Enhanced Integrin Activation of PLD2-Deficient Platelets Accelerates Inflammation after Myocardial Infarction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
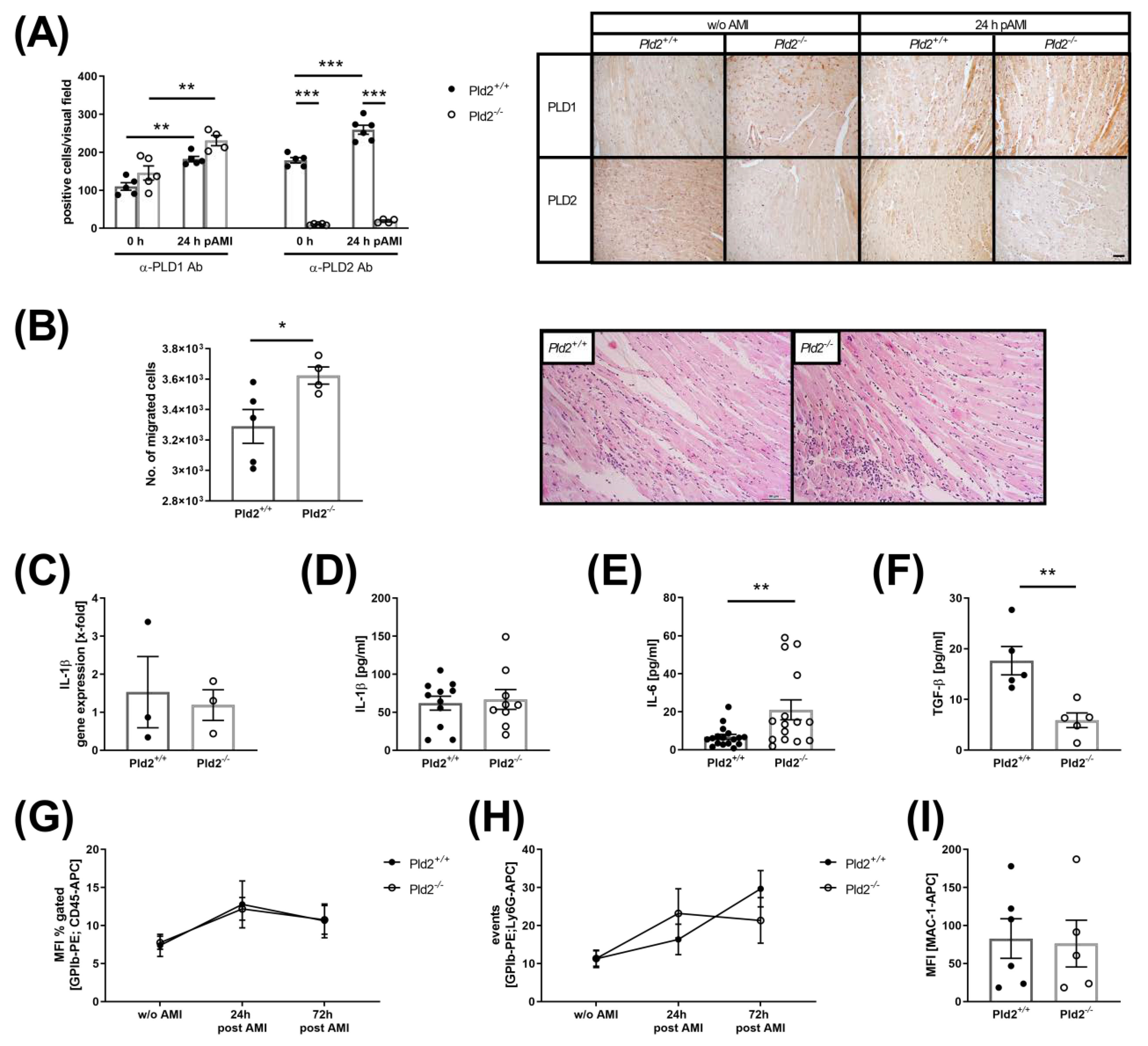
2.1. Loss of PLD2 Accelerates Inflammation after Myocardial Infarction
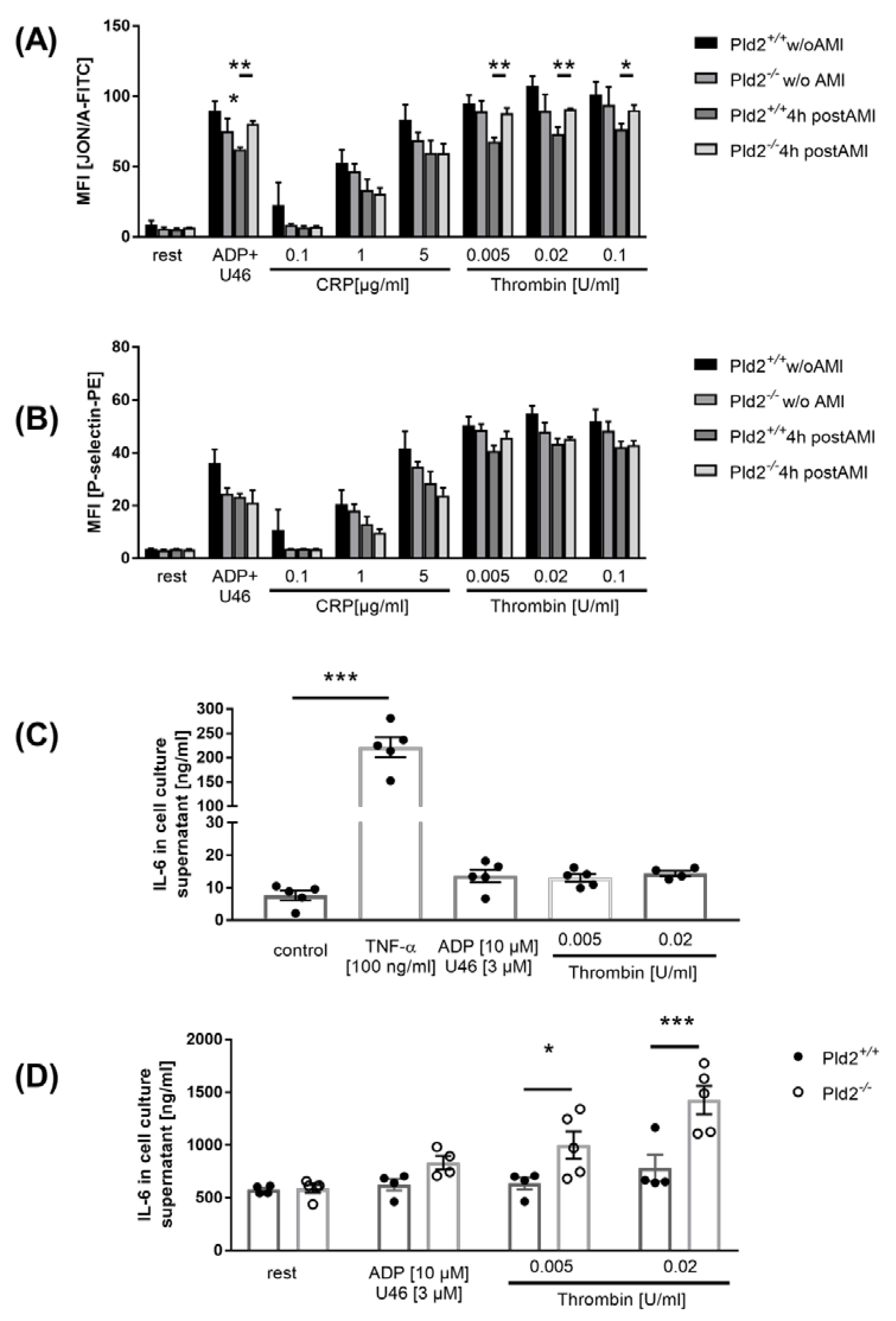
2.2. Enhanced Activation of Platelet Integrin αIIbβ3 in the Acute Phase after Cardiac Ischemia is Responsible for Enhanced IL-6 Release from Endothelial Cells
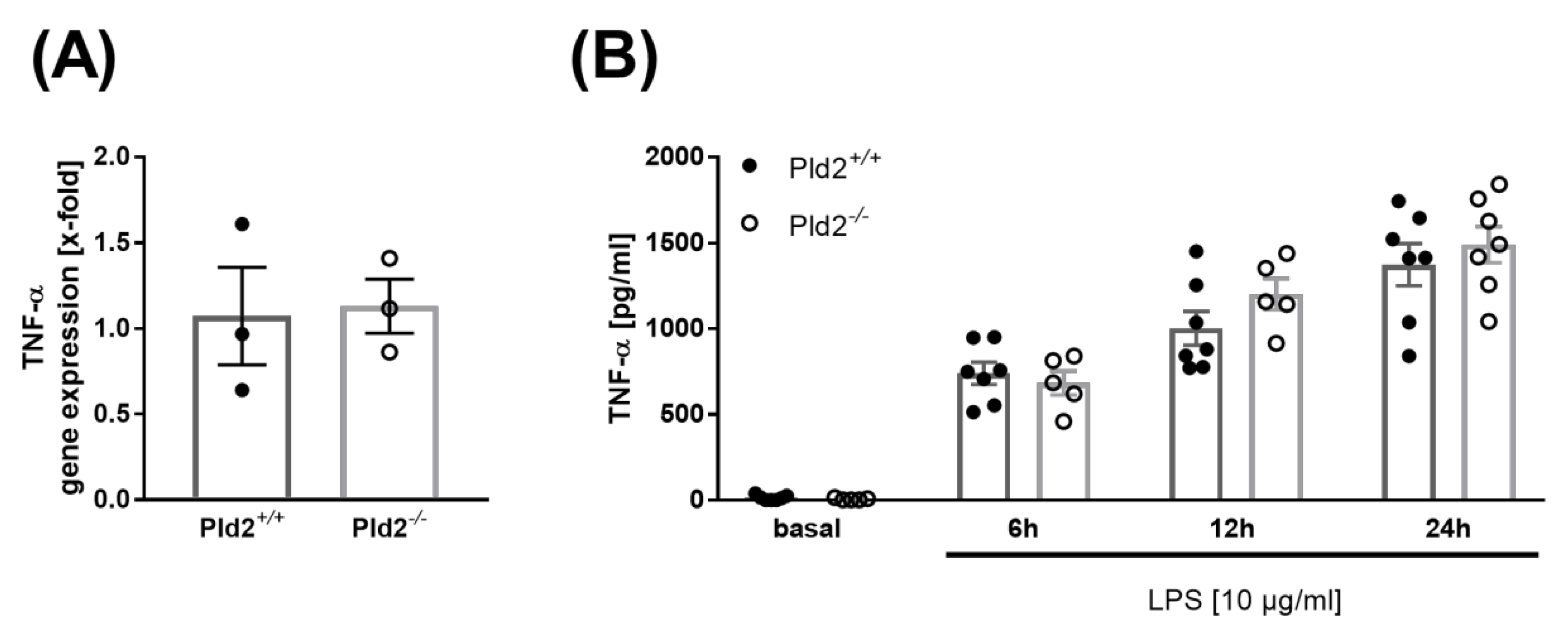
2.3. PLD2 Does Not Regulate TNF-α Expression and Release
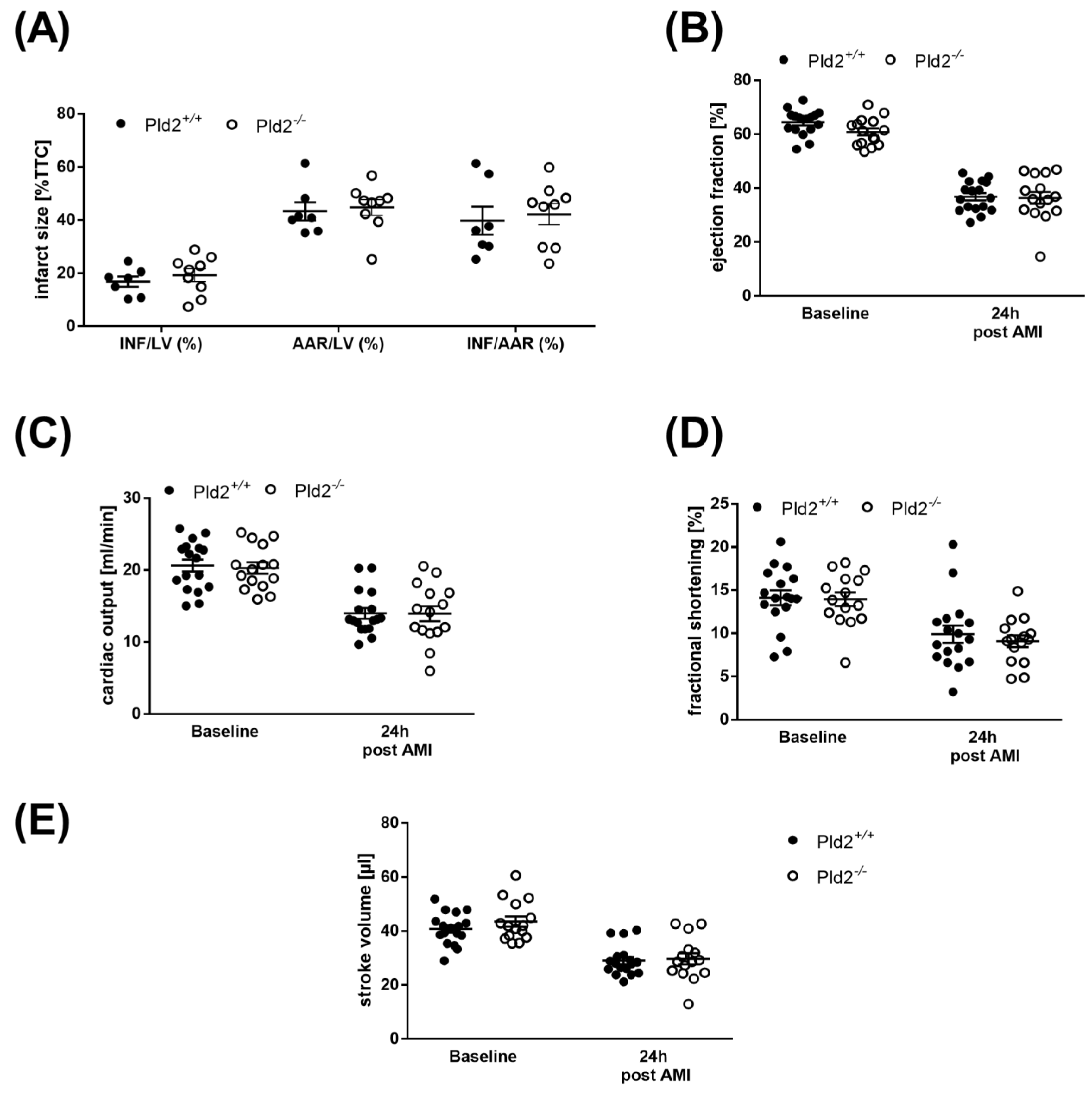
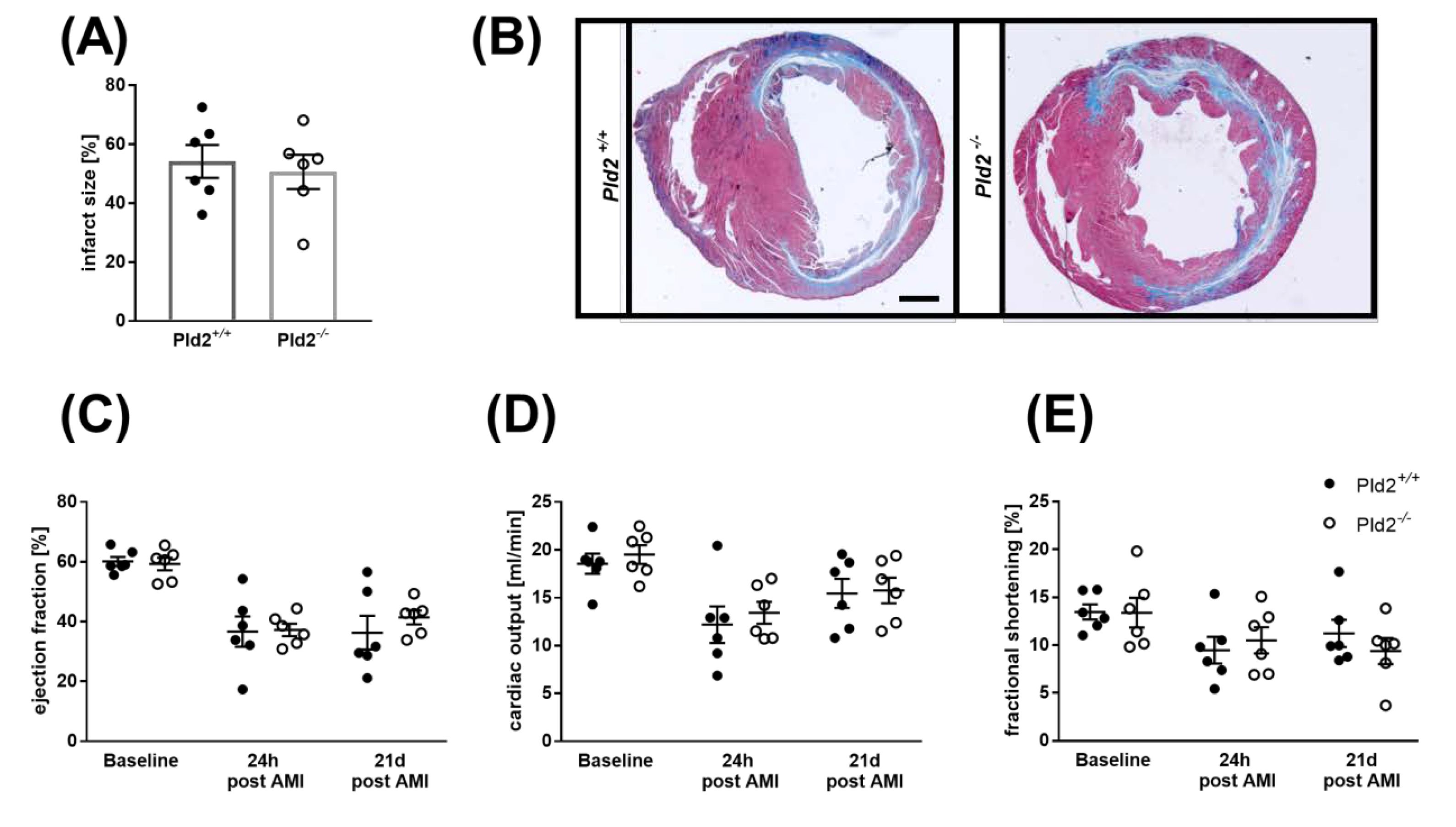
2.4. Unaltered Infarct Size and Cardiac Function in PLD2-Deficient Mice 24 h after Ischemia and Reperfusion
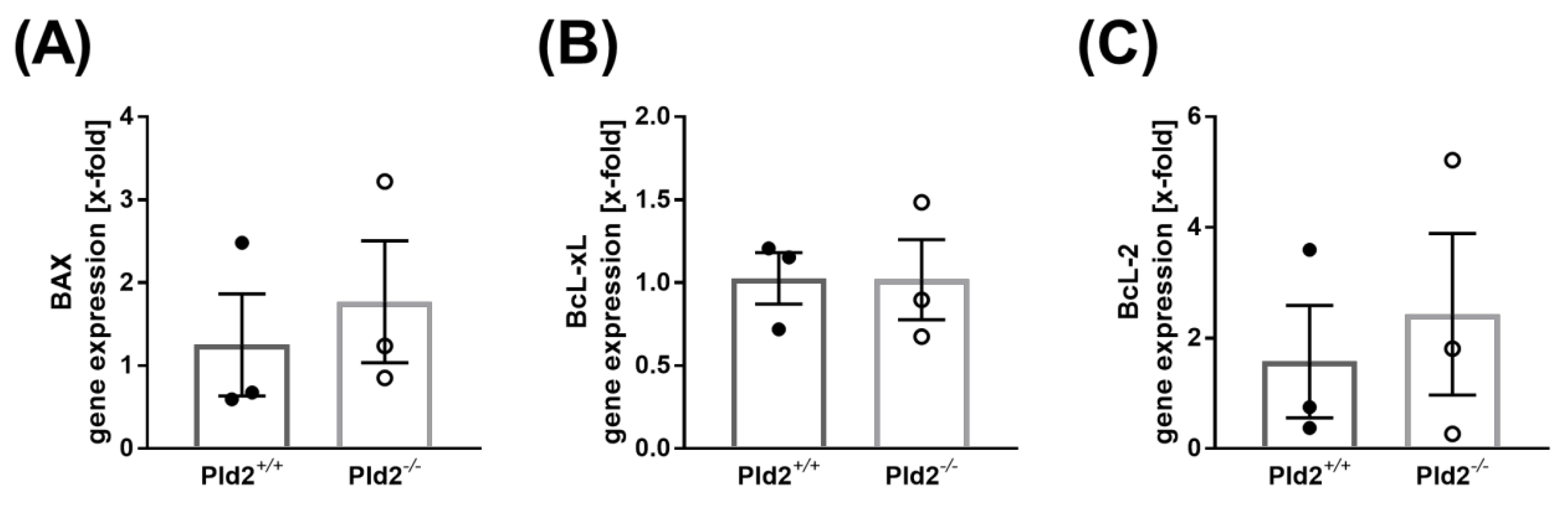
2.5. Cell Survival is Not Altered in PLD2-Deficient Mice after Ischemia and Reperfusion Injury
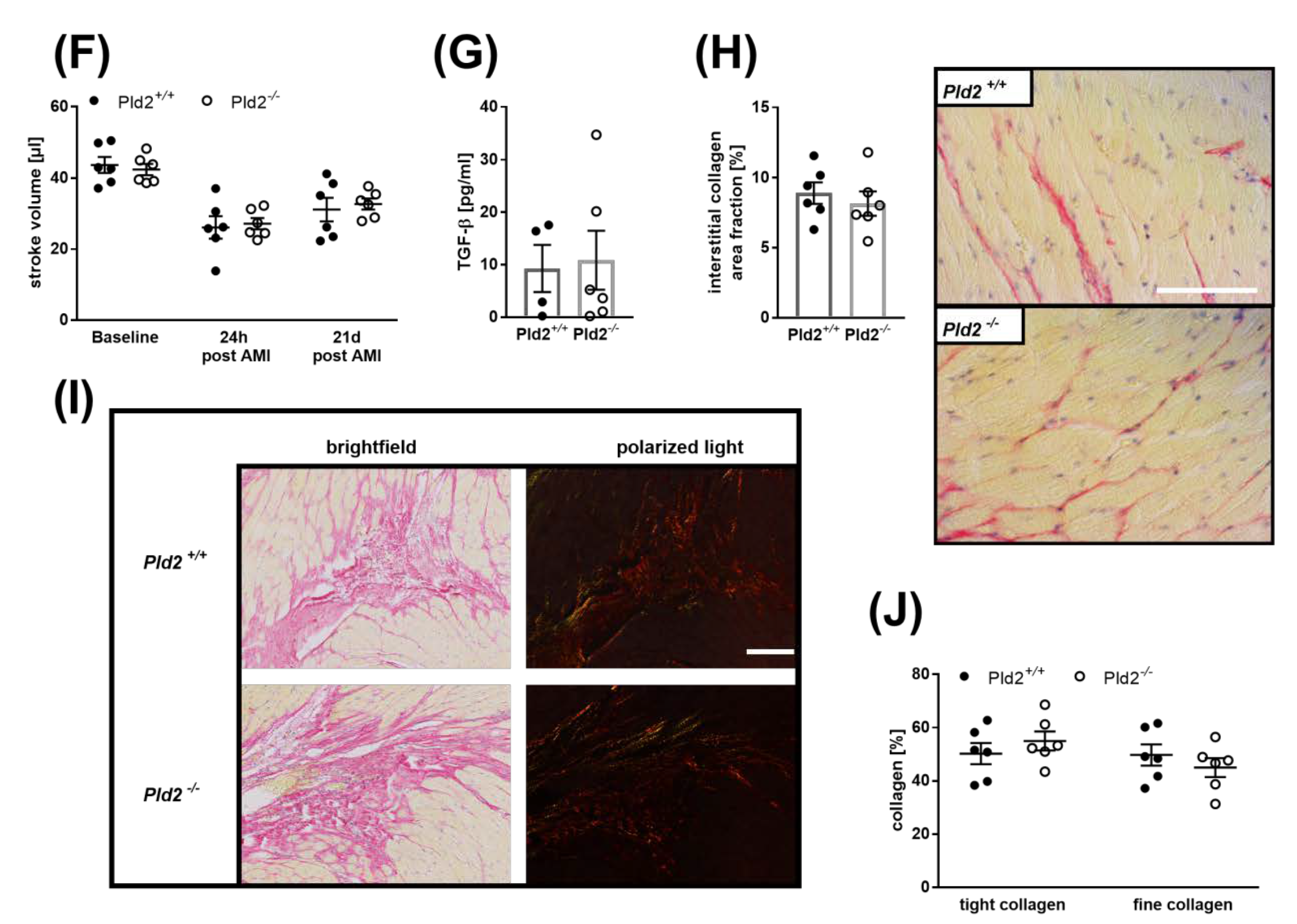
2.6. Loss of PLD2 Does Not Modify Remodeling and Scar Formation 21 Days after Myocardial Infarction
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Myocardial Ischemia and Reperfusion in Mice
4.3. Collagen-Staining of Cardiac Sections
4.4. Immunhistochemistry of Cardiac Sections
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Flow Cytometric Analysis of Platelet-Immune Cell Aggregate Formation, Neutrophil and Platelet Activation
