Post-Translational Modifications of Transcription Factors Harnessing the Etiology and Pathophysiology in Colonic Diseases

Abstract
:1. Introduction
2. Overview of Post-Translational Modifications of Transcription Factors and the Modulatory Effects on Colonic Disease
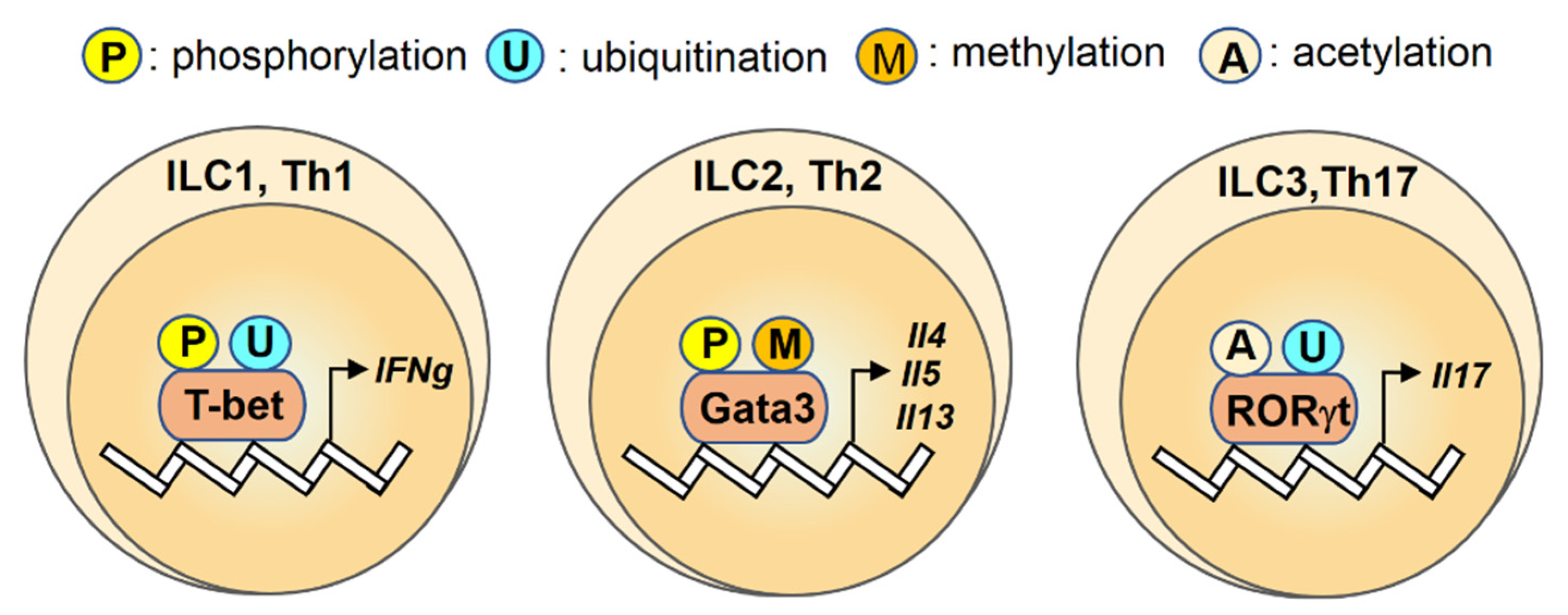
2.1. T-bet
2.2. Gata3
2.3. RORγt
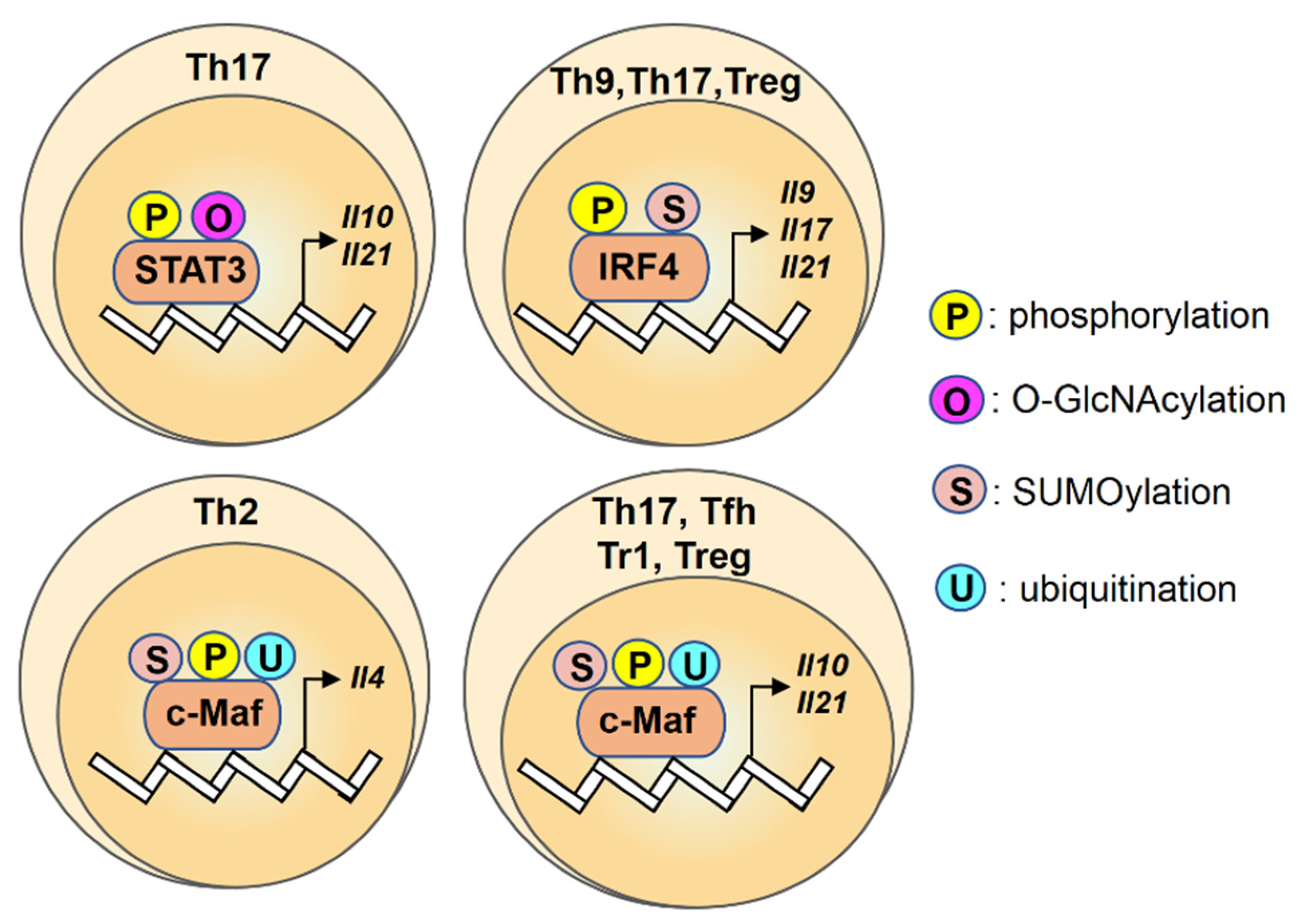
2.4. STAT3
2.5. IRF4
2.6. c-Maf
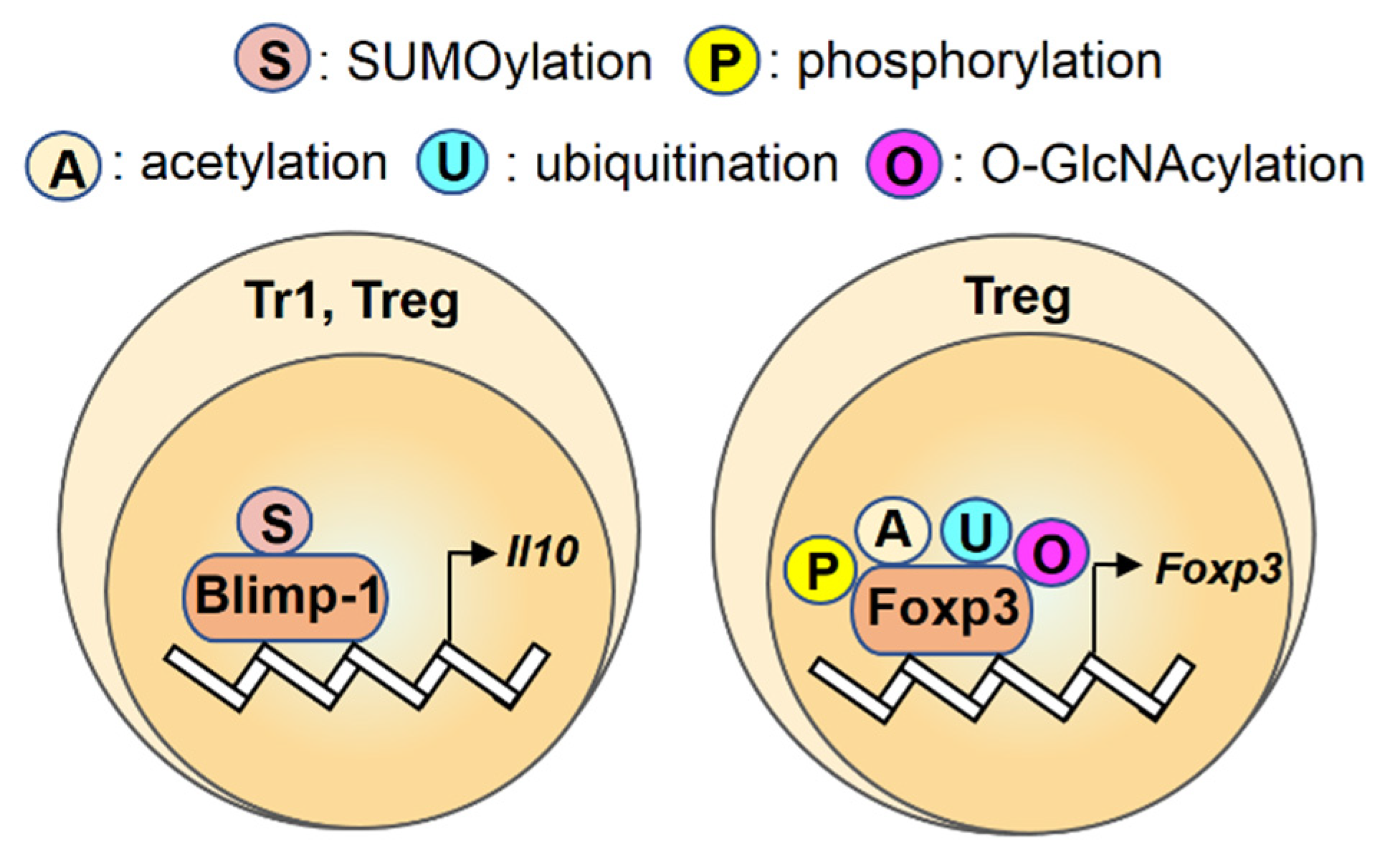
2.7. Blimp-1
2.8. Foxp3
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Neurath, M.F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 2019, 20, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Bouma, G.; Strober, W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 2003, 3, 521–533. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Koch, S.; Nusrat, A. The life and death of epithelia during inflammation: Lessons learned from the gut. Annu. Rev. Pathol. 2012, 7, 35–60. [Google Scholar] [CrossRef]
- Nava, P.; Koch, S.; Laukoetter, M.G.; Lee, W.Y.; Kolegraff, K.; Capaldo, C.T.; Beeman, N.; Addis, C.; Gerner-Smidt, K.; Neumaier, I.; et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity 2010, 32, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.I.; Parkos, C.A.; Nusrat, A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am. J. Pathol. 2010, 177, 512–524. [Google Scholar] [CrossRef]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, M.J.; Lobo, A.J.; Travis, S.P. IBD Section, British Society of Gastroenterology. Guidel. Manag. Inflamm. Bowel Dis. Adults Gut 2004, 53, V1–V16. [Google Scholar] [CrossRef]
- Sica, G.S.; Biancone, L. Surgery for inflammatory bowel disease in the era of laparoscopy. World J. Gastroenterol. 2013, 19, 2445–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Lees, C.W.; Barrett, J.C.; Parkes, M.; Satsangi, J. New IBD genetics: Common pathways with other diseases. Gut 2011, 60, 1739–1753. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Geremia, A.; Arancibia-Carcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Kramer, B.; Goeser, F.; Lutz, P.; Glassner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017, 13, e1006373. [Google Scholar] [CrossRef]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Ann. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.; Yang, K.; Li, Y.; Shaw, T.I.; Wang, Y.; Blanco, D.B.; Wang, X.; Cho, J.H.; Wang, H.; Rankin, S.; et al. Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 2017, 46, 488–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef]
- Han, D.; Huang, M.; Wang, T.; Li, Z.; Chen, Y.; Liu, C.; Lei, Z.; Chu, X. Lysine methylation of transcription factors in cancer. Cell Death Dis. 2019, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Chen, Z.J. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 2011, 12, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.S. Protein modification by SUMO. Ann. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef] [Green Version]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Ann. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Ann. