI3K/Akt pathway, which impairs the activation of caspase-9 and Bad in a gastric cancer cell line [175].
Dynamic intracellular localization processes of HMGA2 mRNA transcripts and translation products have been reported to also influence epithelial tumorigenesis and metastatic potential. Hmga2 expression has been detected in the cell membrane of quiescent non-transformed, post-natal mouse keratinocytes but upon onset of proliferation, membrane-to-nuclear translocation of the protein was observed [176]. Analogous Hmga2 nuclear translocation was also been observed upon ex vivo culture of mouse keratinocytes and during cutaneous carcinogenesis in DMBA and TPA mouse models where Hmga2 induces its own expression in an autoregulatory loop by binding to the Hmga2 promoter [176]. Normal human epithelial prostate cell also exhibited low levels of HMGA2 expression in the plasma membrane which switched to predominantly cytoplasmic then nuclear localization with increasing prostate tumor grade, metastatic potential, and HMGA2 expression [177]. Specific small non-coding circular RNAs (circNSUN2) promotes EMT and human and mouse colorectal cancer (CRC) cell metastasis by stabilizing high levels of cytoplasmic HMGA2 RNA-protein interactions in a circNSUN2/IGF2BP2/HMGA2/ complex and activating the HMGA2 pathway [178]. In this study, lower levels of cytoplasmic HMGA2 mRNA were associated with lower liver metastasis in CRC [178]. HMGA2 mRNA cytoplasmic stability is similarly governed by higher order ribonucleoprotein complexes formed with RNA binding proteins as observed in solid cancers where for instance, HMGA2 mRNA is physically associated with IMP3 RNP in vivo [179]. There is a need for studies of these HMGA2 intracellular translocation processes and their relevance in mesenchymal tumorigenesis.
These studies make it clear that HMGA2 oncogenic mechanisms differ between mesenchymal tumors and their epithelial counterparts and even between tissue-specific tumor subtypes. HMGA2-induced mesenchymal tumorigenesis recapitulates the gene’s embryonic expression signature in mature differentiated tissues using various mechanisms leading to neoplastic growth [17,18,44]. In epithelial tumors, HMGA2 drives epithelial-to-mesenchymal cell transformations (EMT) towards a tumorigenic phenotype [15].
4. Therapeutic Considerations for HMGA2-Induced Neoplasia
High expression of HMGA2 has been associated with highly malignant phenotypes described by resistance to chemotherapeutic agents and metastases [180,181]. Along with let-7 expression, HMGA2 has been determined to be a predictive biomarker of poor clinical outcomes in many epithelial and hematopoietic malignancies including acute myeloid leukemia [182], ovarian and colorectal carcinoma [162,169], and oral squamous cell carcinoma [183]. In some neoplasia, however, disease severity does not always correlate with increased HMGA2 expression levels [184,185], necessitating the resolution of other biomarkers in the HMGA2 pathway. Downstream of HMGA2, IGF2BP2 was found to be overexpressed and correlative to poor survival and induction of EMT in pancreatic ductal adenocarcinoma [186]. Given that many interacting partners in the HMGA2 pathway, including up- and downstream genes IGF2BP2, LIN28 and let-7 have been elucidated, they serve as attractive targets whose modulation could be curative for many cancers involving the HMGA2 pathway. However, most therapeutic strategies adopted to probe targets in this pathway have focused chiefly on perturbing HMGA2 expression in epithelial neoplasms with limited success.
Gene silencing therapy using siRNA suppressed proliferation and growth of ovarian cancer cell lines overexpressing HMGA2 by cell cycle arrest at G1 phase, and decreased the size of tumor xenografts in athymic nude mice treated with a Hmga2-targeting construct [169]. A similar effect was observed when siRNA- and miRNA-mediated silencing of HMGA2 induced apoptosis, G2/M cell cycle arrest, and suppressed proliferation and invasion of human colorectal carcinoma [187,188]. Additionally, p53-induced miR-1249 expression was antagonistic to HMGA2 expression and inhibited HMGA2-induced invasiveness of colorectal cancer cells by stabilizing the epithelial phenotype, decreasing expression of N-cadherin and vimentin and increasing E-cadherin expression [189]. Sustained expression of the epithelial phenotype was also observed in HMGA2-/- prostate cancer cells in which EMT was also inhibited [190]. Similarly, perturbation of HMGA2-HOXA9 signaling arrested the differentiation of human myeloid leukemia cells towards a pathogenic phenotype [191].
In another therapeutic strategy, small molecule inhibitors such as netropsin were found to block the binding of HMGA2 to DNA minor groove in AT sequences in a biosensor-surface plasmon resonance assay designed to screen for potent HMGA2 inhibitors [192]. However, a recent study found that netropsin was not selectively cytocidal to only HMGA2-overexpressing colorectal cancer cells [193], and its potential binding to other AT-hook DNA domains might introduce off-target side-effects, making it therapeutically inefficient. Rather, an antihelminthic drug niclosamide has been repurposed and is selective against HMGA2-overexpressing colorectal cancer cells, reversing the HMGA2-driven gene signature, and inhibiting cell cycle-related genes in these cells [193]. Analogously, successful treatment of poorly differentiated thyroid carcinoma with tyrosine kinase inhibitor selumetinib and histone deacetylase inhibitor panobinostat led to the significant downregulation of HMGA2 expression, correlating with an upregulation or stable expression of associated miRNA let-7b [194].
Supplementary Materials
Supplementary materials can be found at https://0-www-mdpi-com.brum.beds.ac.uk/1422-0067/21/9/3151/s1.
Author Contributions
Conceptualization, U.U., K.C., J.D.; Original draft preparation, U.U., K.C.; Writing—review and editing, U.U., K.C.; Supervision, K.C., J.D.; Funding acquisition, J.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research and APC was funded by the Center for LAM and Rare Lung Disease.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhou, X.; Chada, K. HMGI family proteins: Architectural transcription factors in mammalian development and cancer. Keio J. Med. 1998, 47, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-J. HMGA2 (high mobility group AT-hook 2). Atlas Genet. Cytogenet. Oncol. Haematol. 2016, 20, 403–412. [Google Scholar] [CrossRef]
- Cleynen, I.; Brants, J.R.; Peeters, K.; Deckers, R.; Debiec-Rychter, M.; Sciot, R.; Van de Ven, W.J.; Petit, M.M. HMGA2 regulates transcription of the Imp2 gene via an intronic regulatory element in cooperation with nuclear factor-kappaB. Mol. Cancer Res. 2007, 5, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Benson, K.F.; Ashar, H.R.; Chada, K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 1995, 376, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gilbert, J.A.; Zhang, Y.; Zhang, M.; Qiu, Q.; Ramanujan, K.; Shavlakadze, T.; Eash, J.K.; Scaramozza, A.; Goddeeris, M.M.; et al. An HMGA2-IGF2BP2 axis regulates myoblast proliferation and myogenesis. Dev. Cell 2012, 23, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, O.; Li, J.; Dröge, P. DNA architectural factor and proto-oncogene HMGA2 regulates key developmental genes in pluripotent human embryonic stem cells. FEBS Lett. 2007, 581, 3533–3537. [Google Scholar] [CrossRef] [Green Version]