4.7. Quantitative Real-Time PCR
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAR | Area at risk |
| ADP | Adenosine diphosphate |
| AMI | Acute myocardial infarction |
| CRP | Collagen-related peptide |
| ELISA | Enzym-Linked Immunosorbent Assay |
| FIPI | 5-Fluoro-2-Indolyl Deschlorohalopemide |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GP Ib/VI | Glycoprotein (GP) Ib/VI |
| HE | Hematoxylin/Eosin |
| I/R | Ischemia/reperfusion |
| IL-6 | Interleukin 6 |
| INF | Infarct |
| LAD | Left anterior descending artery |
| LPS | Lipopolysaccharide |
| LV | Left ventricle |
| Mac-1 | Macrophage-1 antigen |
| MFI | Mean of fluorescence |
| MHEC5-T | Mouse Heart Endothelial Cell clone 5-Transformed |
| MI | Myocardial infarction |
| min | Minute |
| PA | Phosphatidic acid |
| PCR | (real-time) Polymerase Chain Reaction |
| PKC | Protein kinase C |
| PLD | Phospholipase D |
| rest | Resting |
| TGF-β | Transforming growth factor β |
| Thr | Thrombin |
| TNF-α | Tumor necrosis factor α |
| TTC | 2,3,5-triphenyltetrazolium chloride |
| U46619 | Thromboxane analog |
References
- McDermott, M.; Wakelam, M.J.; Morris, A.J. Phospholipase D. Biochem. Cell Biol. 2004, 82, 225–253. [Google Scholar] [CrossRef] [PubMed]