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F.; Weigmann, B.; Finotto, S.; Glickman, J.; Nieuwenhuis, E.; Iijima, H.; Mizoguchi, A.; Mizoguchi, E.; Mudter, J.; Galle, P.R.; et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J. Exp. Med. 2002, 195, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krausgruber, T.; Schiering, C.; Adelmann, K.; Harrison, O.J.; Chomka, A.; Pearson, C.; Ahern, P.P.; Shale, M.; Oukka, M.; Powrie, F. T-bet is a key modulator of IL-23-driven pathogenic CD4(+) T cell responses in the intestine. Nat. Commun. 2016, 7, 11627. [Google Scholar] [CrossRef]
- Garrett, W.S.; Lord, G.M.; Punit, S.; Lugo-Villarino, G.; Mazmanian, S.K.; Ito, S.; Glickman, J.N.; Glimcher, L.H. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 2007, 131, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.S.; Szabo, S.J.; Schwartzberg, P.L.; Glimcher, L.H. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science 2005, 307, 430–433. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.S.; Hong, J.H.; Glimcher, L.H. IL-2 production in developing Th1 cells is regulated by heterodimerization of RelA and T-bet and requires T-bet serine residue 508. J. Exp. Med. 2005, 202, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.J.; Park, H.R.; Hong, J.H.; Hwang, E.S. Lysine 313 of T-box is crucial for modulation of protein stability, DNA binding, and threonine phosphorylation of T-bet. J. Immunol. 2013, 190, 5764–5770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, K.; Peng, C.; Zhang, Y.; Zykova, T.A.; Lee, M.H.; Lee, S.Y.; Rao, E.; Chen, H.; Ryu, J.; Wang, L.; et al. RSK2 phosphorylates T-bet to attenuate colon cancer metastasis and growth. Proc. Natl. Acad. Sci. USA 2017, 114, 12791–12796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chornoguz, O.; Hagan, R.S.; Haile, A.; Arwood, M.L.; Gamper, C.J.; Banerjee, A.; Powell, J.D. mTORC1 Promotes T-bet Phosphorylation to Regulate Th1 Differentiation. J. Immunol. 2017, 198, 3939–3948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Lee, S.M.; Gao, B.; Shannon, S.; Zhu, Z.; Fang, D. c-Abl-mediated tyrosine phosphorylation of the T-bet DNA-binding domain regulates CD4+ T-cell differentiation and allergic lung inflammation. Mol. Cell. Biol. 2011, 31, 3445–3456. [Google Scholar] [CrossRef] [Green Version]
- Lazarevic, V.; Chen, X.; Shim, J.H.; Hwang, E.S.; Jang, E.; Bolm, A.N.; Oukka, M.; Kuchroo, V.K.; Glimcher, L.H. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat. Immunol. 2011, 12, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, K.; Ohtsuka, Y.; Ikuse, T.; Baba, Y.; Yamakawa, Y.; Aoyagi, Y.; Fujii, T.; Kudo, T.; Nagata, S.; Shimizu, T. Increased mucosal expression of GATA-3 and STAT-4 in pediatric ulcerative colitis. Pediatr. Int. 2010, 52, 584–589. [Google Scholar] [CrossRef]
- Popp, V.; Gerlach, K.; Mott, S.; Turowska, A.; Garn, H.; Atreya, R.; Lehr, H.A.; Ho, I.C.; Renz, H.; Weigmann, B.; et al. Rectal Delivery of a DNAzyme That Specifically Blocks the Transcription Factor GATA3 and Reduces Colitis in Mice. Gastroenterology 2017, 152, 176–192. [Google Scholar] [CrossRef]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef]