- Hirning-Folz, U.; Wilda, M.; Rippe, V.; Bullerdiek, J.; Hameister, H. The expression pattern of the Hmgic gene during development. Genes Chromosomes Cancer 1998, 23, 350–357. [Google Scholar] [CrossRef]
- Gattas, G.J.; Quade, B.J.; Nowak, R.A.; Morton, C.C. HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes Chromosomes Cancer 1999, 25, 316–322. [Google Scholar] [CrossRef]
- Nishino, J.; Kim, I.; Chada, K.; Morrison, S.J. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell 2008, 135, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Lund, T.; Holtlund, J.; Fredriksen, M.; Laland, S.G. On the presence of two new high mobility group-like proteins in HeLa S3 cells. FEBS Lett. 1983, 152, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, V.; Berlingieri, M.T.; DiFiore, P.P.; Fusco, A.; Vecchio, G.; Crane-Robinson, C. Changes in Nuclear Proteins on Transformation of Rat Epithelial Thyroid Cells by a Murine Sarcoma Retrovirus. Cancer Res. 1985, 45(Pt. 1), 6051. [Google Scholar]
- Giancotti, V.; Buratti, E.; Perissin, L.; Zorzet, S.; Balmain, A.; Portella, G.; Fusco, A.; Goodwin, G.H. Analysis of the HMGI nuclear proteins in mouse neoplastic cells induced by different procedures. Exp. Cell Res. 1989, 184, 538–545. [Google Scholar] [CrossRef]
- Wood, L.J.; Maher, J.F.; Bunton, T.E.; Resar, L.M. The oncogenic properties of the HMG-I gene family. Cancer Res. 2000, 60, 4256–4261. [Google Scholar] [PubMed]
- D’Armiento, J.; Imai, K.; Schiltz, J.; Kolesnekova, N.; Sternberg, D.; Benson, K.; Pardo, A.; Selman, M.; Smolarek, T.; Vundavalli, M.; et al. Identification of the Benign Mesenchymal Tumor Gene HMGA2 in Lymphangiomyomatosis. Cancer Res. 2007, 67, 1902–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, A.; Zaidi, M.R.; Mitoro, A.; Sankarasharma, D.; Szabolcs, M.; Okada, Y.; D’Armiento, J.; Chada, K. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013, 73, 4289–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Li, W.; Liao, H. HMGA2 induces epithelial-to-mesenchymal transition in human hepatocellular carcinoma cells. Oncol. Lett. 2013, 5, 1353–1356. [Google Scholar] [CrossRef]
- Zaidi, M.R.; Okada, Y.; Chada, K.K. Misexpression of Full-length HMGA2 Induces Benign Mesenchymal Tumors in Mice. Cancer Res. 2006, 66, 7453. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.; Narita, M. Oncogenic HMGA2: Short or small? Genes Dev. 2007, 21, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Wei, X.; Zheng, L.; Zeng, J.; Liu, H.; Yang, S.; Tan, H. Amplified HMGA2 promotes cell growth by regulating Akt pathway in AML. J. Cancer Res. Clin. Oncol. 2016, 142, 389–399. [Google Scholar] [CrossRef]
- Tessari, M.A.; Gostissa, M.; Altamura, S.; Sgarra, R.; Rustighi, A.; Salvagno, C.; Caretti, G.; Imbriano, C.; Mantovani, R.; Del Sal, G. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol. Cell. Biol. 2003, 23, 9104–9116. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Peng, L.; Seto, E. Histone Deacetylase 10 Regulates the Cell Cycle G2M Phase Transition via a Novel Let-7–HMGA2–Cyclin A2 Pathway. Mol. Cell. Biol. 2015, 35, 3547. [Google Scholar] [PubMed] [Green Version]
- Yu, H.; Lim, H.H.; Tjokro, N.O.; Sathiyanathan, P.; Natarajan, S.; Chew, T.W.; Klonisch, T.; Goodman, S.D.; Surana, U.; Droge, P. Chaperoning HMGA2 protein protects stalled replication forks in stem and cancer cells. Cell Rep. 2014, 6, 684–697. [Google Scholar] [CrossRef] [Green Version]
- Borrmann, L.; Schwanbeck, R.; Heyduk, T.; Seebeck, B.; Rogalla, P.; Bullerdiek, J.; Wisniewski, J.R. High mobility group A2 protein and its derivatives bind a specific region of the promoter of DNA repair gene ERCC1 and modulate its activity. Nucleic Acids Res. 2003, 31, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- Fujikane, R.; Komori, K.; Sekiguchi, M.; Hidaka, M. Function of high-mobility group A proteins in the DNA damage signaling for the induction of apoptosis. Sci. Rep. 2016, 6, 31714. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Shirjang, S.; Baradaran, B. HMGI-C suppressing induces P53/caspase9 axis to regulate apoptosis in breast adenocarcinoma cells. Cell Cycle 2016, 15, 2585–2592. [Google Scholar] [CrossRef]
- Markowski, D.N.; Winter, N.; Meyer, F.; von Ahsen, I.; Wenk, H.; Nolte, I.; Bullerdiek, J. p14Arf acts as an antagonist of HMGA2 in senescence of mesenchymal stem cells—Implications for benign tumorigenesis. Genes Chromosomes Cancer 2011, 50, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Tian, B.; Ma, C.; Liu, L.; Zhang, N.; Na, Y.; Li, J.; Lu, J.; Qiao, Y. GSK3β activity is essential for senescence-associated heterochromatin foci (SAHF) formation induced by HMGA2 in WI38 cells. Am. J. Transl. Res. 2017, 9, 167–174. [Google Scholar]
- Zhang, S.; Mo, Q.; Wang, X. Oncological role of HMGA2 (Review). Int. J. Oncol. 2019, 55, 775–788. [Google Scholar] [CrossRef]
- Bartuma, H.; Hallor, K.H.; Panagopoulos, I.; Collin, A.; Rydholm, A.; Gustafson, P.; Bauer, H.C.; Brosjo, O.; Domanski, H.A.; Mandahl, N.; et al. Assessment of the clinical and molecular impact of different cytogenetic subgroups in a series of 272 lipomas with abnormal karyotype. Genes Chromosomes Cancer 2007, 46, 594–606. [Google Scholar] [CrossRef]
- Bianchini, L.; Birtwisle, L.; Saada, E.; Bazin, A.; Long, E.; Roussel, J.F.; Michiels, J.F.; Forest, F.; Dani, C.; Myklebost, O.; et al. Identification of PPAP2B as a novel recurrent translocation partner gene of HMGA2 in lipomas. Genes Chromosomes Cancer 2013, 52, 580–590. [Google Scholar] [CrossRef]
- Galindo, L.J.; Hernández-Beeftink, T.; Salas, A.; Jung, Y.; Reyes, R.; de Oca, F.M.; Hernández, M.; Almeida, T.A. HMGA2 and MED12 alterations frequently co-occur in uterine leiomyomas. Gynecol. Oncol. 2018, 150, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.S.; Klotzbücher, M.; Kugoh, H.; Cai, S.-L.; Mullen, J.P.; Manfioletti, G.; Fuhrman, U.; Walker, C.L. Aberrant Expression of HMGA2 in Uterine Leiomyoma Associated with Loss of TSC2 Tumor Suppressor Gene Function. Cancer Res. 2002, 62, 3766–3772. [Google Scholar] [PubMed]
- Mello, J.B.H.; Barros-Filho, M.C.; Abreu, F.B.; Cirilo, P.D.R.; Domingues, M.A.C.; Pontes, A.; Rogatto, S.R. MicroRNAs involved in the HMGA2 deregulation and its co-occurrence with MED12 mutation in uterine leiomyoma. Mol. Hum. Reprod. 2018, 24, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Tallini, G.; Vanni, R.; Manfioletti, G.; Kazmierczak, B.; Faa, G.; Pauwels, P.; Bullerdiek, J.; Giancotti, V.; Van Den Berghe, H.; Dal Cin, P. HMGI-C and HMGI(Y) immunoreactivity correlates with cytogenetic abnormalities in lipomas, pulmonary chondroid hamartomas, endometrial polyps, and uterine leiomyomas and is compatible with rearrangement of the HMGI-C and HMGI(Y) genes. Lab. Investig. J. Tech. Methods Pathol. 2000, 80, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, I.; Rogalla, P.; Bullerdiek, J. Expression patterns of the LPP-HMGA2 fusion transcript in pulmonary chondroid hamartomas with t(3;12)(q27 approximately 28;q14 approximately 15). Cancer Genet. Cytogenet. 2005, 163, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Bol, S.; Wanschura, S.; Thode, B.; Deichert, U.; Van de Ven, W.J.M.; Bartnitzke, S.; Bullerdiek, J. An endometrial polyp with a rearrangement of HMGI-C underlying a complex cytogenetic rearrangement involving chromosomes 2 and 12. Cancer Genet. Cytogenet. 1996, 90, 88–90. [Google Scholar] [CrossRef]
