Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration





Abstract
:
1. Oxidative Stress and Neurodegenerative Diseases
1.1. Alzheimer’s Disease
1.2. Parkinson’s Disease
1.3. Amyotrophic Lateral Sclerosis
1.4. Oxidative Damage of Retinal Ganglion Cells
1.5. ROS and Ataxia
2. Mesenchymal Stem Cells Potential in Neuronal Differentiation
3. Mesenchymal Stem Cells Contrast Neuronal Cell Damage Induced by Oxidative stress
3.1. Mesenchymal Stem Cells Modulate Redox State in Alzheimer’s Disease
3.2. Antioxidant Effects of Mesenchymal Stem Cells in Parkinson’s Disease
3.3. Mesenchymal Stem Cells Counteract Oxidative Stress in Other Brain Disorders
4. Conclusions
- -
- enhance antioxidant capacity;
- -
- increase neurotrophin expression;
- -
- inhibit pro-inflammatory cytokine secretion; and
- -
- counteract microglial ROS production.
Funding
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| AD | Alzheimer’s disease |
| ADSC | adipose-derived stem cell |
| AGEs | advanced glycation end products |
| ALS | amyotrophic lateral sclerosis |
| APP | amyloid-protein precursor |
| ARE | antioxidant response element |
| ASCs | adult stem cells |
| BDNF | brain-derived neurotrophic factor |
| BME | β-mercaptoethanol |
| BM-MSCs | bone marrow-derived mesenchymal stem cells |
| CM | conditioned medium |
| CNS | central nervous system |
| CVC | chorionic villi cells |
| DA | dopamine |
| DAT | DA transporter |
| DCFH-DA | 2′-7′dichlorofluorescin diacetate |
| DPSC | dental pulp stem cells |
| e-CSF | embryonic cerebrospinal fluid |
| ESCs | embryonic stem cells |
| FA | Friedreich’s ataxia |
| FSCs | fetal stem cells |
| GSH | glutathione |
| HD | Huntington’s disease |
| HIF-1α | hypoxia inducible factor 1 alpha |
| iPSCs | induced pluripotent stem cells |
| MAP-2 | microtubule-associated protein 2 |
| MDA | malondialdehyde |
| MenSC | menstrual blood-derived endometrial stem cell |
| MSCs | mesenchymal stem cells |
| NeuroD | neuronal differentiation 1 |
| NFTs | neurofibrillary tangles |
| NGF | nerve growth factor |
| Ngn1 | neurogenin 1 |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NOXs | NADPH oxidases |
| NPCs | neural precursor cells |
| NPs | neuronal precursors |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
References
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017. [Google Scholar] [CrossRef]
- Corbett, G.T.; Wang, Z.; Hong, W.; Colom-Cadena, M.; Rose, J.; Liao, M.; Asfaw, A.; Hall, T.C.; Ding, L.; DeSousa, A.; et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020, 139, 503–526. [Google Scholar] [CrossRef] [Green Version]
- Albers, D.S.; Flint Beal, M. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J. Neural Transm. Suppl. 2000, 59, 133–154. [Google Scholar]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, T.; Prata, C.; Marrazzo, P.; Hrelia, S.; Angeloni, C. Natural Compounds as a Strategy to Optimize “In Vitro” Expansion of Stem Cells. Rejuvenation Res. 2019. [Google Scholar] [CrossRef]
- Angeloni, C.; Giusti, L.; Hrelia, S. New neuroprotective perspectives in fighting oxidative stress and improving cellular energy metabolism by oleocanthal. Neural Regen. Res. 2019, 14, 1217–1218. [Google Scholar] [PubMed]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: Reaction pathways and antioxidant protection. Arterioscler. Thrombosis Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Lee, S.J.; Kim, S.G. Molecular mechanism of Nrf2 activation by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, A.; Yamamoto, M. Cis-element architecture of Nrf2-sMaf heterodimer binding sites and its relation to diseases. Arch. Pharmacal Res. 2020, 43, 275–285. [Google Scholar] [CrossRef]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Cerebrovascular and neurological disorders: Protective role of NRF2. Int. J. Mol. Sci. 2019, 20, 3433. [Google Scholar] [CrossRef] [Green Version]
- Nam, L.B.; Keum, Y.S. Binding partners of NRF2: Functions and regulatory mechanisms. Arch. Biochem. Biophys. 2019, 678, 108184. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Tufekci, K.U.; Civi Bayin, E.; Genc, S.; Genc, K. The Nrf2/ARE pathway: A promising target to counteract mitochondrial dysfunction in Parkinson’s disease. Parkinson’s Dis. 2011. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.L.H.; Wu, C.W.; Chao, Y.M.; Hung, C.Y.; Chan, J.Y.H. Impaired Nrf2 regulation of mitochondrial biogenesis in rostral ventrolateral medulla on hypertension induced by systemic inflammation. Free Radic. Biol. Med. 2016, 97, 58–74. [Google Scholar] [CrossRef]
- de Oliveira, M.R.; Brasil, F.B.; Fürstenau, C.R. Nrf2 Mediates the Anti-apoptotic and Anti-inflammatory Effects Induced by Gastrodin in Hydrogen Peroxide-Treated SH-SY5Y Cells. J. Mol. Neurosci. 2019, 69, 115–122. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Disease. ASN Neuro 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xing, S.; Chen, Y.; Liao, Q.; Li, Q.; Liu, Y.; He, S.; Feng, F.; Chen, Y.; Zhang, J.; et al. Reasonably activating Nrf2: A long-term, effective and controllable strategy for neurodegenerative diseases. Eur. J. Med. Chem. 2020, 185, 111862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ashford, J.W. Advances in screening instruments for Alzheimer’s disease. Aging Med. 2019, 2, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalingam, K.B.; Radhakrishnan, A.; Ping, N.S.; Haleagrahara, N. Current Concepts of Neurodegenerative Mechanisms in Alzheimer’s Disease. BioMed. Res. Int. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of alzheimer’s disease. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
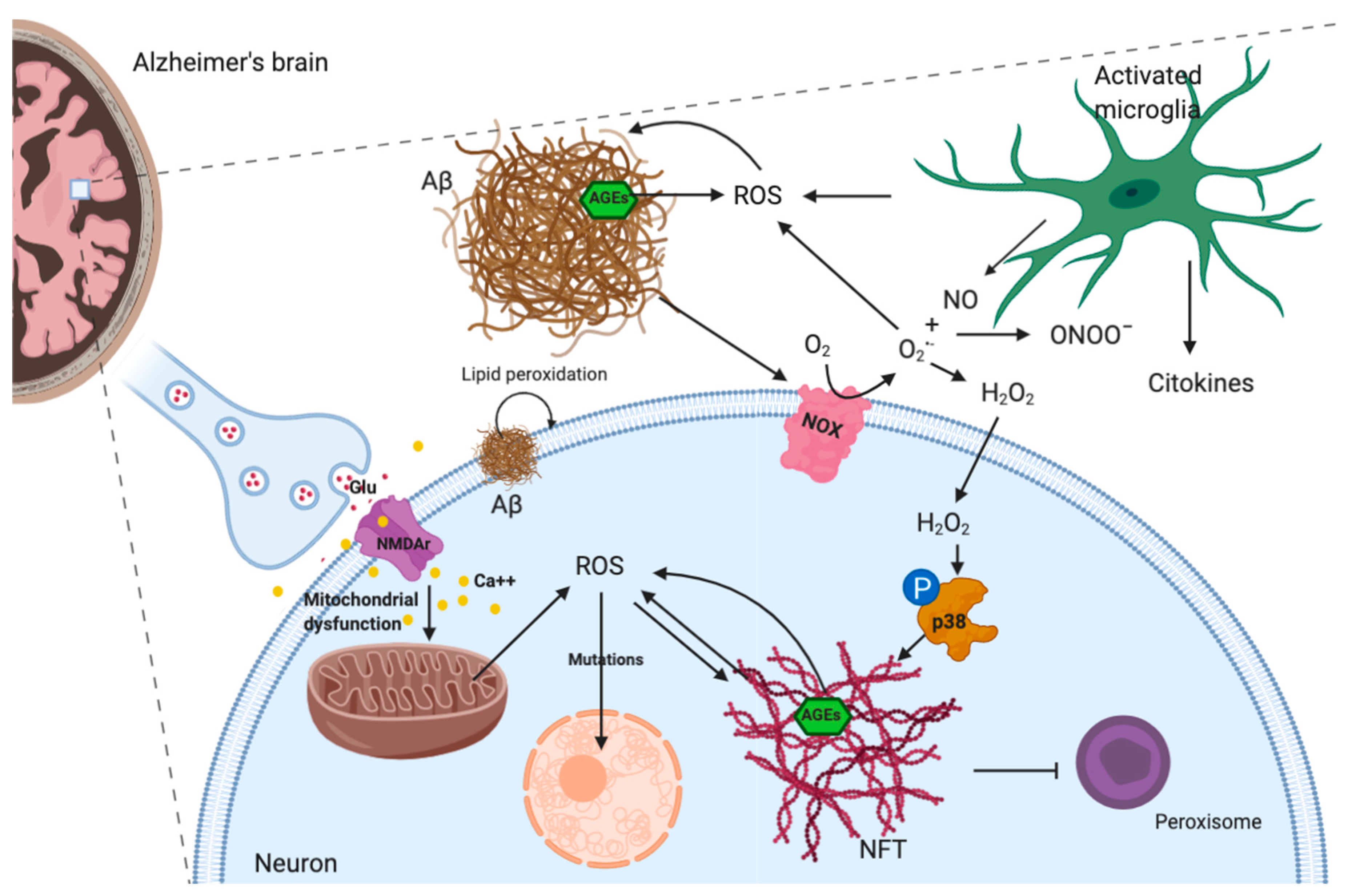
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-Peptide (1-42)-induced oxidative stress in alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M.D. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ramos, A.; Díaz-Nido, J.; Smith, M.A.; Perry, G.; Avila, J. Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells. J. Neurosci. Res. 2003, 71, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [Green Version]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of methylglyoxal in alzheimer’s disease. BioMed. Res. Int. 2014. [Google Scholar] [CrossRef] [Green Version]
