Oxidative Phosphorylation Dysfunction Modifies the Cell Secretome

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Phosphorylation System
3. OXPHOS Dysfunction and Disease
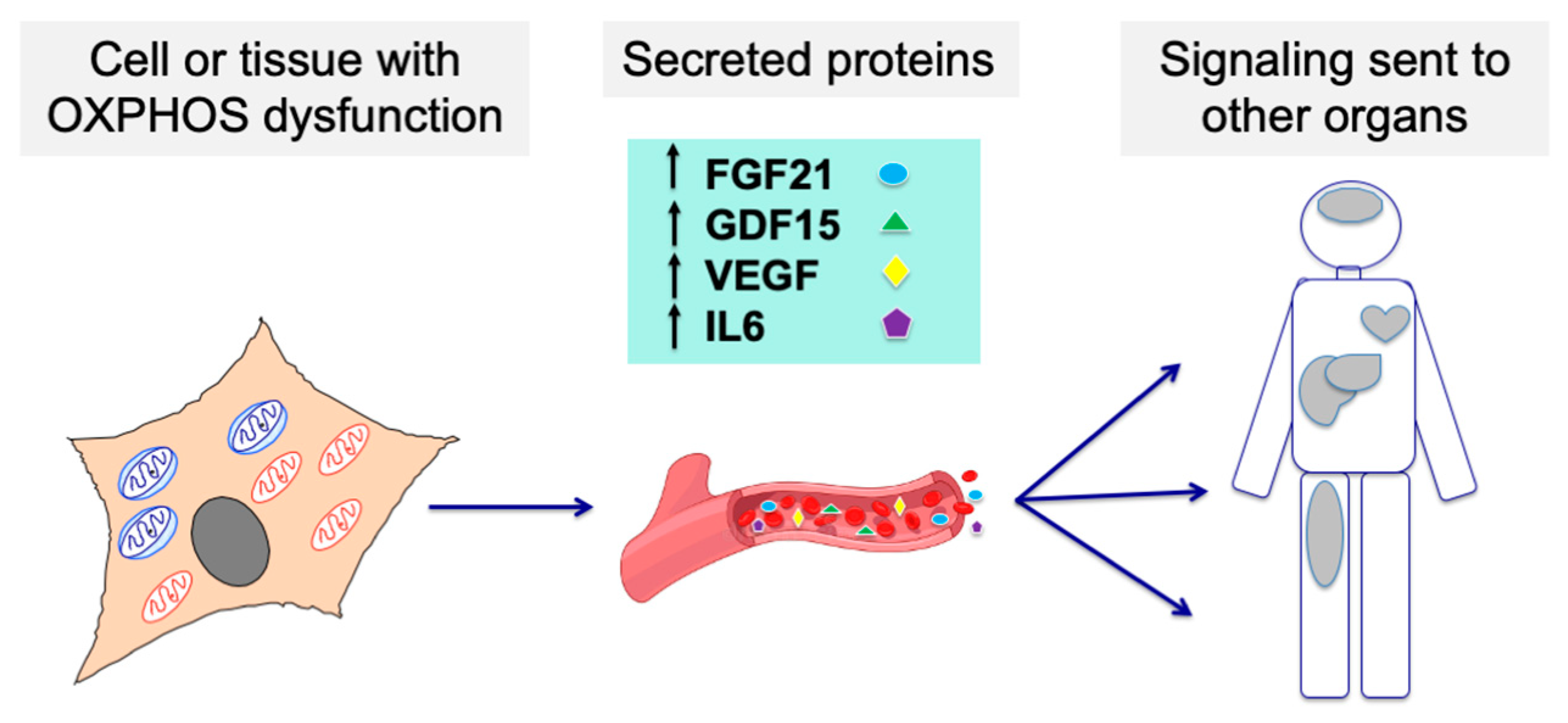
4. OXPHOS Dysfunction Modifies the Protein Secretion by the Cells
5. OXPHOS Dysfunction and Secretomic Studies
6. Biomarkers for OXPHOS Dysfunction
7. Fibroblast Growth Factor 21
8. Growth Differentiation Factor 15
9. Fibroblast Growth Factor 21 vs Growth Differentiation Factor 15
10. Vascular Endothelial Growth Factor
11. Interleukin-6
12. Biosignatures
13. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kapur, S.K.; Katz, A.J. Review of the adipose derived stem cell secretome. Biochimie 2013, 95, 2222–2228. [Google Scholar] [CrossRef]
- Lehr, S.; Hartwig, S.; Lamers, D.; Famulla, S.; Müller, S.; Hanisch, F.G.; Cuvelier, C.; Ruige, J.; Eckardt, K.; Ouwens, D.M.; et al. Identification and validation of novel adipokines released from primary human adipocytes. Mol. Cell. Proteomics 2012, 11, M111.010504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stastna, M.; Van Eyk, J.E. Secreted proteins as a fundamental source for biomarker discovery. Proteomics 2012, 12, 722–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhan, H.; Rabouille, C. Signalling to and from the secretory pathway. J. Cell Sci. 2011, 124, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Kwon, Y.; Joo, J.Y.; Kim, D.G.; Yoon, J.H. Secretomics to discover regulators in diseases. Int. J. Mol. Sci. 2019, 20, 3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjo, S.I.; Manadas, B. A translational view of cells’ secretome analysis - from untargeted proteomics to potential circulating biomarkers. Biochimie 2018, 155, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kim, G.; Kabir, M.H.; Park, S.J.; Lee, S.T.; Lee, C. Use of composite protein database including search result sequences for mass spectrometric analysis of cell secretome. PLoS ONE 2015, 10, e0121692. [Google Scholar] [CrossRef]