- Dahlén, A.; Mertens, F.; Rydholm, A.; Brosjö, O.; Wejde, J.; Mandahl, N.; Panagopoulos, I. Fusion, Disruption, and Expression of HMGA2 in Bone and Soft Tissue Chondromas. Mod. Pathol. 2003, 16, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Petit, M.M.R.; Schoenmakers, E.F.P.M.; Huysmans, C.; Geurts, J.M.W.; Mandahl, N.; Van de Ven, W.J.M. LHFP, a Novel Translocation Partner Gene ofHMGICin a Lipoma, Is a Member of a New Family ofLHFP-like Genes. Genomics 1999, 57, 438–441. [Google Scholar] [CrossRef]
- Schoenmakers, E.F.; Wanschura, S.; Mols, R.; Bullerdiek, J.; Van den Berghe, H.; Van de Ven, W.J. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat. Genet. 1995, 10, 436–444. [Google Scholar] [CrossRef]
- Geurts, J.M.; Schoenmakers, E.F.; Van de Ven, W.J. Molecular characterization of a complex chromosomal rearrangement in a pleomorphic salivary gland adenoma involving the 3′-UTR of HMGIC. Cancer Genet. Cytogenet. 1997, 95, 198–205. [Google Scholar] [CrossRef]
- Fejzo, M.S.; Ashar, H.R.; Krauter, K.S.; Powell, W.L.; Rein, M.S.; Weremowicz, S.; Yoon, S.-J.; Kucherlapati, R.S.; Chada, K.; Morton, C.C. Translocation breakpoints upstream of the HMGIC gene in uterine leiomyomata suggest dysregulation of this gene by a mechanism different from that in lipomas. Genes Chromosomes Cancer 1996, 17, 1–6. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Gorunova, L.; Bjerkehagen, B.; Lobmaier, I.; Heim, S. The recurrent chromosomal translocation t(12;18)(q14~15;q12~21) causes the fusion gene HMGA2-SETBP1 and HMGA2 expression in lipoma and osteochondrolipoma. Int. J. Oncol. 2015, 47, 884–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartuma, H.; Panagopoulos, I.; Collin, A.; Trombetta, D.; Domanski, H.A.; Mandahl, N.; Mertens, F. Expression levels of HMGA2 in adipocytic tumors correlate with morphologic and cytogenetic subgroups. Mol. Cancer 2009, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkachenko, A.; Ashar, H.R.; Meloni, A.M.; Sandberg, A.A.; Chada, K.K. Misexpression of Disrupted HMGI Architectural Factors Activates Alternative Pathways of Tumorigenesis. Cancer Res. 1997, 57, 2276. [Google Scholar] [PubMed]
- Kazmierczak, B.; Dal Cin, P.; Wanschura, S.; Borrmann, L.; Fusco, A.; Van den Berghe, H.; Bullerdiek, J. HMGIY is the target of 6p21.3 rearrangements in various benign mesenchymal tumors. Genes Chromosomes Cancer 1998, 23, 279–285. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Andersen, M.T.; Andersen, M.K. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2007, 2007, 392–397. [Google Scholar] [CrossRef]
- Fröhling, S.; Döhner, H. Chromosomal Abnormalities in Cancer. N. Engl. J. Med. 2008, 359, 722–734. [Google Scholar] [CrossRef]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef]
- Mertens, F.; Antonescu, C.R.; Mitelman, F. Gene fusions in soft tissue tumors: Recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 2016, 55, 291–310. [Google Scholar] [CrossRef] [Green Version]
- Velagaleti, G.V.N.; Tonk, V.S.; Hakim, N.M.; Wang, X.; Zhang, H.; Erickson-Johnson, M.R.; Medeiros, F.; Oliveira, A.M. Fusion of HMGA2 to COG5 in uterine leiomyoma. Cancer Genet. Cytogenet. 2010, 202, 11–16. [Google Scholar] [CrossRef]
- Asher, H.R.; Schoenberg Fejzo, M.; Tkachenko, A.; Zhou, X.; Fletcher, J.A.; Weremowicz, S.; Morton, C.C.; Chada, K. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell 1995, 82, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Kazmierczak, B.; Wanschura, S.; Rosigkeit, J.; Meyer-Bolte, K.; Uschinsky, K.; Haupt, R.; Schoenmakers, E.F.; Bartnitzke, S.; Van de Ven, W.J.; Bullerdiek, J. Molecular characterization of 12q14-15 rearrangements in three pulmonary chondroid hamartomas. Cancer Res. 1995, 55, 2497–2499. [Google Scholar] [PubMed]
- Schoenmakers, E.F.; Huysmans, C.; Van de Ven, W.J. Allelic knockout of novel splice variants of human recombination repair gene RAD51B in t(12;14) uterine leiomyomas. Cancer Res. 1999, 59, 19–23. [Google Scholar] [PubMed]
- Mine, N.; Kurose, K.; Konishi, H.; Araki, T.; Nagai, H.; Emi, M. Fusion of a sequence from HEI10 (14q11) to the HMGIC gene at 12q15 in a uterine leiomyoma. Jpn. J. Cancer Res. Gann 2001, 92, 135–139. [Google Scholar] [CrossRef]
- Mine, N.; Kurose, K.; Nagai, H.; Doi, D.; Ota, Y.; Yoneyama, K.; Konishi, H.; Araki, T.; Emi, M. Gene fusion involving HMGIC is a frequent aberration in uterine leiomyomas. J. Hum. Genet. 2001, 46, 408–412. [Google Scholar] [CrossRef]
- Kurose, K.; Mine, N.; Doi, D.; Ota, Y.; Yoneyama, K.; Konishi, H.; Araki, T.; Emi, M. Novel gene fusion of COX6C at 8q22–23 to HMGIC at 12q15 in a uterine leiomyoma. Genes Chromosomes Cancer 2000, 27, 303–307. [Google Scholar] [CrossRef]
- Fedele, M.; Berlingieri, M.T.; Scala, S.; Chiariotti, L.; Viglietto, G.; Rippel, V.; Bullerdiek, J.; Santoro, M.; Fusco, A. Truncated and chimeric HMGI-C genes induce neoplastic transformation of NIH3T3 murine fibroblasts. Oncogene 1998, 17, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Wanschura, S.; Cin, P.D.; Kazmierczak, B.; Bartnitzke, S.; Van den Berghe, H.; Bullerdiek, J. Hidden paracentric inversions of chromosome arm 12q affecting the HMGIC gene. Genes Chromosomes Cancer 1997, 18, 322–323. [Google Scholar] [CrossRef]
- Sarhadi, V.K.; Wikman, H.; Salmenkivi, K.; Kuosma, E.; Sioris, T.; Salo, J.; Karjalainen, A.; Knuutila, S.; Anttila, S. Increased expression of high mobility group A proteins in lung cancer. J. Pathol. 2006, 209, 206–212. [Google Scholar] [CrossRef]
- Pedeutour, F.; Forus, A.; Coindre, J.-M.; Berner, J.-M.; Nicolo, G.; Michiels, J.-F.; Terrier, P.; Ranchere-Vince, D.; Collin, F.; Myklebost, O.; et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer 1999, 24, 30–41. [Google Scholar] [CrossRef]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemke, M.; Meyer, A.; Hashemi Nezhad, M.; Belge, G.; Bartnitzke, S.; Bullerdiek, J. Loss of let-7 binding sites resulting from truncations of the 3′ untranslated region of HMGA2 mRNA in uterine leiomyomas. Cancer Genet. Cytogenet. 2010, 196, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Kristjansdottir, K.; Fogarty, E.A.; Grimson, A. Systematic analysis of the Hmga2 3′ UTR identifies many independent regulatory sequences and a novel interaction between distal sites. RNA 2015, 21, 1346–1360. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.C.; Steitz, J.A. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef] [Green Version]
- Borrmann, L.; Wilkening, S.; Bullerdiek, J. The expression of HMGA genes is regulated by their 3′UTR. Oncogene 2001, 20, 4537–4541. [Google Scholar] [CrossRef] [Green Version]
- Kools, P.F.; Van de Ven, W.J. Amplification of a rearranged form of the high-mobility group protein gene HMGIC in OsA-CI osteosarcoma cells. Cancer Genet. Cytogenet. 1996, 91, 1–7. [Google Scholar] [CrossRef]
- Rogalla, P.; Kazmierczak, B.; Meyer-Bolte, K.; Tran, K.H.; Bullerdiek, J. The t(3;12)(q27;q14-q15) with underlying HMGIC-LPP fusion is not determining an adipocytic phenotype. Genes Chromosomes Cancer 1998, 22, 100–104. [Google Scholar] [CrossRef]
- Turc-Carel, C.; Dal Cin, P.; Rao, U.; Karakousis, C.; Sandberg, A.A. Cytogenetic studies of adipose tissue tumors. I. A benign lipoma with reciprocal translocation t(3;12)(q28;q14). Cancer Genet. Cytogenet. 1986, 23, 283–289. [Google Scholar] [CrossRef]