- Oude Weernink, P.A.; Lopez de Jesus, M.; Schmidt, M. Phospholipase D signaling: Orchestration by PIP2 and small GTPases. Naunyn Schmiedebergs Arch. Pharm. 2007, 374, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodaki, T.; Yamashita, S. Cloning, expression, and characterization of a novel phospholipase D complementary DNA from rat brain. J. Biol. Chem. 1997, 272, 11408–11413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvers, M.; Stegner, D.; Hagedorn, I.; Kleinschnitz, C.; Braun, A.; Kuijpers, M.E.; Boesl, M.; Chen, Q.; Heemskerk, J.W.; Stoll, G.; et al. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci. Signal. 2010, 3, ra1. [Google Scholar] [CrossRef] [Green Version]
- Klier, M.; Gowert, N.S.; Jackel, S.; Reinhardt, C.; Elvers, M. Phospholipase D1 is a regulator of platelet-mediated inflammation. Cell Signal. 2017, 38, 171–181. [Google Scholar] [CrossRef]
- Thielmann, I.; Stegner, D.; Kraft, P.; Hagedorn, I.; Krohne, G.; Kleinschnitz, C.; Stoll, G.; Nieswandt, B. Redundant functions of phospholipases D1 and D2 in platelet alpha-granule release. J. Thromb. Haemost. 2012, 10, 2361–2372. [Google Scholar] [CrossRef]
- Stegner, D.; Thielmann, I.; Kraft, P.; Frohman, M.A.; Stoll, G.; Nieswandt, B. Pharmacological inhibition of phospholipase D protects mice from occlusive thrombus formation and ischemic stroke--brief report. Arter. Thromb. Vasc. Biol. 2013, 33, 2212–2217. [Google Scholar] [CrossRef] [Green Version]