- Kubis-Kubiak, A.M.; Rorbach-Dolata, A.; Piwowar, A. Crucial players in Alzheimer’s disease and diabetes mellitus: Friends or foes? Mech. Ageing Dev. 2019, 181, 7–21. [Google Scholar] [CrossRef]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front. Cell. Infect. Microbiol. 2019, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassano, T.; Pace, L.; Bedse, G.; Michele Lavecchia, A.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and Mitochondria: Two Prominent Players in the Oxidative Stress-Induced Neurodegeneration. Curr. Alzheimer Res. 2015, 13, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Spurny, B.; Seiger, R.; Moser, P.; Vanicek, T.; Reed, M.B.; Heckova, E.; Michenthaler, P.; Basaran, A.; Gryglewski, G.; Klöbl, M.; et al. Hippocampal GABA levels correlate with retrieval performance in an associative learning paradigm. NeuroImage 2020, 204, 116244. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Figueroa, C.M.; González-Robles, T.; Delgado, Y.; Molina, A.; Méndez, J.; Morales, M.; Griebenow, K. Activation of caspase-dependent apoptosis by intracellular delivery of cytochrome c-based nanoparticles. J. Nanobiotechnol. 2014, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Vécsei, L. Alzheimer’s Disease: Recent Concepts on the Relation of Mitochondrial Disturbances, Excitotoxicity, Neuroinflammation, and Kynurenines. J. Alzheimer’s Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef] [Green Version]
- Mailly, F.; Marin, P.; Israël, M.; Glowinski, J.; Prémont, J. Increase in external glutamate and NMDA receptor activation contribute to H2O2-induced neuronal apoptosis. J. Neurochem. 1999, 73, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Kish, S.J.; Boileau, I.; Callaghan, R.C.; Tong, J. Brain dopamine neurone ‘damage’: Methamphetamine users vs. Parkinson’s disease-a critical assessment of the evidence. Eur. Neurosci. 2017, 45, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Moustafa, A.A.; Chakravarthy, S.; Phillips, J.R.; Gupta, A.; Keri, S.; Polner, B.; Frank, M.J.; Jahanshahi, M. Motor symptoms in Parkinson’s disease: A unified framework. Neurosci. Biobehav. Rev. 2016, 68, 727–740. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Yu, S.; Zheng, Y.; Yang, H.; Zhang, J. Oxidative Modification and Its Implications for the Neurodegeneration of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 1404–1418. [Google Scholar] [CrossRef]
- Fahn, S.; Cohen, G. The oxidant stress hypothesis in Parkinson’s disease: Evidence supporting it. Ann. Neurol. 1992, 32, 804–812. [Google Scholar] [CrossRef]
- Ishibashi, K.; Ishii, K.; Oda, K.; Kawasaki, K.; Mizusawa, H.; Ishiwata, K. Regional analysis of age-related decline in dopamine transporters and dopamine D2-like receptors in human striatum. Synapse 2009, 63, 282–290. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Mochizuki, H.; Choong, C.J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm. 2020, 127, 181–187. [Google Scholar] [CrossRef]
- Vila, M.; Laguna, A.; Carballo-Carbajal, I. Intracellular crowding by age-dependent neuromelanin accumulation disrupts neuronal proteostasis and triggers Parkinson disease pathology. Autophagy 2019, 15, 2028–2030. [Google Scholar] [CrossRef] [Green Version]
- Anzai, Y.; Gatenby, C.; Friend, S.; Maravilla, K.R.; Hu, S.C.; Cain, K.C.; Agarwal, P. Brain iron concentrations in regions of interest and relation with serum iron levels in Parkinson disease. J. Neurol. Sci. 2017, 378, 38–44. [Google Scholar]
- Wang, J.Y.; Zhuang, Q.Q.; Zhu, L.B.; Zhu, H.; Li, T.; Li, R.; Chen, S.F.; Huang, C.P.; Zhang, X.; Zhu, J.H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dexter, D.T.; Wells, F.R.; Lee, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased Nigral Iron Content and Alterations in Other Metal Ions Occurring in Brain in Parkinson’s Disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A.E.; Collingwood, J.F.; Dobson, J.; Love, G.; Perrott, H.R.; Edwardson, J.A.; Elstner, M.; Morris, C.M. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 2007, 68, 1820–1825. [Google Scholar] [CrossRef]
- Song, L.; Cortopassi, G. Mitochondrial complex i defects increase ubiquitin in substantia nigra. Brain Res. 2015, 1594, 82–91. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeynen, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Reale, M.; Pesce, M.; Priyadarshini, M.; A Kamal, M.; Patruno, A. Mitochondria as an Easy Target to Oxidative Stress Events in Parkinson’s Disease. CNS Neurol. Disord.-Drug Target. 2012, 11, 430–438. [Google Scholar] [CrossRef]
- Homma, T.; Fujii, J. Application of Glutathione as Anti-Oxidative and Anti-Aging Drugs. Curr. Drug Metab. 2015, 16, 560–571. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Sullivan, A.M.; Nolan, Y.M. The influence of microglia on the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2009, 89, 277–287. [Google Scholar] [CrossRef]
- Sun, F.; Deng, Y.; Han, X.; Liu, Q.; Zhang, P.; Manzoor, R.; Ma, H. A secret that underlies Parkinson’s disease: The damaging cycle. Neurochem. Int. 2019, 129, 104484. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Lanuza, M.A.; Just-Borràs, L.; Hurtado, E.; Cilleros-Mañé, V.; Tomàs, M.; Garcia, N.; Tomàs, J. The Impact of Kinases in Amyotrophic Lateral Sclerosis at the Neuromuscular Synapse: Insights into BDNF/TrkB and PKC Signaling. Cells 2019, 8, 1578. [Google Scholar] [CrossRef] [Green Version]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; Van Den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Velde, C.V.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef] [Green Version]
- Bozzo, F.; Mirra, A.; Carrì, M.T. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Cozzolino, M.; Carrì, M.T. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Redox (dys)Regulation. Antioxid. Redox Signal. 2018, 29, 15–36. [Google Scholar] [CrossRef]