- Nalleweg, N.; Chiriac, M.T.; Podstawa, E.; Lehmann, C.; Rau, T.T.; Atreya, R.; Krauss, E.; Hundorfean, G.; Fichtner-Feigl, S.; Hartmann, A.; et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut 2015, 64, 743–755. [Google Scholar] [CrossRef] [Green Version]
- Defendenti, C.; Sarzi-Puttini, P.; Saibeni, S.; Bollani, S.; Bruno, S.; Almasio, P.L.; Declich, P.; Atzeni, F. Significance of serum Il-9 levels in inflammatory bowel disease. Int. J. Immunopathol. Pharmacol. 2015, 28, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Matusiewicz, M.; Neubauer, K.; Bednarz-Misa, I.; Gorska, S.; Krzystek-Korpacka, M. Systemic interleukin-9 in inflammatory bowel disease: Association with mucosal healing in ulcerative colitis. World J. Gastroenterol. 2017, 23, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, H.; Kato, M.; Tohyama, H.; Tamaki, Y.; Endo, Y.; Kimura, M.Y.; Tumes, D.J.; Motohashi, S.; Matsumoto, M.; Nakayama, K.I.; et al. Methylation of Gata3 protein at Arg-261 regulates transactivation of the Il5 gene in T helper 2 cells. J. Biol. Chem. 2015, 290, 13095–13103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, H.; Tanaka, T.; Endo, Y.; Kato, M.; Shinoda, K.; Suzuki, A.; Motohashi, S.; Matsumoto, M.; Nakayama, K.I.; Nakayama, T. Akt1-mediated Gata3 phosphorylation controls the repression of IFNgamma in memory-type Th2 cells. Nat. Commun. 2016, 7, 11289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, J.; Moro, K.; Motomura, Y.; Okamoto, K.; Zhu, J.; Takayanagi, H.; Kubo, M.; Koyasu, S. Critical role of p38 and GATA3 in natural helper cell function. J. Immunol. 2013, 191, 1818–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Zheng, B.; Huang, Y.; Yang, D.; Katzman, S.; Chang, C.; Fowell, D.; Zeng, W.P. Interaction between GATA-3 and the transcriptional coregulator Pias1 is important for the regulation of Th2 immune responses. J. Immunol. 2007, 179, 8297–8304. [Google Scholar] [CrossRef] [Green Version]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [Green Version]
- Kathania, M.; Khare, P.; Zeng, M.; Cantarel, B.; Zhang, H.; Ueno, H.; Venuprasad, K. Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-gammat ubiquitination. Nat. Immunol. 2016, 17, 997–1004. [Google Scholar] [CrossRef]
- Rutz, S.; Kayagaki, N.; Phung, Q.T.; Eidenschenk, C.; Noubade, R.; Wang, X.; Lesch, J.; Lu, R.; Newton, K.; Huang, O.W.; et al. Deubiquitinase DUBA is a post-translational brake on interleukin-17 production in T cells. Nature 2015, 518, 417–421. [Google Scholar] [CrossRef]
- Wang, X.; Yang, J.; Han, L.; Zhao, K.; Wu, Q.; Bao, L.; Li, Z.; Lv, L.; Li, B. TRAF5-mediated Lys-63-linked Polyubiquitination Plays an Essential Role in Positive Regulation of RORgammat in Promoting IL-17A Expression. J. Biol. Chem. 2015, 290, 29086–29094. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Xu, P.; Han, L.; Guo, Z.; Wang, X.; Chen, Z.; Nie, J.; Yin, S.; Piccioni, M.; Tsun, A.; et al. Cutting edge: Ubiquitin-specific protease 4 promotes Th17 cell function under inflammation by deubiquitinating and stabilizing RORgammat. J. Immunol. 