- Schonberger, T.; Jurgens, T.; Muller, J.; Armbruster, N.; Niermann, C.; Gorressen, S.; Sommer, J.; Tian, H.; di Paolo, G.; Scheller, J.; et al. Pivotal role of phospholipase D1 in tumor necrosis factor-alpha-mediated inflammation and scar formation after myocardial ischemia and reperfusion in mice. Am. J. Pathol 2014, 184, 2450–2464. [Google Scholar] [CrossRef]
- Klier, M.; Gorressen, S.; Urbahn, M.A.; Barbosa, D.; Ouwens, M.; Fischer, J.W.; Elvers, M. Enzymatic Activity Is Not Required for Phospholipase D Mediated TNF-alpha Regulation and Myocardial Healing. Front. Physiol 2018, 9, 1698. [Google Scholar] [CrossRef]
- Bombeli, T.; Schwartz, B.R.; Harlan, J.M. Adhesion of activated platelets to endothelial cells: Evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J. Exp. Med. 1998, 187, 329–339. [Google Scholar] [CrossRef]
- Tian, M.; Yuan, Y.C.; Li, J.Y.; Gionfriddo, M.R.; Huang, R.C. Tumor necrosis factor-alpha and its role as a mediator in myocardial infarction: A brief review. Chronic Dis. Transl. Med. 2015, 1, 18–26. [Google Scholar]
- Urbahn, M.A.; Kaup, S.C.; Reusswig, F.; Kruger, I.; Spelleken, M.; Jurk, K.; Klier, M.; Lang, P.A.; Elvers, M. Phospholipase D1 regulation of TNF-alpha protects against responses to LPS. Sci. Rep. 2018, 8, 10006. [Google Scholar] [CrossRef]