- Bonafede, R.; Mariotti, R. ALS pathogenesis and therapeutic approaches: The role of mesenchymal stem cells and extracellular vesicles. Front. Cell. Neurosci. 2017, 11, 80. [Google Scholar] [CrossRef]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Casoni, F.; Basso, M.; Massignan, T.; Gianazzail, E.; Cheroni, C.; Salmona, M.; Bendotti, C.; Bonetto, V. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: Possible multifunctional role in the pathogenesis. J. Biol. Chem. 2005, 280, 16295–16304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell. Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Yan, P.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Ince, P.; Stout, N.; Shaw, P.; Slade, J.; Hunziker, W.; Heizmann, C.W.; Baimbridge, K.G. Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol. Appl. Neurobiol. 1993, 19, 291–299. [Google Scholar] [CrossRef]
- Payne, B.A.I.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta-Bioenerg. 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [Green Version]
- Atsumi, T. The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1981, 55, 193–198. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Almer, G.; Guégan, C.; Teismann, P.; Naini, A.; Rosoklija, G.; Hays, A.P.; Chen, C.; Przedborski, S. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann. Neurol. 2001, 49, 176–185. [Google Scholar] [CrossRef]
- Sekizawa, T.; Openshaw, H.; Ohbo, K.; Sugamura, K.; Itoyama, Y.; Niland, J.C. Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: Immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J. Neurol. Sci. 1998, 154, 194–199. [Google Scholar] [CrossRef]
- Elliott, J.L. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Mol. Brain Res. 2001, 95, 172–178. [Google Scholar] [CrossRef]
- Hensley, K.; Abdel-Moaty, H.; Hunter, J.; Mhatre, M.; Mhou, S.; Nguyen, K.; Potapova, T.; Pye, Q.N.; Qi, M.; Rice, H.; et al. Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J. Neuroinflamm. 2006, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Kuhle, J.; Lindberg, R.L.P.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [CrossRef]
- Chiu, I.M.; Phatnani, H.; Kuligowski, M.; Tapia, J.C.; Carrasco, M.A.; Zhang, M.; Maniatis, T.; Carrolla, M.C. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20960–20965. [Google Scholar] [CrossRef] [Green Version]
- Graber, D.J.; Hickey, W.F.; Harris, B.T. Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J. Neuroinflamm. 2010, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Seredenina, T.; Nayernia, Z.; Sorce, S.; Maghzal, G.J.; Filippova, A.; Ling, S.C.; Basset, O.; Plastre, O.; Daali, Y.; Rushing, E.J.; et al. Evaluation of NADPH oxidases as drug targets in a mouse model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2016, 97, 95–108. [Google Scholar] [CrossRef]
- Boillée, S.; Cleveland, D.W. Revisiting oxidative damage in ALS: Microglia, Nox, and mutant SOD1. J. Clin. Investig. 2008, 118, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Sanes, J.R.; Masland, R.H. The types of retinal ganglion cells: Current status and implications for neuronal classification. Annu. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, L.; Zhao, J.; Zhang, H.; Tian, Y.; Liang, H.; Ma, Q. Serum Response Factor Protects Retinal Ganglion Cells Against High-Glucose Damage. J. Mol. Neurosci.: MN 2016, 59, 232–240. [Google Scholar] [CrossRef]
- Kern, T.S.; Barber, A.J. Retinal ganglion cells in diabetes. Proc. J. Phys. 2008, 586, 4401–4408. [Google Scholar] [CrossRef]
- Wang, C.; Ren, Y.-L.; Zhai, J.; Zhou, X.-Y.; Wu, J. Down-regulated LAMA4 inhibits oxidative stress-induced apoptosis of retinal ganglion cells through the MAPK signaling pathway in rats with glaucoma. Cell Cycle (Georget. Tex.) 2019, 18, 932–948. [Google Scholar] [CrossRef]
- Morrone, L.A.; Rombolà, L.; Corasaniti, M.T.; Bagetta, G.; Nucci, C.; Russo, R. Natural compounds and retinal ganglion cell neuroprotection. Prog. Brain Res. 2015, 220, 257–281. [Google Scholar]
- Li, L.; Du, G.; Wang, D.; Zhou, J.; Jiang, G.; Jiang, H. Overexpression of Heme Oxygenase-1 in Mesenchymal Stem Cells Augments Their Protection on Retinal Cells In Vitro and Attenuates Retinal Ischemia/Reperfusion Injury In Vivo against Oxidative Stress. Stem Cells Int. 2017, 2017, 4985323. [Google Scholar] [CrossRef]
- Wu, J.; Wang, R.; Yang, D.; Tang, W.; Chen, Z.; Sun, Q.; Liu, L.; Zang, R. Hydrogen postconditioning promotes survival of rat retinal ganglion cells against ischemia/reperfusion injury through the PI3K/Akt pathway. Biochem. Biophys. Res. Commun. 2018, 495, 2462–2468. [Google Scholar] [CrossRef]
- Himori, N.; Yamamoto, K.; Maruyama, K.; Ryu, M.; Taguchi, K.; Yamamoto, M.; Nakazawa, T. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J. Neurochem. 2013, 127, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Li, X.; Wang, C.J.; Li, P.; Yang, B.; Wang, C.B.; Wang, L.X. Role of NF-E2-related factor 2 in neuroprotective effect of l-carnitine against high glucose-induced oxidative stress in the retinal ganglion cells. Biomed. Pharmacother. 2015, 69, 345–348. [Google Scholar] [CrossRef]
- Cho, H.; Hartsock, M.J.; Xu, Z.; He, M.; Duh, E.J. Monomethyl fumarate promotes Nrf2-dependent neuroprotection in retinal ischemia-reperfusion. J.Neuroinflamm. 2015, 12, 239. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-F.; Zhou, D.-D.; Xie, T.; Hao, J.-L.; Malik, T.H.; Lu, C.-B.; Qi, J.; Pant, O.P.; Lu, C.-W. The Nrf2 Signaling in Retinal Ganglion Cells under Oxidative Stress in Ocular Neurodegenerative Diseases. Int. J. Biol. Sci. 2018, 14, 1090–1098. [Google Scholar] [CrossRef]
- Alsina, D.; Purroy, R.; Ros, J.; Tamarit, J. Iron in Friedreich Ataxia: A Central Role in the Pathophysiology or an Epiphenomenon? Pharmaceuticals (Basel, Switzerland) 2018, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Tamarit, J.; Obis, È.; Ros, J. Oxidative stress and altered lipid metabolism in Friedreich ataxia. Free Radic. Biol. Med. 2016, 100, 138–146. [Google Scholar] [CrossRef]