2015, 194, 4094–4097. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, H.; Zhong, B.; Blonska, M.; Gorjestani, S.; Yan, M.; Tian, Q.; Zhang, D.E.; Lin, X.; Dong, C. USP18 inhibits NF-kappaB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J. Exp. Med. 2013, 210, 1575–1590. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Liu, X.; Wang, X.; Chang, S.H.; Liu, X.; Wang, A.; Reynolds, J.M.; Dong, C. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat. Immunol. 2012, 13, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.S.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnolzer, M.; et al. SIRT1 deacetylates RORgammat and enhances Th17 cell generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Nie, J.; Gao, Y.; Xu, P.; Sun, Q.; Yang, J.; Han, L.; Chen, Z.; Wang, X.; Lv, L.; et al. Reciprocal regulation of RORgammat acetylation and function by p300 and HDAC1. Sci. Rep. 2015, 5, 16355. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Z.; Li, L.; Gong, W.; Lazenby, A.J.; Swanson, B.J.; Herring, L.E.; Asara, J.M.; Singer, J.D.; Wen, H. Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J. Exp. Med. 2017, 214, 1093–1109. [Google Scholar] [CrossRef] [Green Version]
- Mudter, J.; Amoussina, L.; Schenk, M.; Yu, J.; Brustle, A.; Weigmann, B.; Atreya, R.; Wirtz, S.; Becker, C.; Hoffman, A.; et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J. Clin. Investig. 2008, 118, 2415–2426. [Google Scholar] [CrossRef] [Green Version]
- Cretney, E.; Xin, A.; Shi, W.; Minnich, M.; Masson, F.; Miasari, M.; Belz, G.T.; Smyth, G.K.; Busslinger, M.; Nutt, S.L.; et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011, 12, 304–311. [Google Scholar] [CrossRef]
- Mudter, J.; Yu, J.; Zufferey, C.; Brustle, A.; Wirtz, S.; Weigmann, B.; Hoffman, A.; Schenk, M.; Galle, P.R.; Lehr, H.A.; et al. IRF4 regulates IL-17A promoter activity and controls RORgammat-dependent Th17 colitis in vivo. Inflamm. Bowel Dis. 2011, 17, 1343–1358. [Google Scholar] [CrossRef]
- Brustle, A.; Heink, S.; Huber, M.; Rosenplanter, C.; Stadelmann, C.; Yu, P.; Arpaia, E.; Mak, T.W.; Kamradt, T.; Lohoff, M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 2007, 8, 958–966. [Google Scholar] [CrossRef]
- Biswas, P.S.; Gupta, S.; Chang, E.; Song, L.; Stirzaker, R.A.; Liao, J.K.; Bhagat, G.; Pernis, A.B. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Investig. 2010, 120, 3280–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Chaudhry, A.; Kas, A.; de Roos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 2009, 458, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wang, A.; Ma, X.; Demarque, M.; Jin, W.; Xin, H.; Dejean, A.; Dong, C. Protein SUMOylation Is Required for Regulatory T Cell Expansion and Function. Cell Rep. 2016, 16, 1055–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, I.C.; Hodge, M.R.; Rooney, J.W.; Glimcher, L.H. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell 1996, 85, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Weigmann, B.; Nemetz, A.; Becker, C.; Schmidt, J.; Strand, D.; Lehr, H.A.; Galle, P.R.; Ho, I.C.; Neurath, M.F. A critical regulatory role of leucin zipper transcription factor c-Maf in Th1-mediated experimental colitis. J. Immunol. 2004, 173, 3446–3455. [Google Scholar] [CrossRef] [Green Version]