- Janumyan, Y.M.; Sansam, C.G.; Chattopadhyay, A.; Cheng, N.; Soucie, E.L.; Penn, L.Z.; Andrews, D.; Knudson, C.M.; Yang, E. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. Embo. J. 2003, 22, 5459–5470. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Moraru, I.I.; Popescu, L.M.; Maulik, N.; Liu, X.; Das, D.K. Phospholipase D signaling in ischemic heart. Biochim. Et Biophys. Acta 1992, 1139, 148–154. [Google Scholar] [CrossRef]
- Cohen, M.V.; Liu, Y.; Liu, G.S.; Wang, P.; Weinbrenner, C.; Cordis, G.A.; Das, D.K.; Downey, J.M. Phospholipase D plays a role in ischemic preconditioning in rabbit heart. Circulation 1996, 94, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Dent, M.R.; Singal, T.; Dhalla, N.S.; Tappia, P.S. Expression of phospholipase D isozymes in scar and viable tissue in congestive heart failure due to myocardial infarction. J. Cell. Mol. Med. 2004, 8, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Tappia, P.S.; Dent, M.R.; Dhalla, N.S. Oxidative stress and redox regulation of phospholipase D in myocardial disease. Free Radic. Biol. Med. 2006, 41, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Abdulnour, R.E.; Howrylak, J.A.; Tavares, A.H.; Douda, D.N.; Henkels, K.M.; Miller, T.E.; Fredenburgh, L.E.; Baron, R.M.; Gomez-Cambronero, J.; Levy, B.D. Phospholipase D isoforms differentially regulate leukocyte responses to acute lung injury. J. Leukoc. Biol. 2018, 103, 919–932. [Google Scholar] [CrossRef]