- Bayot, A.; Santos, R.; Camadro, J.-M.; Rustin, P. Friedreich’s ataxia: The vicious circle hypothesis revisited. BMC Med. 2011, 9, 112. [Google Scholar] [CrossRef] [Green Version]
- Sturm, B.; Bistrich, U.; Schranzhofer, M.; Sarsero, J.P.; Rauen, U.; Scheiber-Mojdehkar, B.; de Groot, H.; Ioannou, P.; Petrat, F. Friedreich’s ataxia, no changes in mitochondrial labile iron in human lymphoblasts and fibroblasts: A decrease in antioxidative capacity? J. Biol. Chem. 2005, 280, 6701–6708. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef]
- Lodi, R.; Rajagopalan, B.; Blamire, A.M.; Cooper, J.M.; Davies, C.H.; Bradley, J.L.; Styles, P.; Schapira, A.H.V. Cardiac energetics are abnormal in Friedreich ataxia patients in the absence of cardiac dysfunction and hypertrophy: An in vivo 31P magnetic resonance spectroscopy study. Cardiovasc. Res. 2001, 52, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.S.; Khdour, O.; Hecht, S.M. Does oxidative stress contribute to the pathology of Friedreich’s ataxia? A radical question. FASEB J. 2010, 24, 2152–2163. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Mikoluc, B.; Pietrucha, B.; Heropolitanska-Pliszka, E.; Pac, M.; Motkowski, R.; Car, H. Oxidative stress, mitochondrial abnormalities and antioxidant defense in Ataxia-telangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biol. 2017, 11, 375–383. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Lloret, A.; Lebel, M.; d’Ischia, M.; Cogger, V.C.; Le Couteur, D.G.; Gadaleta, M.N.; Castello, G.; Pagano, G. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: Ataxia-Telangiectasia, Down Syndrome, Fanconi Anaemia and Werner Syndrome. Biogerontology 2010, 11, 401–419. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Heropolitanska-Pliszka, E.; Pietrucha, B.; Sawicka-Powierza, J.; Bernatowska, E.; Wolska-Kusnierz, B.; Pac, M.; Car, H.; Zalewska, A.; Mikoluc, B. Antioxidant Defense, Redox Homeostasis, and Oxidative Damage in Children With Ataxia Telangiectasia and Nijmegen Breakage Syndrome. Front. Immun. 2019, 10, 2322. [Google Scholar] [CrossRef]
- Hernández, R.; Jiménez-Luna, C.; Perales-Adán, J.; Perazzoli, G.; Melguizo, C.; Prados, J. Differentiation of Human Mesenchymal Stem Cells towards Neuronal Lineage: Clinical Trials in Nervous System Disorders. Biomol. Ther. 2019. [Google Scholar] [CrossRef]
- Baharvand, H.; Ashtiani, S.K.; Taee, A.; Massumi, M.; Valojerdi, M.R.; Yazdi, P.E.; Moradi, S.Z.; Farrokhi, A. Generation of new human embryonic stem cell lines with diploid and triploid karyotypes. Dev. Growth Differ. 2006, 48, 117–128. [Google Scholar] [CrossRef]
- Sierra, R.A.; Hoverter, N.P.; Ramirez, R.N.; Vuong, L.M.; Mortazavi, A.; Merrill, B.J.; Waterman, M.L.; Donovan, P.J. TCF7L1 suppresses primitive streak gene expression to support human embryonic stem cell pluripotency. Development (Cambridge, England) 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Macrin, D.; Joseph, J.P.; Pillai, A.A.; Devi, A. Eminent Sources of Adult Mesenchymal Stem Cells and Their Therapeutic Imminence. Stem Cell Rev. Rep. 2017, 13, 741–756. [Google Scholar] [CrossRef]
- Rami, F.; Beni, S.N.; Kahnamooi, M.M.; Rahimmanesh, I.; Salehi, A.R.; Salehi, R. Recent Advances in Therapeutic Applications of Induced Pluripotent Stem Cells. Cell. Reprogram. 2017, 19, 65–74. [Google Scholar] [CrossRef]
- Youssef, A.A.; Ross, E.G.; Bolli, R.; Pepine, C.J.; Leeper, N.J.; Yang, P.C. The Promise and Challenge of Induced Pluripotent Stem Cells for Cardiovascular Applications. JACC: Basic Transl. Sci. 2016, 1, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Ilic, D.; Polak, J.M. Stem cells in regenerative medicine: Introduction. Br. Med. Bull. 2011, 98, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Cananzi, M.; De Coppi, P. CD117+ amniotic fluid stem cells: State of the art and future perspectives. Organogenesis 2012, 8, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Balbi, C.; Bollini, S. Fetal and perinatal stem cells in cardiac regeneration: Moving forward to the paracrine era. Placenta 2017, 59, 96–106. [Google Scholar] [CrossRef]
- Gurusamy, N.; Alsayari, A.; Rajasingh, S.; Rajasingh, J. Adult Stem Cells for Regenerative Therapy. In Progress in Molecular Biology and Translational Science; Elsevier, B.V.: New York, NY, USA, 2018; Volume 160, pp. 1–22. ISBN 9780128162378. [Google Scholar]
- Behrens, A.; van Deursen, J.M.; Rudolph, K.L.; Schumacher, B. Impact of genomic damage and ageing on stem cell function. Nat. Cell Biol. 2014, 16, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Kopach, O. Monitoring maturation of neural stem cell grafts within a host microenvironment. World J. Stem Cells 2019, 11, 982–989. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef]
- Nogueira, A.B.; Sogayar, M.C.; Colquhoun, A.; Siqueira, S.A.; Nogueira, A.B.; Marchiori, P.E.; Teixeira, M.J. Existence of a potential neurogenic system in the adult human brain. J. Transl. Med. 2014, 12, 75. [Google Scholar] [CrossRef] [Green Version]
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 2018, 22, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, L.; Gardin, C.; Tocco-Tussardi, I.; Epis, R.; Casadei, A.; Vindigni, V.; Mucci, G.; Zavan, B. Potential for neural differentiation of mesenchymal stem cells. Adv. Biochem. Eng./Biotechnol. 2013, 129, 89–115. [Google Scholar]
- Simerman, A.A.; Perone, M.J.; Gimeno, M.L.; Dumesic, D.A.; Chazenbalk, G.D. A mystery unraveled: Nontumorigenic pluripotent stem cells in human adult tissues. Expert Opin. Biol. Ther. 2014, 14, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qu, X.; Zhao, R.C. Clinical applications of mesenchymal stem cells. J. Hematol. Oncol. 2012, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Caprnda, M.; Kubatka, P.; Gazdikova, K.; Gasparova, I.; Valentova, V.; Stollarova, N.; La Rocca, G.; Kobyliak, N.; Dragasek, J.; Mozos, I.; et al. Immunomodulatory effects of stem cells: Therapeutic option for neurodegenerative disorders. Biomed. Pharmacother. 2017, 91, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Petrou, P.; Gothelf, Y.; Argov, Z.; Gotkine, M.; Levy, Y.S.; Kassis, I.; Vaknin-Dembinsky, A.; Ben-Hur, T.; Offen, D.; Abramsky, O.; et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis. JAMA Neurol. 2016, 73, 337–344. [Google Scholar] [CrossRef]