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.Y.; Yeh, L.T.; Fu, S.H.; Chien, M.W.; Liu, Y.W.; Miaw, S.C.; Chang, D.M.; Sytwu, H.K. SUMO-defective c-Maf preferentially transactivates Il21 to exacerbate autoimmune diabetes. J. Clin. Investig. 2018, 128, 3779–3793. [Google Scholar] [CrossRef]
- Xu, M.; Pokrovskii, M.; Ding, Y.; Yi, R.; Au, C.; Harrison, O.J.; Galan, C.; Belkaid, Y.; Bonneau, R.; Littman, D.R. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018, 554, 373–377. [Google Scholar] [CrossRef]
- Neumann, C.; Blume, J.; Roy, U.; Teh, P.P.; Vasanthakumar, A.; Beller, A.; Liao, Y.; Heinrich, F.; Arenzana, T.L.; Hackney, J.A.; et al. c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat. Immunol. 2019, 20, 471–481. [Google Scholar] [CrossRef]
- Blonska, M.; Joo, D.; Nurieva, R.I.; Zhao, X.; Chiao, P.; Sun, S.C.; Dong, C.; Lin, X. Activation of the transcription factor c-Maf in T cells is dependent on the CARMA1-IKKbeta signaling cascade. Sci. Signal. 2013, 6, ra110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.C.; Lai, C.Y.; Yen, W.F.; Lin, Y.H.; Chang, H.H.; Tai, T.S.; Lu, Y.J.; Tsao, H.W.; Ho, I.C.; Miaw, S.C. Reciprocal regulation of C-Maf tyrosine phosphorylation by Tec and Ptpn22. PLoS ONE 2015, 10, e0127617. [Google Scholar] [CrossRef] [PubMed]
- Leavenworth, J.W.; Ma, X.; Mo, Y.Y.; Pauza, M.E. SUMO conjugation contributes to immune deviation in nonobese diabetic mice by suppressing c-Maf transactivation of IL-4. J. Immunol. 2009, 183, 1110–1119. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.S.; Tsai, P.Y.; Hsieh, W.Y.; Tsao, H.W.; Liu, M.W.; Grenningloh, R.; Wang, L.F.; Ho, I.C.; Miaw, S.C. SUMOylation attenuates c-Maf-dependent IL-4 expression. Eur. J. Immunol. 2010, 40, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xu, X.; Tong, J.; Han, K.; Zhang, Z.; Tang, J.; Li, S.; Yang, C.; Li, J.; Cao, B.; et al. Ubiquitination of the transcription factor c-MAF is mediated by multiple lysine residues. Int. J. Biochem. Cell Biol. 2014, 57, 157–166. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Z.; Li, J.; Tong, J.; Cao, B.; Taylor, P.; Tang, X.; Wu, D.; Moran, M.F.; Zeng, Y.; et al. The ubiquitin-conjugating enzyme UBE2O modulates c-Maf stability and induces myeloma cell apoptosis. J. Hematol. Oncol. 2017, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tong, J.; Tang, X.; Juan, J.; Cao, B.; Hurren, R.; Chen, G.; Taylor, P.; Xu, X.; Shi, C.X.; et al. The ubiquitin ligase HERC4 mediates c-Maf ubiquitination and delays the growth of multiple myeloma xenografts in nude mice. Blood 2016, 127, 1676–1686. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Zhang, H.; Zeissig, S.; Lipinski, S.; Till, A.; Jiang, T.; Stade, B.; Bromberg, Y.; Ellinghaus, E.; Keller, A.; et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 2013, 145, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.H.; Lin, M.H.; Yeh, L.T.; Wang, Y.L.; Chien, M.W.; Lin, S.H.; Chang, D.M.; Sytwu, H.K. Targeting tumour necrosis factor receptor 1 assembly reverses Th17-mediated colitis through boosting a Th2 response. Gut 2015, 64, 765–775. [Google Scholar] [CrossRef]