- Sreekantaiah, C.; Leong, S.P.; Karakousis, C.P.; McGee, D.L.; Rappaport, W.D.; Villar, H.V.; Neal, D.; Fleming, S.; Wankel, A.; Herrington, P.N.; et al. Cytogenetic profile of 109 lipomas. Cancer Res. 1991, 51, 422–433. [Google Scholar]
- Dal Cin, P.; Turc-Carel, C.; Sandberg, A.A. Consistent involvement of band 12q14 in two different translocations in three lipomas from the same patient. Cancer Genet. Cytogenet. 1988, 31, 237–240. [Google Scholar] [CrossRef]
- Bartuma, H.; Nord, K.H.; Macchia, G.; Isaksson, M.; Nilsson, J.; Domanski, H.A.; Mandahl, N.; Mertens, F. Gene expression and single nucleotide polymorphism array analyses of spindle cell lipomas and conventional lipomas with 13q14 deletion. Genes Chromosomes Cancer 2011, 50, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Petit, M.M.R.; Swarts, S.; Bridge, J.A.; Van de Ven, W.J.M. Expression of Reciprocal Fusion Transcripts of the HMGIC and LPP Genes in Parosteal Lipoma. Cancer Genet. Cytogenet. 1998, 106, 18–23. [Google Scholar] [CrossRef]
- Petit, M.M.R.; Mols, R.; Schoenmakers, E.F.P.M.; Mandahl, N.; Van de Ven, W.J.M. LPP, the Preferred Fusion Partner Gene of HMGIC in Lipomas, Is a Novel Member of the LIM Protein Gene Family. Genomics 1996, 36, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Broberg, K.; Zhang, M.; Strombeck, B.; Isaksson, M.; Nilsson, M.; Mertens, F.; Mandahl, N.; Panagopoulos, I. Fusion of RDC1 with HMGA2 in lipomas as the result of chromosome aberrations involving 2q35-37 and 12q13-15. Int. J. Oncol. 2002, 21, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Morita, T.; Ogose, A.; Hotta, T.; Kobayashi, H.; Segawa, H.; Uchiyama, T.; Takenouchi, T.; Sato, T. Clinicopathological Features of Lipomas with Gene Fusions Involving HMGA2. Anticancer Res. 2008, 28, 535–538. [Google Scholar]
- Nilsson, M.; Mertens, F.; Hoglund, M.; Mandahl, N.; Panagopoulos, I. Truncation and fusion of HMGA2 in lipomas with rearrangements of 5q32-->q33 and 12q14-->q15. Cytogenet. Genome Res. 2006, 112, 60–66. [Google Scholar] [CrossRef]
- Italiano, A.; Ebran, N.; Attias, R.; Chevallier, A.; Monticelli, I.; Mainguene, C.; Benchimol, D.; Pedeutour, F. NFIB rearrangement in superficial, retroperitoneal, and colonic lipomas with aberrations involving chromosome band 9p22. Genes Chromosomes Cancer 2008, 47, 971–977. [Google Scholar] [CrossRef]
- Lacaria, M.; El Demellawy, D.; McGowan-Jordan, J. A rare case of pediatric lipoma with t(9;12)(p22;q14) and evidence of HMGA2-NFIB gene fusion. Cancer Genet. 2017, 216–217, 100–104. [Google Scholar] [CrossRef]
- Pierron, A.; Fernandez, C.; Saada, E.; Keslair, F.; Hery, G.; Zattara, H.; Pedeutour, F. HMGA2-NFIB fusion in a pediatric intramuscular lipoma: A novel case of NFIB alteration in a large deep-seated adipocytic tumor. Cancer Genet. Cytogenet. 2009, 195, 66–70. [Google Scholar] [CrossRef]
- Nilsson, M.; Panagopoulos, I.; Mertens, F.; Mandahl, N. Fusion of the HMGA2 and NFIB genes in lipoma. Virchows Arch. 2005, 447, 855–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazmierczak, B.; Pohnke, Y.; Bullerdiek, J. Fusion transcripts between the HMGIC gene and RTVL-H-related sequences in mesenchymal tumors without cytogenetic aberrations. Genomics 1996, 38, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Polito, P.; Dal Cin, P.; Kazmierczak, B.; Rogalla, P.; Bullerdiek, J.; Van den Berghe, H. Deletion of HMG17 in uterine leiomyomas with ring chromosome 1. Cancer Genet. Cytogenet. 1999, 108, 107–109. [Google Scholar] [CrossRef]
- Quade, B.J.; Weremowicz, S.; Neskey, D.M.; Vanni, R.; Ladd, C.; Dal Cin, P.; Morton, C.C. Fusion transcripts involving HMGA2 are not a common molecular mechanism in uterine leiomyomata with rearrangements in 12q15. Cancer Res. 2003, 63, 1351–1358. [Google Scholar] [PubMed]
- Takahashi, T.; Nagai, N.; Oda, H.; Ohama, K.; Kamada, N.; Miyagawa, K. Evidence for RAD51L1/HMGIC fusion in the pathogenesis of uterine leiomyoma. Genes Chromosomes Cancer 2001, 30, 196–201. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Gorunova, L.; Agostini, A.; Lobmaier, I.; Bjerkehagen, B.; Heim, S. Fusion of the HMGA2 and C9orf92 genes in myolipoma with t(9;12)(p22;q14). Diagn. Pathol. 2016, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, F.; Erickson-Johnson, M.R.; Keeney, G.L.; Clayton, A.C.; Nascimento, A.G.; Wang, X.; Oliveira, A.M. Frequency and characterization of HMGA2 and HMGA1 rearrangements in mesenchymal tumors of the lower genital tract. Genes Chromosomes Cancer 2007, 46, 981–990. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Bjerkehagen, B.; Gorunova, L.; Taksdal, I.; Heim, S. Rearrangement of chromosome bands 12q14~15 causing HMGA2-SOX5 gene fusion and HMGA2 expression in extraskeletal osteochondroma. Oncol. Rep. 2015, 34, 577–584. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Bjerkehagen, B.; Gorunova, L.; Berner, J.M.; Boye, K.; Heim, S. Several fusion genes identified by whole transcriptome sequencing in a spindle cell sarcoma with rearrangements of chromosome arm 12q and MDM2 amplification. Int. J. Oncol. 2014, 45, 1829–1836. [Google Scholar] [CrossRef] [Green Version]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef]
- Wei, J.; Li, H.; Wang, S.; Li, T.; Fan, J.; Liang, X.; Li, J.; Han, Q.; Zhu, L.; Fan, L.; et al. Let-7 enhances osteogenesis and bone formation while repressing adipogenesis of human stromal/mesenchymal stem cells by regulating HMGA2. Stem Cells Dev. 2014, 23, 1452–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Laser, J.; Shi, G.; Mittal, K.; Melamed, J.; Lee, P.; Wei, J.J. Antiproliferative effects by Let-7 repression of high-mobility group A2 in uterine leiomyoma. Mol. Cancer Res. 2008, 6, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlotta, P.; Tai, A.K.; Manfioletti, G.; Clifford, C.; Jay, G.; Ono, S.J. Transgenic mice expressing a truncated form of the high mobility group I-C protein develop adiposity and an abnormally high prevalence of lipomas. J. Biol. Chem. 2000, 275, 14394–14400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, J.; Stabell, M.; Meza-Zepeda, L.A.; Lauvrak, S.A.; Kassem, M.; Myklebost, O. Identification of target genes for wild type and truncated HMGA2 in mesenchymal stem-like cells. BMC Cancer 2010, 10, 329. [Google Scholar] [CrossRef] [Green Version]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Hao, D.; Liu, X.; Wang, D.; Ning, N.; Li, X. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol. Cancer 2015, 14, 125. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Chen, Y.; Ito, H.; Watanabe, A.; Ge, X.; Kodama, T.; Aburatani, H. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene 2006, 384, 51–61. [Google Scholar] [CrossRef]
- Jonson, L.; Christiansen, J.; Hansen, T.V.O.; Vikesa, J.; Yamamoto, Y.; Nielsen, F.C. IMP3 RNP safe houses prevent miRNA-directed HMGA2 mRNA decay in cancer and development. Cell Rep. 2014, 7, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Madison, B.B.; Jeganathan, A.N.; Mizuno, R.; Winslow, M.M.; Castells, A.; Cuatrecasas, M.; Rustgi, A.K. Let-7 Represses Carcinogenesis and a Stem Cell Phenotype in the Intestine via Regulation of Hmga2. PLoS Genet. 2015, 11, e1005408. [Google Scholar] [CrossRef] [Green Version]