- Speranza, F.J.; Mahankali, M.; Gomez-Cambronero, J. Macrophage migration arrest due to a winning balance of Rac2/Sp1 repression over beta-catenin-induced PLD expression. J. Leukoc. Biol. 2013, 94, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Dawood, F.; Wen, W.H.; Chen, M.; Dixon, I.; Kirshenbaum, L.A.; Liu, P.P. Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction. Circulation 2004, 110, 3221–3228. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.K.; Ya-Ping, J.; Gadbery, J.; Abedeen, D.; Sampson, N. Phospholipase D2 loss results in increased blood pressure via inhibition of the endothelial nitric oxide synthase pathway. Sci. Rep. 2017, 7, 9112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.K.; Kim, S.D.; Kook, M.; Lee, H.Y.; Ghim, J.; Choi, Y.; Zabel, B.A.; Ryu, S.H.; Bae, Y.S. Phospholipase D2 drives mortality in sepsis by inhibiting neutrophil extracellular trap formation and down-regulating CXCR2. J. Exp. Med. 2015, 212, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvers, M.; Grenegard, M.; Khoshjabinzadeh, H.; Munzer, P.; Borst, O.; Tian, H.; Di Paolo, G.; Lang, F.; Gawaz, M.; Lindahl, T.L.; et al. A novel role for phospholipase D as an endogenous negative regulator of platelet sensitivity. Cell. Signal. 