- Neumann, C.; Heinrich, F.; Neumann, K.; Junghans, V.; Mashreghi, M.F.; Ahlers, J.; Janke, M.; Rudolph, C.; Mockel-Tenbrinck, N.; Kuhl, A.A.; et al. Role of Blimp-1 in programing Th effector cells into IL-10 producers. J. Exp. Med. 2014, 211, 1807–1819. [Google Scholar] [CrossRef]
- Martins, G.A.; Cimmino, L.; Shapiro-Shelef, M.; Szabolcs, M.; Herron, A.; Magnusdottir, E.; Calame, K. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat. Immunol. 2006, 7, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Kallies, A.; Hawkins, E.D.; Belz, G.T.; Metcalf, D.; Hommel, M.; Corcoran, L.M.; Hodgkin, P.D.; Nutt, S.L. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat. Immunol. 2006, 7, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Angelin-Duclos, C.; Greenwood, J.; Liao, J.; Calame, K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol. Cell. Biol. 2000, 20, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimshon, L.; Michaeli, A.; Hadar, R.; Nutt, S.L.; David, Y.; Navon, A.; Waisman, A.; Tirosh, B. SUMOylation of Blimp-1 promotes its proteasomal degradation. FEBS Lett. 2011, 585, 2405–2409. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.Y.; Su, S.T.; Hsu, P.H.; Chang, C.C.; Lin, I.Y.; Tseng, Y.H.; Tsai, M.D.; Shih, H.M.; Lin, K.I. SUMOylation of Blimp-1 is critical for plasma cell differentiation. EMBO Rep. 2012, 13, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Chunder, N.; Wang, L.; Chen, C.; Hancock, W.W.; Wells, A.D. Cyclin-dependent kinase 2 controls peripheral immune tolerance. J. Immunol. 2012, 189, 5659–5666. [Google Scholar] [CrossRef] [Green Version]
- Morawski, P.A.; Mehra, P.; Chen, C.; Bhatti, T.; Wells, A.D. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J. Biol. Chem. 2013, 288, 24494–24502. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lin, F.; Zhuo, C.; Deng, G.; Chen, Z.; Yin, S.; Gao, Z.; Piccioni, M.; Tsun, A.; Cai, S.; et al. PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J. Biol. Chem. 2014, 289, 26872–26881. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Nagai, Y.; Xiao, Y.; Li, Z.; Dai, S.; Ohtani, T.; Banham, A.; Li, B.; Wu, S.L.; Hancock, W.; et al. Pim-2 Kinase Influences Regulatory T Cell Function and Stability by Mediating Foxp3 Protein N-terminal Phosphorylation. J. Biol. Chem. 2015, 290, 20211–20220. [Google Scholar] [CrossRef] [Green Version]
- Nie, H.; Zheng, Y.; Li, R.; Guo, T.B.; He, D.; Fang, L.; Liu, X.; Xiao, L.; Chen, X.; Wan, B.; et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat. Med. 2013, 19, 322–328. [Google Scholar] [CrossRef]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- De Zoeten, E.F.; Wang, L.; Sai, H.; Dillmann, W.H.; Hancock, W.W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010, 138, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, U.H.; Akimova, T.; Liu, Y.; Wang, L.; Hancock, W.W. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr. Opin. Immunol. 2011, 23, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.S.; Lim, H.W.; Wu, J.; Schnolzer, M.; Verdin, E.; Ott, M. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J. Immunol. 2012, 188, 2712–2721. [Google Scholar] [CrossRef] [Green Version]