- Berner, J.M.; Meza-Zepeda, L.; Kools, P.F.; Forus, A.; Schoenmakers, E.; Van de Ven, W.J.; Fodstad, O.; Myklebost, O. HMGIC, the gene for an architectural transcription factor is amplified and rearranged in a subset of human sarcomas. Oncogene 1997, 14, 2935–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A.; Meister, G.; Hermeking, H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Gisselsson, D.; Hoglund, M.; Mertens, F.; Mitelman, F.; Mandahl, N. Chromosomal organization of amplified chromosome 12 sequences in mesenchymal tumors detected by fluorescence in situ hybridization. Genes Chromosomes Cancer 1998, 23, 203–212. [Google Scholar] [CrossRef]
- Ozturk, N.; Singh, I.; Mehta, A.; Braun, T.; Barreto, G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2014, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868. [Google Scholar] [CrossRef]
- Luo, R.X.; Dean, D.C. Chromatin remodeling and transcriptional regulation. J. Natl. Cancer Inst. 1999, 91, 1288–1294. [Google Scholar] [CrossRef]
- Zhang, Y.; Fatima, N.; Dufau, M.L. Coordinated changes in DNA methylation and histone modifications regulate silencing/derepression of luteinizing hormone receptor gene transcription. Mol. Cell. Biol. 2005, 25, 7929–7939. [Google Scholar] [CrossRef] [Green Version]
- Karlić, R.; Chung, H.-R.; Lasserre, J.; Vlahoviček, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhang, H.; Yu, L. HMGA2 promotes glioma invasion and poor prognosis via a long-range chromatin interaction. Cancer Med. 2018, 7, 3226–3239. [Google Scholar] [CrossRef]
- Thomas, J.O. Histone H1: Location and role. Curr. Opin. Cell Biol. 1999, 11, 312–317. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Khochbin, S.; Dimitrov, S. What do linker histones do in chromatin? Bioessays 1997, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Li, O.; Vasudevan, D.; Davey, C.A.; Droge, P. High-level expression of DNA architectural factor HMGA2 and its association with nucleosomes in human embryonic stem cells. Genesis 2006, 44, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Leng, F. Specific recognition of AT-rich DNA sequences by the mammalian high mobility group protein AT-hook 2: A SELEX study. Biochemistry 2007, 46, 13059–13066. [Google Scholar] [CrossRef]
- Catez, F.; Yang, H.; Tracey, K.J.; Reeves, R.; Misteli, T.; Bustin, M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol. Cell. Biol. 2004, 24, 4321–4328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.; Ramani, P.D.; Wong, S.Q.R.; Zhao, X.; Ivanyi-Nagy, R.; Leong, T.C.; Chua, C.; Li, Z.; Hentze, H.; Tan, I.B.; et al. The chromatin structuring protein HMGA2 influences human subtelomere stability and cancer chemosensitivity. PLoS ONE 2019, 14, e0215696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strumberg, D.; Pilon, A.A.; Smith, M.; Hickey, R.; Malkas, L.; Pommier, Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 2000, 20, 3977–3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.; Dröge, P. Oncofetal HMGA2 attenuates genotoxic damage induced by topoisomerase II target compounds through the regulation of local DNA topology. Mol. Oncol. 2019, 13, 2062–2078. [Google Scholar] [CrossRef] [Green Version]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT Facilitates Transcription-Dependent Nucleosome Alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef] [Green Version]
- Schwabish, M.A.; Struhl, K. Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol. Cell 2006, 22, 415–422. [Google Scholar] [CrossRef]
- Matsui, K.; Tatsuguchi, A.; Valencia, J.; Yu, Z.; Bechtle, J.; Beasley, M.B.; Avila, N.; Travis, W.D.; Moss, J.; Ferrans, V.J. Extrapulmonary lymphangioleiomyomatosis (LAM): Clinicopathologic features in 22 cases. Hum. Pathol. 2000, 31, 1242–1248. [Google Scholar] [CrossRef] [Green Version]
- Urban, T.; Lazor, R.; Lacronique, J.; Murris, M.; Labrune, S.; Valeyre, D.; Cordier, J.F. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM“O”P). Medicine 1999, 78, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Kitaichi, M.; Nishimura, K.; Itoh, H.; Izumi, T. Pulmonary lymphangioleiomyomatosis: A report of 46 patients including a clinicopathologic study of prognostic factors. Am. J. Respir. Crit. Care Med. 1995, 151, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Anton, E.; Casanova, A.; Xaubet, A.; Roman, A.; Villena, V.; Montero, M.C.; Molina-Molina, M.; Perez-Sanchez, E.; Sueiro, A.; Morell, F.; et al. Lymphangioleiomyomatosis: A study of 72 patients from the Spanish registry. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 85–91. [Google Scholar]
- Taveira-DaSilva, A.M.; Moss, J. Clinical features, epidemiology, and therapy of lymphangioleiomyomatosis. Clin. Epidemiol. 2015, 7, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, F.X. Lymphangioleiomyomatosis: A clinical update. Chest 2008, 133, 507–516. [Google Scholar] [CrossRef]
- Henske, E.P.; McCormack, F.X. Lymphangioleiomyomatosis—A wolf in sheep’s clothing. J. Clin. Investig. 2012, 122, 3807–3816. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.C.; Shyamsundar, M.M.; Thomas, M.W.; Maynard, J.; Idziaszczyk, S.; Tomkins, S.; Sampson, J.R.; Cheadle, J.P. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 1999, 64, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Björnsson, J.; King, B.F.; Kumar, R.; Zincke, H.; Edell, E.S.; Wilson, T.O.; Hattery, R.R.; Gomez, M.R. Extrapulmonary Lymphangioleiomyomatosis and Lymphangiomatous Cysts in Tuberous Sclerosis Complex. Mayo Clin. Proc. 1995, 70, 641–648. [Google Scholar] [CrossRef]
- Northrup, H.; Krueger, D.A. On behalf of the International Tuberous Sclerosis Complex Consensus, G., Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Cudzilo, C.J.; Szczesniak, R.D.; Brody, A.S.; Rattan, M.S.; Krueger, D.A.; Bissler, J.J.; Franz, D.N.; McCormack, F.X.; Young, L.R. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest 2013, 144, 578–585. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valensi, Q.J. Pulmonary Lymphangiomyoma, A Probable Forme Frust of Tuberous Sclerosis. Am. Rev. Respir. Dis. 1973, 108, 1411–1415. [Google Scholar] [PubMed]
- Carsillo, T.; Astrinidis, A.; Henske, E.P. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. USA 2000, 97, 6085–6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, T.N.; Pacheco-Rodriguez, G.; Gorio, A.; Lesma, E.; Walker, C.; Moss, J. Lymphangioleiomyomatosis and TSC2-/- cells. Lymphat. Res. Biol. 2010, 8, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbowniczek, M.; Yu, J.; Henske, E.P. Renal angiomyolipomas from patients with sporadic lymphangiomyomatosis contain both neoplastic and non-neoplastic vascular structures. Am. J. Pathol. 2003, 162, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Niida, Y.; Stemmer-Rachamimov, A.O.; Logrip, M.; Tapon, D.; Perez, R.; Kwiatkowski, D.J.; Sims, K.; MacCollin, M.; Louis, D.N.; Ramesh, V. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am. J. Hum. Genet. 2001, 69, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Badri, K.R.; Gao, L.; Hyjek, E.; Schuger, N.; Schuger, L.; Qin, W.; Chekaluk, Y.; Kwiatkowski, D.J.; Zhe, X. Exonic mutations of TSC2/TSC1 are common but not seen in all sporadic pulmonary lymphangioleiomyomatosis. Am. J. Respir. Crit. Care Med. 2013, 187, 663–665. [Google Scholar] [CrossRef]
- Johnson, S.R.; Clelland, C.A.; Ronan, J.; Tattersfield, A.E.; Knox, A.J. The TSC-2 product tuberin is expressed in lymphangioleiomyomatosis and angiomyolipoma. Histopathology 2002, 40, 458–463. [Google Scholar] [CrossRef]