- Cho, S.R.; Suh, H.; Yu, J.H.; Kim, H.H.; Seo, J.H.; Seo, C.H. Astroglial activation by an enriched environment after transplantation of mesenchymal stem cells enhances angiogenesis after hypoxic-ischemic brain injury. Int. J. Mol. Sci. 2016, 17, 1550. [Google Scholar] [CrossRef] [Green Version]
- Mesentier-Louro, L.A.; Teixeira-Pinheiro, L.C.; Gubert, F.; Vasques, J.F.; Silva-Junior, A.J.; Chimeli-Ormonde, L.; Nascimento-Dos-Santos, G.; Mendez-Otero, R.; Santiago, M.F. Long-term neuronal survival, regeneration, and transient target reconnection after optic nerve crush and mesenchymal stem cell transplantation. Stem Cell Res. Ther. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Nagahama, H.; Nakazaki, M.; Sasaki, M.; Kataoka-Sasaki, Y.; Namioka, T.; Namioka, A.; Oka, S.; Onodera, R.; Suzuki, J.; Sasaki, Y.; et al. Preservation of interhemispheric cortical connections through corpus callosum following intravenous infusion of mesenchymal stem cells in a rat model of cerebral infarction. Brain Res. 2018, 1695, 37–44. [Google Scholar] [CrossRef]
- Harrell, C.R.; Jankovic, M.G.; Fellabaum, C.; Volarevic, A.; Djonov, V.; Arsenijevic, A.; Volarevic, V. Molecular mechanisms responsible for anti-inflammatory and immunosuppressive effects of mesenchymal stem cell-derived factors. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2019; Volume 1084, pp. 187–206. [Google Scholar]
- Wang, L.; Qing, L.; Liu, H.; Liu, N.; Qiao, J.; Cui, C.; He, T.; Zhao, R.; Liu, F.; Yan, F.; et al. Mesenchymal stromal cells ameliorate oxidative stress-induced islet endothelium apoptosis and functional impairment via Wnt4-β-catenin signaling. Stem Cell Res. Ther. 2017, 8, 188. [Google Scholar] [CrossRef] [Green Version]
- Tomasoni, S.; Longaretti, L.; Rota, C.; Morigi, M.; Conti, S.; Gotti, E.; Capelli, C.; Introna, M.; Remuzzi, G.; Benigni, A. Transfer of growth factor receptor mRNA via exosomes unravels the regenerative effect of mesenchymal stem cells. Stem Cells Dev. 2013, 22, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Sugaya, K.; Vaidya, M. Stem cell therapies for neurodegenerative diseases. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2018; Volume 1056, pp. 61–84. [Google Scholar]
- Gunawardena, T.N.A.; Rahman, M.T.; Abdullah, B.J.J.; Abu Kasim, N.H. Conditioned media derived from mesenchymal stem cell cultures: The next generation for regenerative medicine. J. Tissue Eng. Regen. Med. 2019, 13, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, L.; Jiang, C.; Liu, Y.; George, J.; Ye, H.; Cui, Z. Electrophysiological properties and synaptic function of mesenchymal stem cells during neurogenic differentiation—A mini-review. Int. J. Artif. Organs 2012, 35, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Momin, E.N.; Mohyeldin, A.; Zaidi, H.A.; Vela, G.; Quiñones-Hinojosa, A. Mesenchymal stem cells: New approaches for the treatment of neurological diseases. Curr. Stem Cell Res. Ther. 2010, 5, 326–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gögel, S.; Gubernator, M.; Minger, S.L. Progress and prospects: Stem cells and neurological diseases. Gene Ther. 2011, 18, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Huang, S.; Cheng, B.; Nie, X.; Enhe, J.; Feng, C.; Fu, X. Mesenchymal stem cells: A revolution in therapeutic strategies of age-related diseases. Ageing Res. Rev. 2013, 12, 103–115. [Google Scholar] [CrossRef]
- Hong, H.E.; Kim, O.H.; Kwak, B.J.; Choi, H.J.; Kim, K.H.; Ahn, J.; Kim, S.J. Antioxidant action of hypoxic conditioned media from adipose-derived stem cells in the hepatic injury of expressing higher reactive oxygen species. Ann. Surg. Treat. Res. 2019, 97, 159–167. [Google Scholar] [CrossRef]
- Bellei, B.; Papaccio, F.; Filoni, A.; Caputo, S.; Lopez, G.; Migliano, E.; Picardo, M. Extracellular fraction of adipose tissue as an innovative regenerative approach for vitiligo treatment. Exp. Dermatol. 2019, 28, 695–703. [Google Scholar] [CrossRef]
- Baez-Jurado, E.; Guio-Vega, G.; Hidalgo-Lanussa, O.; González, J.; Echeverria, V.; Ashraf, G.M.; Sahebkar, A.; Barreto, G.E. Mitochondrial Neuroglobin Is Necessary for Protection Induced by Conditioned Medium from Human Adipose-Derived Mesenchymal Stem Cells in Astrocytic Cells Subjected to Scratch and Metabolic Injury. Mol. Neurobiol. 2019, 56, 5167–5187. [Google Scholar] [CrossRef]
- Mawrie, D.; Bhattacharjee, K.; Sharma, A.; Sharma, R.; Bhattacharyya, J.; Bhattacharjee, H.; Deori, N.; Kumar, A.; Jaganathan, B.G. Human orbital adipose tissue-derived mesenchymal stem cells possess neuroectodermal differentiation and repair ability. Cell Tissue Res. 2019, 378, 531–542. [Google Scholar] [CrossRef]
- Wen, C.; Huang, C.; Yang, M.; Fan, C.; Li, Q.; Zhao, J.; Gan, D.; Li, A.; Zhu, L.; Lu, D. The Secretion from Bone Marrow Mesenchymal Stem Cells Pretreated with Berberine Rescues Neurons with Oxidative Damage Through Activation of the Keap1-Nrf2-HO-1 Signaling Pathway. Neurotoxic. Res. 2020. [Google Scholar] [CrossRef]
- Palomares, T.; Cordero, M.; Bruzos-Cidon, C.; Torrecilla, M.; Ugedo, L.; Alonso-Varona, A. The neuroprotective effect of conditioned medium from human adipose-derived mesenchymal stem Cells is impaired by N-acetyl cysteine supplementation. Mol. Neurobiol. 2018, 55, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.P.; Bernardi, A.; Luiz Frozza, R.; Grudzinski, P.B.; Hoppe, J.B.; De Souza, L.F.; Chagastelles, P.; De Souza Wyse, A.T.; Bernard, E.A.; Battastini, A.M.O.; et al. Mesenchymal stem cell-conditioned medium triggers neuroinflammation and reactive species generation in organotypic cultures of rat hippocampus. Stem Cells Dev. 2011, 20, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Qiao, H.; He, D.; Qin, X.; Zhang, Q.; Zhou, Y. Mesenchymal stem cells inhibited the inflammation and oxidative stress in LPS-activated microglial cells through AMPK pathway. Neural Trans. (Vienna, Austria: 1996) 2019, 126, 1589–1597. [Google Scholar] [CrossRef]