- Van Loosdregt, J.; Fleskens, V.; Fu, J.; Brenkman, A.B.; Bekker, C.P.; Pals, C.E.; Meerding, J.; Berkers, C.R.; Barbi, J.; Grone, A.; et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity 2013, 39, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Kumar, S.; Dahiya, S.; Wang, F.; Wu, J.; Newick, K.; Han, R.; Samanta, A.; Beier, U.H.; Akimova, T.; et al. Ubiquitin-specific Protease-7 Inhibition Impairs Tip60-dependent Foxp3+ T-regulatory Cell Function and Promotes Antitumor Immunity. EBioMedicine 2016, 13, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lu, Y.; Wang, S.; Han, Z.; Zhu, F.; Ni, Y.; Liang, R.; Zhang, Y.; Leng, Q.; Wei, G.; et al. USP21 prevents the generation of T-helper-1-like Treg cells. Nat. Commun. 2016, 7, 13559. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Xiong, X.; Ren, K.; Xu, B.; Cheng, M.; Sahu, C.; Wu, K.; Nie, Y.; Huang, Z.; Blumberg, R.S.; et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Sun, Q.H.; Wang, Y.S.; Liu, G.; Zhou, H.L.; Jian, Y.P.; Liu, M.D.; Zhang, D.; Ding, Q.; Zhao, R.X.; Chen, J.F.; et al. Enhanced O-linked Glcnacylation in Crohn’s disease promotes intestinal inflammation. EBioMedicine 2020, 53, 102693. [Google Scholar] [CrossRef]
- Liu, B.; Salgado, O.C.; Singh, S.; Hippen, K.L.; Maynard, J.C.; Burlingame, A.L.; Ball, L.E.; Blazar, B.R.; Farrar, M.A.; Hogquist, K.A.; et al. The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nat. Commun. 2019, 10, 354. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Transcription Factor | Position | Modification | Physiological Effect | Ref. |
|---|---|---|---|---|
| T-bet | Tyr525 | phosphorylation | inhibition of Gata3 binding ability and promotion of IFNγ-associated inflammation | [39] |
| Ser508 | phosphorylation | inhibition of IL-2 production | [40] | |
| Thr302 | phosphorylation | interaction with NFAT1 and regulation of IFNγ-associated inflammation | [41] | |
| Ser498/Ser502 | phosphorylation | promoting IFNγ production for inhibition of colon cancer metastasis | [42] | |
| Tyr219/Tyr265/Tyr304 | phosphorylation | modulation of the binding ability to promote IFNγ-associated inflammation | [44] | |
| Tyr304 | phosphorylation | inhibition of Th17 cell development and IL-17-associated inflammation | [45] | |
| Lys313 | ubiquitination | regulation of T-bet protein stability and IFNγ-associated inflammation | [41] | |
| Gata3 | Arg261 | methylation | regulation of IL-5 production to promote Th2 inflammation | [52] |
| Ser308/Thr315/Ser316 | phosphorylation | inhibition of T-bet-mediated and IFN-γ-associated inflammation | [53] | |
| phosphorylation | regulation of IL-6 production | [54] | ||
| SUMOylation | promotion of Gata3 binding ability to enhance Th2 inflammation | [55] | ||