- Henske, E.P.; Scheithauer, B.W.; Short, M.P.; Wollmann, R.; Nahmias, J.; Hornigold, N.; van Slegtenhorst, M.; Welsh, C.T.; Kwiatkowski, D.J. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am. J. Hum. Genet. 1996, 59, 400–406. [Google Scholar]
- Davies, D.M.; de Vries, P.J.; Johnson, S.R.; McCartney, D.L.; Cox, J.A.; Serra, A.L.; Watson, P.C.; Howe, C.J.; Doyle, T.; Pointon, K.; et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: A phase 2 trial. Clin. Cancer Res. 2011, 17, 4071–4081. [Google Scholar] [CrossRef] [Green Version]
- Barrera, P.; Simons, S.O.; Luijk, B.; Wessels, M.J.; Heijdra, Y.F. Efficacy of sirolimus therapy for chylous effusions in lymphangioleiomyomatosis. Ann. Am. Thorac. Soc. 2013, 10, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Taveira-DaSilva, A.M.; Jones, A.M.; Julien-Williams, P.; Stylianou, M.; Moss, J. Sustained Effects of Sirolimus on Lung Function and Cystic Lung Lesions in Lymphangioleiomyomatosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1273–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, F.X.; Inoue, Y.; Moss, J.; Singer, L.G.; Strange, C.; Nakata, K.; Barker, A.F.; Chapman, J.T.; Brantly, M.L.; Stocks, J.M.; et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med. 2011, 364, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- D’Armiento, J.; Shiomi, T.; Marks, S.; Geraghty, P.; Sankarasharma, D.; Chada, K. Mesenchymal Tumorigenesis Driven by TSC2 Haploinsufficiency Requires HMGA2 and Is Independent of mTOR Pathway Activation. Cancer Res. 2016, 76, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Kazmierczak, B.; Rosigkeit, J.; Wanschura, S.; Meyer-Bolte, K.; Van de Ven, W.J.; Kayser, K.; Krieghoff, B.; Kastendiek, H.; Bartnitzke, S.; Bullerdiek, J. HMGI-C rearrangements as the molecular basis for the majority of pulmonary chondroid hamartomas: A survey of 30 tumors. Oncogene 1996, 12, 515–521. [Google Scholar]
- Howe, S.R.; Gottardis, M.M.; Everitt, J.I.; Goldsworthy, T.L.; Wolf, D.C.; Walker, C. Rodent model of reproductive tract leiomyomata. Establishment and characterization of tumor-derived cell lines. Am. J. Pathol. 1995, 146, 1568–1579. [Google Scholar]
- Li, Z.; Zhang, Y.; Ramanujan, K.; Ma, Y.; Kirsch, D.G.; Glass, D.J. Oncogenic NRAS, required for pathogenesis of embryonic rhabdomyosarcoma, relies upon the HMGA2-IGF2BP2 pathway. Cancer Res. 2013, 73, 3041–3050. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, D.J. Animal Models of Lymphangioleiomyomatosis (LAM) and Tuberous Sclerosis Complex (TSC). Lymphat. Res. Biol. 2010, 8, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, J.; Mitoro, A.; Kawashiri, S.; Chada, K.K.; Imai, K. Expression of mesenchyme-specific gene HMGA2 in squamous cell carcinomas of the oral cavity. Cancer Res. 2004, 64, 2024–2029. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Hlubek, F.; Spaderna, S.; Schmalhofer, O.; Hiendlmeyer, E.; Jung, A.; Kirchner, T. Invasion and metastasis in colorectal cancer: Epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs 2005, 179, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Chen, C.C.; Chang, L.C. Gene expressions of HMGI-C and HMGI(Y) are associated with stage and metastasis in colorectal cancer. Int. J. Colorectal. Dis. 2009, 24, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Z.; Zha, L.; Kong, D.; Liao, G.; Li, H. HMGA2 regulates epithelial-mesenchymal transition and the acquisition of tumor stem cell properties through TWIST1 in gastric cancer. Oncol. Rep. 2017, 37, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Ueda, Y.; Akaboshi, S.-I.; Hino, Y.; Sekita, Y.; Nakao, M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am. J. Pathol. 2009, 174, 854–868. [Google Scholar] [CrossRef] [Green Version]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.; Moustakas, A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127(Pt. 2), 2021–2036. [Google Scholar] [CrossRef] [Green Version]
- Thuault, S.; Tan, E.J.; Peinado, H.; Cano, A.; Heldin, C.H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar]
- Tan, E.J.; Thuault, S.; Caja, L.; Carletti, T.; Heldin, C.H.; Moustakas, A. Regulation of transcription factor Twist expression by the DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transition. J. Biol. Chem. 2012, 287, 7134–7145. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.J.; Kahata, K.; Idås, O.; Thuault, S.; Heldin, C.-H.; Moustakas, A. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2014, 43, 162–178. [Google Scholar] [CrossRef] [Green Version]
- Shell, S.; Park, S.M.; Radjabi, A.R.; Schickel, R.; Kistner, E.O.; Jewell, D.A.; Feig, C.; Lengyel, E.; Peter, M.E. Let-7 expression defines two differentiation stages of cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 11400–11405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, B.; Ishinaga, H.; Midorikawa, K.; Nakamura, S.; Hiraku, Y.; Oikawa, S.; Ma, N.; Takeuchi, K.; Murata, M. Let-7c inhibits migration and epithelial–mesenchymal transition in head and neck squamous cell carcinoma by targeting IGF1R and HMGA2. Oncotarget 2018, 9, 8927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Wu, K.; Li, J.; Mo, Y.; Lin, Y.; Wang, Y.; Shen, X.; Li, S.; Li, L.; Yang, Z. Let-7a inhibits migration, invasion and epithelial-mesenchymal transition by targeting HMGA2 in nasopharyngeal carcinoma. J. Transl. Med. 2015, 13, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.A.; Thomson, J.M.; Hammond, S.M. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 2008, 14, 1539–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangi-Garimella, S.; Yun, J.; Eves, E.M.; Newman, M.; Erkeland, S.J.; Hammond, S.M.; Minn, A.J.; Rosner, M.R. Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009, 28, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Gomes, S.; Chen, P.; Frankenberger, C.A.; Sankarasharma, D.; Chung, C.H.; Chada, K.K.; Rosner, M.R. RKIP and HMGA2 regulate breast tumor survival and metastasis through lysyl oxidase and syndecan-2. Oncogene 2014, 33, 3528–3537. [Google Scholar] [CrossRef] [Green Version]
- Shajari, N.; Davudian, S.; Kazemi, T.; Mansoori, B.; Salehi, S.; Khaze Shahgoli, V.; Shanehbandi, D.; Mohammadi, A.; Duijf, P.H.G.; Baradaran, B. Silencing of BACH1 inhibits invasion and migration of prostate cancer cells by altering metastasis-related gene expression. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1495–1504. [Google Scholar] [CrossRef]
- Malek, A.; Bakhidze, E.; Noske, A.; Sers, C.; Aigner, A.; Schäfer, R.; Tchernitsa, O. HMGA2 gene is a promising target for ovarian cancer silencing therapy. Int. J. Cancer 2008, 123, 348–356. [Google Scholar] [CrossRef]
- Masai, H.; Tanaka, T.; Kohda, D. Stalled replication forks: making ends meet for recognition and stabilization. Bioessays 2010, 32, 687–697. [Google Scholar] [CrossRef]
- Vallone, D.; Battista, S.; Pierantoni, G.M.; Fedele, M.; Casalino, L.; Santoro, M.; Viglietto, G.; Fusco, A.; Verde, P. Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. EMBO J. 1997, 16, 5310–5321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, L.; Wang, J.; Li, X.; Sahengbieke, S.; Wu, J.; Lai, M. HMGA2 promotes intestinal tumorigenesis by facilitating MDM2-mediated ubiquitination and degradation of p53. J. Pathol. 2018, 246, 508–518. [Google Scholar] [CrossRef]