2012, 24, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, X.M.; Fang, L.; Jennings, N.L.; Su, Y.; Samson, A.L.; Kiriazis, H.; Wang, X.F.; Shan, L. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Pachel, C.; Mathes, D.; Arias-Loza, A.P.; Heitzmann, W.; Nordbeck, P.; Deppermann, C.; Lorenz, V.; Hofmann, U.; Nieswandt, B.; Frantz, S. Inhibition of Platelet GPVI Protects Against Myocardial Ischemia-Reperfusion Injury. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Baumer, Y.; Leder, C.; Ziegler, M.; Schonberger, T.; Ochmann, C.; Perk, A.; Degen, H.; Schmid-Horch, B.; Elvers, M.; Munch, G.; et al. The recombinant bifunctional protein alphaCD133-GPVI promotes repair of the infarcted myocardium in mice. J. Thromb. Haemost. 2012, 10, 1152–1164. [Google Scholar] [CrossRef]
- Schonberger, T.; Ziegler, M.; Borst, O.; Konrad, I.; Nieswandt, B.; Massberg, S.; Ochmann, C.; Jurgens, T.; Seizer, P.; Langer, H.; et al. The dimeric platelet collagen receptor GPVI-Fc reduces platelet adhesion to activated endothelium and preserves myocardial function after transient ischemia in mice. Am. J. Physiol. Cell Physiol. 2012, 303, C757–C766. [Google Scholar] [CrossRef]
- Ziegler, M.; Elvers, M.; Baumer, Y.; Leder, C.; Ochmann, C.; Schonberger, T.; Jurgens, T.; Geisler, T.; Schlosshauer, B.; Lunov, O.; et al. The bispecific SDF1-GPVI fusion protein preserves myocardial function after transient ischemia in mice. Circulation 2012, 125, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-beta in the infarcted myocardium. J. Thorac. Dis. 2017, 9, S52–S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, T.G.; Chan, R.B.; Tian, H.; Laredo, M.; Shui, G.; Staniszewski, A.; Zhang, H.; Wang, L.; Kim, T.W.; Duff, K.E.; et al. Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J. Neurosci. 2010, 30, 16419–16428. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klose, A.M.; Klier, M.; Gorressen, S.; Elvers, M. Enhanced Integrin Activation of PLD2-Deficient Platelets Accelerates Inflammation after Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 3210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093210
Klose AM, Klier M, Gorressen S, Elvers M. Enhanced Integrin Activation of PLD2-Deficient Platelets Accelerates Inflammation after Myocardial Infarction. International Journal of Molecular Sciences. 2020; 21(9):3210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093210
Chicago/Turabian StyleKlose, Aglaia Maria, Meike Klier, Simone Gorressen, and Margitta Elvers. 2020. "Enhanced Integrin Activation of PLD2-Deficient Platelets Accelerates Inflammation after Myocardial Infarction" International Journal of Molecular Sciences 21, no. 9: 3210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093210