| RORγt | ubiquitination | regulation of RORγt protein stability for inhibition of Th17 inflammation | [57,58] | |
| Lys63 | ubiquitination | regulation of IL-17 production for promotion of Th17 inflammation | [59] | |
| Lys48 | ubiquitination | regulation of RORγt protein stability for inhibition of Th17 inflammation | [60] | |
| Lys69/Lys81/Lys99/Lys112 | acetylation | regulation of RORγt binding ability for inhibition of Th17 inflammation | [63] | |
| Lys81 | acetylation | regulation of IL-17-mediated inflammation | [64] | |
| STAT3 | Tyr705 and Ser727 | phosphorylation | regulation of STAT3 activation | [65] |
| Thr717 | O-GlcNAcylation | inhibition of IL-10 production for promotion of colonic inflammation | [66] | |
| IRF4 | phosphorylation | promotion of Th17 inflammation | [71] | |
| Lys349 | SUMOylation | regulation of IRF4 protein stability in Treg for inhibition of inflammation | [73] | |
| c-Maf | phosphorylation | regulation of c-Maf binding ability for promotion of IL-21-mediated inflammation | [81,82] | |
| Lys33 | SUMOylation | regulation of c-Maf binding ability for inhibition of IL-21-mediated inflammation | [78,83,84] | |
| Lys331/Lys345 | ubiquitination | regulation of c-Maf protein stability | [86] | |
| Lys85/Lys297 | ubiquitination | regulation of c-Maf protein stability | [87] | |
| Blimp-1 | Lys816 | SUMOylation | regulation of Blimp-1 transcriptional activity and association with colonic inflammation | [94,95] |
| Foxp3 | Ser19/Thr175 | phosphorylation | regulation of Foxp3 transcriptional activity for inhibition of inflammation | [96,97] |
| Ser422 | phosphorylation | inhibition of Foxp3 binding ability for promotion of inflammation | [98] | |
| Ser418 | phosphorylation | regulation of Treg population | [100,101] | |
| Lys31/Lys262/Lys267 | acetylation | modulation of Treg function for inhibition of inflammation | [104] | |
| ubiquitination | regulation of Foxp3 protein stability for inhibition of inflammation | [105,106,107] | ||
| O-GlcNAcylation | regulation of Treg cell lineage stability for inhibition of inflammation | [108,110] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-Y.; Fu, S.-H.; Chien, M.-W.; Liu, Y.-W.; Chen, S.-J.; Sytwu, H.-K. Post-Translational Modifications of Transcription Factors Harnessing the Etiology and Pathophysiology in Colonic Diseases. Int. J. Mol. Sci. 2020, 21, 3207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093207
Hsu C-Y, Fu S-H, Chien M-W, Liu Y-W, Chen S-J, Sytwu H-K. Post-Translational Modifications of Transcription Factors Harnessing the Etiology and Pathophysiology in Colonic Diseases. International Journal of Molecular Sciences. 2020; 21(9):3207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093207
Chicago/Turabian StyleHsu, Chao-Yuan, Shin-Huei Fu, Ming-Wei Chien, Yu-Wen Liu, Shyi-Jou Chen, and Huey-Kang Sytwu. 2020. "Post-Translational Modifications of Transcription Factors Harnessing the Etiology and Pathophysiology in Colonic Diseases" International Journal of Molecular Sciences 21, no. 9: 3207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093207