- Yu, K.R.; Park, S.B.; Jung, J.W.; Seo, M.S.; Hong, I.S.; Kim, H.S.; Seo, Y.; Kang, T.W.; Lee, J.Y.; Kurtz, A.; et al. HMGA2 regulates the in vitro aging and proliferation of human umbilical cord blood-derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell Res. 2013, 10, 156–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, M.; Visone, R.; De Martino, I.; Troncone, G.; Palmieri, D.; Battista, S.; Ciarmiello, A.; Pallante, P.; Arra, C.; Melillo, R.M.; et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 2006, 9, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.-H.; Wei, L.-X.; Lai, M.-Y.; Chen, J.-Z.; Mo, X.-J. Effect of silencing of high mobility group A2 gene on gastric cancer MKN-45 cells. World J. Gastroenterol. 2013, 19, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pi, X.-Y.; Boland, K.; Lad, S.; Johnson, K.; Verfaillie, C.; Morris, R.J. Hmga2 translocation induced in skin tumorigenesis. Oncotarget 2017, 8, 30019–30029. [Google Scholar] [CrossRef] [PubMed]
- Hawsawi, O.; Henderson, V.; Burton, L.J.; Dougan, J.; Nagappan, P.; Odero-Marah, V. High mobility group A2 (HMGA2) promotes EMT via MAPK pathway in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 196–202. [Google Scholar] [CrossRef]
- Chen, R.-X.; Chen, X.; Xia, L.-P.; Zhang, J.-X.; Pan, Z.-Z.; Ma, X.-D.; Han, K.; Chen, J.-W.; Judde, J.-G.; Deas, O.; et al. N6-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat. Commun. 2019, 10, 4695. [Google Scholar] [CrossRef] [Green Version]
- Degrauwe, N.; Suvà, M.-L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef] [Green Version]
- Pallante, P.; Sepe, R.; Puca, F.; Fusco, A. High mobility group a proteins as tumor markers. Front. Med. 2015, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, Y.; Deng, H.; Liu, C.; Wu, J.; Lai, M. HMGA2 enhances 5-fluorouracil chemoresistance in colorectal cancer via the Dvl2/Wnt pathway. Oncotarget 2018, 9, 9963–9974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquis, M.; Beaubois, C.; Lavallée, V.-P.; Abrahamowicz, M.; Danieli, C.; Lemieux, S.; Ahmad, I.; Wei, A.; Ting, S.B.; Fleming, S.; et al. High expression of HMGA2 independently predicts poor clinical outcomes in acute myeloid leukemia. Blood Cancer J. 2018, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.-Y.; Liew, P.-L.; Chen, C.-L.; Lin, Y.-H.; Fang, C.-L.; Chen, W.-Y. High HMGA2 Expression Correlates with Reduced Recurrence-free Survival and Poor Overall Survival in Oral Squamous Cell Carcinoma. Anticancer Res. 2017, 37, 1891–1899. [Google Scholar]
- Helmke, B.M.; Markowski, D.N.; Meyer, A.; Bullerdiek, J. The Expression of HMGA2 Varies Strongly among Colon Carcinomas. Anticancer Res. 2012, 32, 1589–1593. [Google Scholar] [PubMed]
- Chiou, S.-H.; Dorsch, M.; Kusch, E.; Naranjo, S.; Kozak, M.M.; Koong, A.C.; Winslow, M.M.; Grüner, B.M. Hmga2 is dispensable for pancreatic cancer development, metastasis, and therapy resistance. Sci. Rep. 2018, 8, 14008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlem, C.; Barghash, A.; Puchas, P.; Haybaeck, J.; Kessler, S.M. The Insulin-Like Growth Factor 2 mRNA Binding Protein IMP2/IGF2BP2 is Overexpressed and Correlates with Poor Survival in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3204. [Google Scholar] [CrossRef] [Green Version]
- Esmailzadeh, S.; Mansoori, B.; Mohammadi, A.; Shanehbandi, D.; Baradaran, B. siRNA-Mediated Silencing of HMGA2 Induces Apoptosis and Cell Cycle Arrest in Human Colorectal Carcinoma. J. Gastrointest Cancer 2017, 48, 156–163. [Google Scholar] [CrossRef]
- Yu, F.Y.; Tu, Y.; Deng, Y.; Guo, C.; Ning, J.; Zhu, Y.; Lv, X.; Ye, H. MiR-4500 is epigenetically downregulated in colorectal cancer and functions as a novel tumor suppressor by regulating HMGA2. Cancer Biol. Ther. 2016, 17, 1149–1157. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zeng, K.; Xu, M.; Liu, X.; Hu, X.; Xu, T.; He, B.; Pan, Y.; Sun, H.; Wang, S. P53-induced miR-1249 inhibits tumor growth, metastasis, and angiogenesis by targeting VEGFA and HMGA2. Cell Death Dis. 2019, 10, 131. [Google Scholar] [CrossRef]
- Cai, J.; Shen, G.; Liu, S.; Meng, Q. Downregulation of HMGA2 inhibits cellular proliferation and invasion, improves cellular apoptosis in prostate cancer. Tumor Biol. 2016, 37, 699–707. [Google Scholar] [CrossRef]
- Tan, L.; Xu, H.; Chen, G.; Wei, X.; Yu, B.; Ye, J.; Xu, L.; Tan, H. Silencing of HMGA2 reverses retardance of cell differentiation in human myeloid leukaemia. Br. J. Cancer 2018, 118, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; Cui, T.; Leng, F.; Wilson, W.D. Inhibition of high-mobility-group A2 protein binding to DNA by netropsin: A biosensor-surface plasmon resonance assay. Anal. Biochem. 2008, 374, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, S.W.; Chou, C.J.; Huang, T.C.; Yang, P.M. An Integrated Bioinformatics Analysis Repurposes an Antihelminthic Drug Niclosamide for Treating HMGA2-Overexpressing Human Colorectal Cancer. Cancers 2019, 11, 1482. [Google Scholar] [CrossRef] [Green Version]
- Wachter, S.; Wunderlich, A.; Roth, S.; Mintziras, I.; Maurer, E.; Hoffmann, S.; Verburg, F.A.; Fellinger, S.A.; Holzer, K.; Bartsch, D.K.; et al. Individualised Multimodal Treatment Strategies for Anaplastic and Poorly Differentiated Thyroid Cancer. J. Clin. Med. 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
let-7 regulatory mechanisms govern HMGA2 misexpression, driving benign mesenchymal tumorigenesis. (A) Absence of tumor suppressor let-7 miRNA expression in the undifferentiated mesenchyme ensures ubiquitous HMGA2 expression during mammalian embryogenesis. (B) Increased let-7 expression as mesenchymal tissues mature and differentiate inhibits HMGA2 expression by binding to sites in the 3’ UTR of HMGA2 mRNA leading to transcript degradation. (C) Abundant HMGA2 transcription in many uterine leiomyomas and mammary fibroadenomas yield enough mRNA possessing multiple 3’ UTR binding sites that can soak up available let-7 allowing for remnant HMGA2 misexpression of full-length transcripts. (D) Intragenic chromosomal breaks at preferred locations between exons III and IV and (E) balanced translocations with chromosomal partners forming chimeric HMGA2 fused to ectopic sequences, are two mechanisms ensuring loss of HMGA2 3’ UTR and its let-7 binding sites allowing for misexpression of full-length HMGA2 transcripts and activation of the HMGA2 pathway in differentiated benign mesenchymal tumors.
Figure 1.
let-7 regulatory mechanisms govern HMGA2 misexpression, driving benign mesenchymal tumorigenesis. (A) Absence of tumor suppressor let-7 miRNA expression in the undifferentiated mesenchyme ensures ubiquitous HMGA2 expression during mammalian embryogenesis. (B) Increased let-7 expression as mesenchymal tissues mature and differentiate inhibits HMGA2 expression by binding to sites in the 3’ UTR of HMGA2 mRNA leading to transcript degradation. (C) Abundant HMGA2 transcription in many uterine leiomyomas and mammary fibroadenomas yield enough mRNA possessing multiple 3’ UTR binding sites that can soak up available let-7 allowing for remnant HMGA2 misexpression of full-length transcripts. (D) Intragenic chromosomal breaks at preferred locations between exons III and IV and (E) balanced translocations with chromosomal partners forming chimeric HMGA2 fused to ectopic sequences, are two mechanisms ensuring loss of HMGA2 3’ UTR and its let-7 binding sites allowing for misexpression of full-length HMGA2 transcripts and activation of the HMGA2 pathway in differentiated benign mesenchymal tumors.