- Marrazzo, P.; Angeloni, C.; Freschi, M.; Lorenzini, A.; Prata, C.; Maraldi, T.; Hrelia, S. Combination of epigallocatechin gallate and sulforaphane counteracts in vitro oxidative stress and delays stemness loss of amniotic fluid stem cells. Oxid. Med. Cell. Longev. 2018, 2018, 5263985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Lee, J.K.; Lee, H.; Shin, J.W.; Carter, J.E.; Sakamoto, T.; Jin, H.K.; Bae, J.S. The therapeutic potential of human umbilical cord blood-derived mesenchymal stem cells in Alzheimer’s disease. J. Neurosci. Lett. 2010, 481, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yue, C.; Yang, H.; Xie, Z.; Hu, H.; Wei, L.; Wang, P.; Zhao, C.; Bi, J. Intravenous administration of human umbilical cord mesenchymal stem cells improves cognitive impairments and reduces amyloid-beta deposition in an AβPP/PS1 transgenic mouse model. Neurochem. Res. 2013, 38, 2474–2482. [Google Scholar] [CrossRef] [PubMed]
- Bodart-Santos, V.; de Carvalho, L.R.P.; de Godoy, M.A.; Batista, A.F.; Saraiva, L.M.; Lima, L.G.; Abreu, C.A.; De Felice, F.G.; Galina, A.; Mendez-Otero, R.; et al. Extracellular vesicles derived from human Wharton’s jelly mesenchymal stem cells protect hippocampal neurons from oxidative stress and synapse damage induced by amyloid-β oligomers. Stem Cell Res. Ther. 2019, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Yokokawa, K.; Iwahara, N.; Hisahara, S.; Emoto, M.C.; Saito, T.; Suzuki, H.; Manabe, T.; Matsumura, A.; Matsushita, T.; Suzuki, S.; et al. Transplantation of Mesenchymal Stem Cells Improves Amyloid-β Pathology by Modifying Microglial Function and Suppressing Oxidative Stress. J. Alzheimer’s Dis. 2019, 72, 867–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, G., III; Gamez, N.; Armijo, E.; Kramm, C.; Morales, R.; Taylor-Presse, K.; Schulz, P.E.; Soto, C.; Moreno-Gonzalez, I. Peripheral Delivery of Neural Precursor Cells Ameliorates Parkinson’s Disease-Associated Pathology. Cells 2019, 8, 1359. [Google Scholar] [CrossRef] [Green Version]
- Chierchia, A.; Chirico, N.; Boeri, L.; Raimondi, I.; Riva, G.A.; Raimondi, M.T.; Tunesi, M.; Giordano, C.; Forloni, G.; Albani, D. Secretome released from hydrogel-embedded adipose mesenchymal stem cells protects against the Parkinson’s disease related toxin 6-hydroxydopamine. Eur. J. Pharm. Biopharm. 2017, 121, 113–120. [Google Scholar] [CrossRef]
- Calzarossa, C.; Bossolasco, P.; Besana, A.; Manca, M.P.; De Grada, L.; De Coppi, P.; Giardino, D.; Silani, V.; Cova, L. Neurorescue effects and stem properties of chorionic villi and amniotic progenitor cells. Neuroscience 2013, 234, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yahaya, B.H.; Ng, W.H.; Yusoff, N.M.; Lin, J. Conditioned Medium of Human Menstrual Blood-Derived Endometrial Stem Cells Protects Against MPP+-Induced Cytotoxicity in vitro. Front. Mol. Neurosci. 2019, 12, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezquer, M.; Urzua, C.A.; Montecino, S.; Leal, K.; Conget, P.; Ezquer, F. Intravitreal administration of multipotent mesenchymal stromal cells triggers a cytoprotective microenvironment in the retina of diabetic mice. Stem Cell Res. Ther. 2016, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Xu, N.; Xu, W.; Xu, G. Mesenchymal stem cells attenuate hydrogen peroxide-induced oxidative stress and enhance neuroprotective effects in retinal ganglion cells. In Vitro Cell. Dev. Biol.-Anim. 2017, 53, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Oses, C.; Olivares, B.; Ezquer, M.; Acosta, C.; Bosch, P.; Donoso, M.; Léniz, P.; Ezquer, F. Preconditioning of adipose tissue-derived mesenchymal stem cells with deferoxamine increases the production of pro-angiogenic, neuroprotective and anti-inflammatory factors: Potential application in the treatment of diabetic neuropathy. PLoS ONE 2017, 12, e0178011. [Google Scholar] [CrossRef] [PubMed]
- Bonafede, R.; Scambi, I.; Peroni, D.; Potrich, V.; Boschi, F.; Benati, D.; Bonetti, B.; Mariotti, R. Exosome derived from murine adipose-derived stromal cells: Neuroprotective effect on in vitro model of amyotrophic lateral sclerosis. Exp. Cell Res. 2016, 340, 150–158. [Google Scholar] [CrossRef]
- Maguire, G.; Paler, L.; Green, L.; Mella, R.; Valcarcel, M.; Villace, P. Rescue of degenerating neurons and cells by stem cell released molecules: Using a physiological renormalization strategy. Physiolog. Rep. 2019, 7, e14072. [Google Scholar] [CrossRef]
- Jones, J.; Estirado, A.; Redondo, C.; Bueno, C.; Martínez, S. Human adipose stem cell-conditioned medium increases survival of Friedreich’s ataxia cells submitted to oxidative stress. Stem Cells Dev. 2012, 21, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, R.; Bagher, Z.; Kamrava, S.K.; Falah, M.; Ghasemi Hamidabadi, H.; Eskandarian Boroujeni, M.; Mohammadi, F.; Khodaverdi, S.; Zare-Sadeghi, A.; Olya, A.; et al. Differentiation of human mesenchymal stem cells (MSC) to dopaminergic neurons: A comparison between Wharton’s Jelly and olfactory mucosa as sources of MSCs. J. Chem. Neuroanat. 2019, 96, 126–133. [Google Scholar] [CrossRef]
- Faghih, H.; Javeri, A.; Amini, H.; Taha, M.F. Directed differentiation of human adipose tissue-derived stem cells to dopaminergic neurons in low-serum and serum-free conditions. Neurosci. Lett. 2019, 708, 134353. [Google Scholar] [CrossRef]
- Hazeri, Y.; Irani, S.; Zandi, M.; Pezeshki-Modaress, M. Polyvinyl alcohol/sulfated alginate nanofibers induced the neuronal differentiation of human bone marrow stem cells. Int. J. Biol. Macromol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hei, W.-H.; Kim, S.; Park, J.-C.; Seo, Y.-K.; Kim, S.-M.; Jahng, J.W.; Lee, J.-H. Schwann-like cells differentiated from human dental pulp stem cells combined with a pulsed electromagnetic field can improve peripheral nerve regeneration. Bioelectromagnetics 2016, 37, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Chun, S.Y.; Park, J.-S.; Chung, J.-W.; Ha, Y.-S.; Lee, J.N.; Kwon, T.G. Laminin and Platelet-Derived Growth Factor-BB Promote Neuronal Differentiation of Human Urine-Derived Stem Cells. Tissue Eng. Regen. Med. 2018, 15, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mao, X.; Zhou, X.; Su, Y.; Zhou, X.; Shi, K.; Zhao, S. An optimized method for neuronal differentiation of embryonic stem cells in vitro. J. Neurosci. Methods 2019, 330, 108486. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, J.; Pineda, J.R.; Irastorza, I.; Uribe-Etxebarria, V.; García-Gallastegui, P.; Encinas, J.M.; Chamero, P.; Unda, F.; Ibarretxe, G. BDNF and NT3 reprogram human ectomesenchymal dental pulp stem cells to neurogenic and gliogenic neural crest progenitors cultured in serum-free medium. Cell. Physiol. Biochem. 2019, 52, 1361–1380. [Google Scholar]
- Maraldi, T.; Bertoni, L.; Riccio, M.; Zavatti, M.; Carnevale, G.; Resca, E.; Guida, M.; Beretti, F.; La Sala, G.B.; De Pol, A. Human amniotic fluid stem cells: Neural differentiation in vitro and in vivo. Cell Tissue Res. 2014, 357, 1–13. [Google Scholar] [CrossRef]
- Marei, H.E.S.; El-Gamal, A.; Althani, A.; Afifi, N.; Abd-Elmaksoud, A.; Farag, A.; Cenciarelli, C.; Thomas, C.; Anwarul, H. Cholinergic and dopaminergic neuronal differentiation of human adipose tissue derived mesenchymal stem cells. J. Cell. Phys. 2018, 233, 936–945. [Google Scholar] [CrossRef]
- Naghdi, M.; Tiraihi, T.; Mesbah-Namin, S.A.; Arabkheradmand, J. Induction of bone marrow stromal cells into cholinergic-like cells by nerve growth factor. Iran. Biomed. J. 2009, 13, 117–123. [Google Scholar]
- Phonchai, R.; Phermthai, T.; Kitiyanant, N.; Suwanjang, W.; Kotchabhakdi, N.; Chetsawang, B. Potential effects and molecular mechanisms of melatonin on the dopaminergic neuronal differentiation of human amniotic fluid mesenchymal stem cells. Neurochem. Int. 2019, 124, 82–93. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, F.; Song, G.; Sun, X.; Jiang, R.; Chen, M.; Ge, J. Cholinergic neuronal differentiation of bone marrow mesenchymal stem cells in rhesus monkeys. Sci. China Life Sci. 2010, 53, 573–580. [Google Scholar] [CrossRef]
- Santos, J.; Milthorpe, B.K.; Padula, M.P. Proteomic analysis of cyclic ketamine compounds ability to induce neural differentiation in human adult mesenchymal stem cells. Int. J. Mol. Sci. 2019, 20, 523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Kakkar, A.; Sharma, R.; Kharbanda, O.P.; Monga, N.; Kumar, M.; Chowdhary, S.; Airan, B.; Mohanty, S. Synergistic Effect of BDNF and FGF2 in Efficient Generation of Functional Dopaminergic Neurons from human Mesenchymal Stem Cells. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Dai, J.; Li, H.; Li, Y.; Hao, W.; Zhang, Y.; Zhang, Y.; Su, L.; Wei, H. Anti-aging effects exerted by Tetramethylpyrazine enhances self-renewal and neuronal differentiation of rat bMSCs by suppressing NF-kB signaling. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi-Mahdiabadi-Hasani, M.-H.; Nabiuni, M.; Parivar, K.; Yari, S.; Sahebi, A.R.; Miyan, J. The Effects of Embryonic Cerebrospinal Fluid on The Viability and Neuronal Differentiation of Adipose Tissue-Derived Stem Cells in Wistar Rats. Cell J. 2020, 22, 245–252. [Google Scholar] [PubMed]





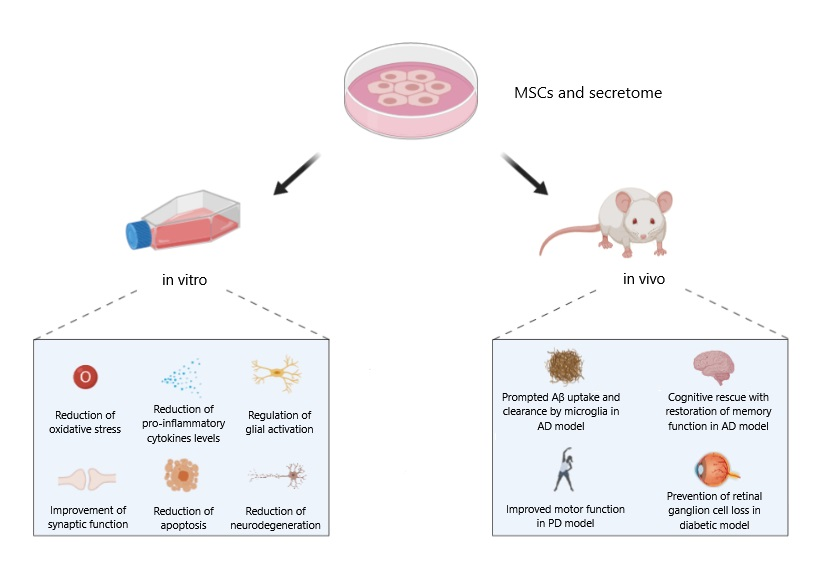{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Neuronal Markers | Types of Neuronal Cells | Types of MSC | Ref. |
|---|---|---|---|---|
| 10% AP-g-CH/GL/AG scaffold + RA, Shh, BDNF, GDNF | Hb-9 Islet-1 ChAT TCP | Motor neuron-like cells | hOE-MSCs | [182] |
| Shh, bFGF, FGF8 and BDNF in free-serum condition | TUJ1 (28–60% of cells) TH NSE EN1 GLI1 NURR1 VMAT2 GIRK2 | Dopaminergic neurons | hADSCs | [183] |
| PVA/SA nanofibers + beta carotene | Morphology MAP-2 | Neural-like cells | hBMSCs | [184] |
| PEMF | Morphology (elongated spindle shape) CD104 S100 GFAP laminin P75NTR | Schwann-like cells | hDPSCs | [185] |
| Lamin and PDGF-BB | Nestin MAP2 β-tubulin III Nerofilament-M | Neuron-like cells | hUSCs | [186] |
| Phase I: trans-RA Phase II: N2B27 | GAPDH Nestin Sox1 β-tubulin III NF200 | Phase I: NPCs Phase II: mature cells of the neural lineage | MESC lines: A2lox and 129 | [187] |
| Serum-free + BDNF + NT-3/neurotrophins | β-tubulin III Nestin Connexin (Cx26, Cx43) Pannexin (Pnx1) NTRK2, NTRK3 NGF, BDNF, NT-3 expression | Central NSC and peripheral neural crest stem cells | hDPSCs | [188] |
| 10% FBS, Retinoic acid | GFAP, β-III tubulin, CNPase, MAP2, NeuN, synapsines, S100, PMP22 | Neuron-like cells, oligodendrocyte-like cells | hAFSC | [189] |
| Heparin mix with FGF | ChAT genes expression β-tubulin III MAP2 Sox1 | Dopaminergic neurons | hADSCs | [190] |
| Retinoic acid mix with EGF, FGF2, BMP-9 | TH genes expression β-tubulin III MAP2 | Cholinergic neurons | hADSCs | [190] |
| BME + NGF | NF-68, NF-160, NF-200 NeuroD ChAT MAP-2 synapsin-1 | Choliergic neurons | mBMSCs | [191] |
| Melatonine | β-tubulin III TH NURR1 GFAPdecrease CD29, CD45, CD73, CD90, CD105decrease DAT | Dopaminergic neurons | hAF-MSCs | [192] |
| SHH + RA | Neuron-like morphology NF-M TuJ-1 vimentin synapsin membrane potential (−70/−65 mV) | Cholinergic neurons | Monkey BMSCs | [193] |
| Cyclic ketamine | Filamin-B Rac1 thioredoxine peroxiredoxin-1 Gelsolin Galectin-3 | Neurons | hADSC | [194] |
| BDNF + FGF2 | MAP2 TH NGN2 PITX3 DAT synaptophysin Kv4.2 SCN5A | Dopaminergic neurons | hDPSCs | [195] |
| TMP | Neuronal morphology MAP-2 NSE Ngn1 NeuroD Mash1 | Neurons | mBMSCs | [196] |
| e-CSF | β-tubulin III neurite outgrowth assay | Neuronal-like cells | m-ADSCs | [197] |
| In Vitro Model | Analyzed Pathways | MSC Sources | Redox Pathway Involved | Ref. |
|---|---|---|---|---|
| Primary hippocampal cultures exposed to Aβ | TUNEL staining DHE staining | hUCB-MSCs co-culture | Reduction of oxidative stress and apoptosis level | [168] |