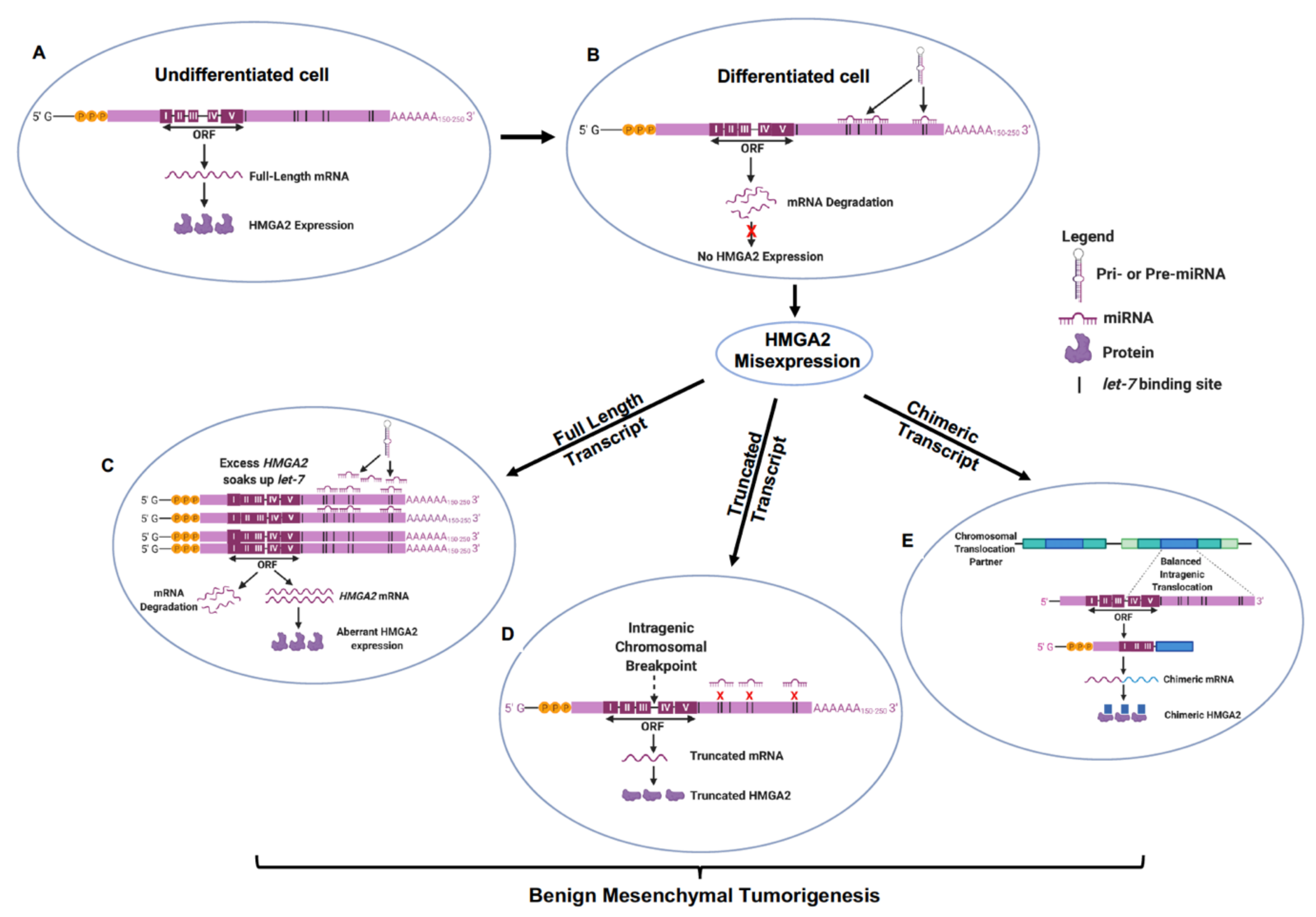{kind=link}
Table 1.
Balanced Chromosomal Rearrangements Forming Hmga2-Induced Gene Fusions in Human Mesenchymal Tumors.
Table 1.
Balanced Chromosomal Rearrangements Forming Hmga2-Induced Gene Fusions in Human Mesenchymal Tumors.
| Tumor Type | [Refs] | Chromosomal Translocation Partners | Chromosomal Rearrangement | Fusion Protein |
|---|---|---|---|---|
| Lipoma | [29,30,43] | 1 | t(1;12)(p32;q14) | HMGA2/PPAP2B |
| [77,78] | 2 | t(2;12)(q37;q14) | HMGA2/ACKR3 | |
| [29,39,69,70,71,72,73,74] | 3 | t(3;12)(q28;q14) | HMGA2/LPP | |
| [29,43,73,79] | 5 | t(5;12)(q33;q15) | HMGA2/EBFEBF/BC058822 | |
| [79] | 5 | t(5;12)(q33;q14) | HMGA2/EBF1 | |
| [29,79] | 5 | ins(5;12)(q33;q14q21) | Partial Genomic Loss | |
| [29,73,79] | 5 | t(5;12)(q33;q15) | No Genomic Loss/Gain | |
| [79] | 5 | t(5;12)(q32;q14) | No Genomic Loss/Gain | |
| [79] | 5 | t(5;12)(q32;q15) | No Genomic Loss/Gain | |
| [29,80] | 5 | ins(12;5)(q15;q33q13) | No Genomic Loss/Gain | |
| [81,82,83] | 9 | t(9;12)(p22;q14) | HMGA2/NFIB | |
| [42] | Intragenic | t(12;12)(q14;q14) | HMGA2/GRIP1 | |
| [38] | 13 | t(12;13)(q14;q13) | HMGA2/LHFP | |
| [51] | 15 | t(12;15)(q14;q24) | HMGA2/Ser-Thr domain | |
| [42] | 18 | t(12;18)(q14;q12) | HMGA2/SETBP1 | |
| [42] | 18 | t(12;18)(q14~q15;q12~q21) | HMGA2/SETBP1 | |
| [42] | 18 | t(12;18)(q14~q15;q12~q21) | HMGA2/GRIP1 | |
| [42] | 18 | t(12;18)(q14~q15;q12~q21) | HMGA2/18q12.3 Sequence | |
| Osteochondrolipoma | [42] | 18 | t(12;18)(q14~q15;q12~q21 | HMGA2/SETBP1 |
| Uterine Leiomyoma | [50] | 7 | t(7;12)(q31;q14) | HMGA2/COG5 |
| [39,55,56] | 8 | t(8;12)(q22;q14) | HMGA2/COX6C | |
| [84] | Intragenic | der(12)(q14) | HMGA2/RTVL-H | |
| [85,86,87] | 14 | t(12;14)(q14;q24) | RAD51l1/HMGA2 | |
| [54] | 14 | t(12;14)(q15;q11) | HMGA2/HE110 | |
| Soft Tissue Chondroma | [37] | 3 | t(3;12)(q27;q15) | HMGA2/LPP |
| Sarcoma | [76] | Intragenic | t(12;12)(q15;q14) | FRS2/HMGA2 |
| [76] | 1 | t(1;12)(p32;q14) | HMGA2/DAB1 | |
| [76] | Intragenic | t(12;12)(q14;q13) | HMGA2/PCBP2 | |
| [76] | Intragenic | t(12;12)(q14;q12) | HMGA2/NELL2 | |
| [76] | Intragenic | t(12;12)(q14;q21) | HMGA2/PPFIA2 | |
| [76] | 1 | t(1;12)(p32;q14) | HMGA2/C1orf87 | |
| [76] | Intragenic | t(12;12)(q14;q13) | HMGA2/SARNP | |
| [76] | 11 | t(11;12)(p11;q14) | HMGA2/ARFGAP2 | |
| [76] | Intragenic | t(12;12)(q14;q22) | HMGA2/NR2C1 | |
| [76] | 6 | t(6;12)(q24;q14) | UTRN/HMGA2 | |
| Myolipoma | [88] | 9 | t(9;12)(p22;q14) | HMGA2/C90RF92 |
| Aggressive angiomyxoma | [89] | 1 | t(1;12)(p32;q15) | HMGA2(3’UTR)/NT032977.8 |
| Extra-skeletal Osteochondroma | [90] | Intragenic | inv(12)(p12q14) | HMGA2/SOX5 |
| Spindle Cell Sarcoma | [91] | Intragenic | t(12;12)(q14;q15) | HMGA2/DYRK2 |
| Pulmonary Chondroid Harmatoma (PCH) | [68] | 3 | t(3;12)(q27;q14;q15) | HMGA2/LPP |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Unachukwu, U.; Chada, K.; D’Armiento, J. High Mobility Group AT-Hook 2 (HMGA2) Oncogenicity in Mesenchymal and Epithelial Neoplasia. Int. J. Mol. Sci. 2020, 21, 3151. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093151
AMA Style
Unachukwu U, Chada K, D’Armiento J. High Mobility Group AT-Hook 2 (HMGA2) Oncogenicity in Mesenchymal and Epithelial Neoplasia. International Journal of Molecular Sciences. 2020; 21(9):3151. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093151
Chicago/Turabian StyleUnachukwu, Uchenna, Kiran Chada, and Jeanine D’Armiento. 2020. "High Mobility Group AT-Hook 2 (HMGA2) Oncogenicity in Mesenchymal and Epithelial Neoplasia" International Journal of Molecular Sciences 21, no. 9: 3151. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093151
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.