| (LPS)-activated microglial cells | AMPK pathway TNF-α, IL-1β, IL-6, PEG2, NADPH oxidase activity, ROS, MDA, SOD, GSH-Px | hBM-MSCs co-culture | Inhibitory effect on the pro-inflammatory mediators and oxidative stress by AMPK pathway | [166] |
| Neuroectodermal lineage cells treated with H2O2 | NeuroD and Nestin expression, Bax, Bcl-2 | OAMSCs-CM containing neurotrophic factors | Abrogation of neuronal cell damage induced by oxidative stress | [162] |
| RGC-5 cells (diabetic retinopathy (DR)) | Cell viability IL-1β and TNF-α MDA and antioxidant enzymes (such as SOD) BDNF CNTF | mBMSCs co-culture | Reduction of H2O2-induced inflammatory factor, downregulation of intracellular oxidant factor (MDA) and upregulation of intracellular antioxidant factor (SOD), increase of neurotrophins expression | [177] |
| 6-OHDA-induced PD cell model | Cell viability | hCVC-CM (nestin, connexin 43, VEGF) | Reduction of neurotoxin-induced apoptosis, enhancement of cell-recovery following neuronal damage | [174] |
| NSC-34 cells expressing ALS mutation (in vitro model of ALS) | H2O2-induced cytotoxicity | mADSC-exo | Neuroprotective effect of low concentration of exosomes on cell viability and protection from oxidative damage | [179] |
| FA cells model (periodontal ligament cells from FA patient) | CD44, CD34, CD-90 cleaved caspase-3 Oxidative-stress-related genes (such as frataxin) BDNF | hADSC-CM | Change of transcription levels of oxidative-stress-related genes avoiding cellular degeneration | [181] |
| SH-SY5Y (MPP+ for inducing PD) | Cell viability pro-inflammatory cytokines, mitochondrial membrane potential (Δψm) oxidative stress (DHE assay) cell apoptosis | hMenSC-CM | Increase of cell viability, reduction of MPP+ induced inflammation, Δψm loss, ROS generation and cells numbers in late apoptosis stage | [175] |
| SH-SY5Y (PD) | ROS detection (DCFDA) SOD2 Hsp70 SIRT1 SIRT3 | RAA-MSC-CM | Reduction of mortality and ROS generation Upregulation of the antioxidant enzyme SIRT3 (SIRT1 and Hsp70 unchanged) | [173] |
| SH-SY5Yd exposed to H2O2 | Cell viability assay ROS detection (DCFDA) Morphological analysis | ASC-CM | Moderate reduction of ROS and maintenance of an appropriate redox state to preserve neuronal function | [164] |
| Rat organotypic hippocampal cultures | GFAP Microglial cells detection (isolectin-B4) TNF-α, IL-6 ROS measurement (DCF-DA) iNOS | rBMSC-CM | Induction of glial activation Increase of ROS generation and neuroinflammation | [165] |
| Primary rat hippocampal cultures exposed to AβOs | ROS measurement (DCF-DA) Catalase detection | hMSC-EVs | Protection from oxidative stress and synapse damage induced by AβOs Reduction (via catalase activity) of abnormal ROS production and propagation of astrocyte neurotoxic effects induced by AβOs | [170] |
| Human recombinant TDP-43-tGFP U2OS cell line exposed to sodium arsenite | FUS TDP-43 Cell viability caspase-3 LDH release | hADSC-CM | Inhibition of stress granule number Rescue from glutamate toxicity, reduction of LDH release and caspase 3/7 activity | [180] |
| T98G cells (human astrocytes) subjected to scratch injury | IL-2, IL-6, IL-8, IL-10, TNF-α ROS determination (DHE) membrane potential (Δψm) Ca2+ intracellular GM-CSF AKT/pAKT ERK1/ERK2/pERK Ngb localization | hMSAC-CM | Modulation of localization and expression of neuroglobin Protective effects through regulation of proteins involved in survival pathways and oxidative stress | [161] |
| Rat DGR neurons | HIF-1α, NGF, neuroptophin-3, glial cell-derived neurotrophic factor, IL-4, IL-5, VEGF, angiopoietin 1 | hAD-MSC secretome | DFX (iron chelator deferoxamine) preconditioning significantly increased the total antioxidant capacity of the MSC secretome | [178] |
| In Vivo Model | Analyzed Pathways | MSC Sources | Redox Pathway Involved | Ref. |
|---|---|---|---|---|
| Diabetic retinopathy mouse model | NGF bFGF GDNF | AD-MSC injection (robust expression of SOD1, SOD2, CAT, GPX1) | Reduction of oxidative damage: small reduction in the ROS level but strong reduction in lipid peroxidation levels | [176] |
| PD mouse model (C57BL/6J MPTP-PD induced model) | Behavioral assessment (Wire Hang and Rotarod) GFAP and BLBP-expression Iba-1 | NPs injection obtained from ESCs and MSCs | Amelioration of the motor symptoms Significantly reduction of reactive astroglia and microglia cells Reduction of astroglia and macrophages | [172] |
| AD model mice (APdE9) | MWM test Aβ deposition Iba-1 CD14-positive microglia Aβ1-40 and Aβ1-42 ELISA assay EPR imaging system | MSC injection (tail vein) | Amelioration of oxidative stress through upregulation of microglia expression Promotion of microglial Aβ uptake and clearance Decrease of Aβ-dependent ROS production, thereby improving the redox balance | [171] |
| AβPP/PS1 mouse model of AD | Reduction of β-secretase 1 and CTFβ, and Aβ deposition | hUCB-MSCs vain infusion | Increased GSH and SOD activity. Decreased malondialdehyde activity and protein carbonyl level | [169] |
| C57BL/6J mice (acute Aβ-induced AD model) | MWM test DHE staining TUNEL staining GFAP staining Iba-1 staining | hUCB-MSCs transplant | Recover of impaired memory function Inhibition of oxidative stress and of glial activation that mediates apoptosis | [168] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeloni, C.; Gatti, M.; Prata, C.; Hrelia, S.; Maraldi, T. Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093299
Angeloni C, Gatti M, Prata C, Hrelia S, Maraldi T. Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration. International Journal of Molecular Sciences. 2020; 21(9):3299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093299
Chicago/Turabian StyleAngeloni, Cristina, Martina Gatti, Cecilia Prata, Silvana Hrelia, and Tullia Maraldi. 2020. "Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration" International Journal of Molecular Sciences 21, no. 9: 3299. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093299