- Shin, J.; Rhim, J.; Kwon, Y.; Choi, S.Y.; Shin, S.; Ha, C.W.; Lee, C. Comparative analysis of differentially secreted proteins in serum-free and serum-containing media by using BONCAT and pulsed SILAC. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, S.; Chen, L.; Huang, P.; Tian, R. Proteomic analysis of secreted proteins from cell microenvironment. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1662, pp. 45–58. [Google Scholar]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 1–22. [Google Scholar] [CrossRef]
- Munnich, A.; Rotig, A.; Cormier-Daire, V.; Rustin, P. Clinical Presentation of Respiratory Chain Deficiency Arnold Munnich. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; MacGraw Hill: New York, NY, USA, 2006. [Google Scholar]
- Schoffner, J. Oxidative Phosphorylation Diseases. In The Online Metabolic and Molecular Bases of Inherited Disease; MacGraw Hill: New York, NY, USA, 2006. [Google Scholar]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Scott, B.; Ong, S.; Walford, G.A.; Sugiana, C.; Boneh, A.; William, K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Mootha, V.K. The Mitochondrial Proteome and Human Disease. Annu. Rev. Genomics Hum. Genet. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Suomalainen, A. Biomarkers for mitochondrial respiratory chain disorders. J. Inherit. Metab. Dis. 2011, 34, 277–282. [Google Scholar] [CrossRef]
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef]
- Prigione, A.; Cortopassi, G. Mitochondrial DNA deletions induce the adenosine monophosphate-activated protein kinase energy stress pathway and result in decreased secretion of some proteins. Aging Cell 2007, 6, 619–630. [Google Scholar] [CrossRef]
- Villarroya, J.; Dorado, B.; Vilà, M.R.; Garcia-Arumí, E.; Domingo, P.; Giralt, M.; Hirano, M.; Villarroya, F. Thymidine kinase 2 deficiency-induced mitochondrial dna depletion causes abnormal development of adipose tissues and adipokine levels in mice. PLoS ONE 2011, 6, e29691. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Wang, C.C.; Huang, H.C.; Wei, Y.H. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050. [Google Scholar] [CrossRef]
- Zhang, Y.; Marsboom, G.; Toth, P.T.; Rehman, J. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS ONE 2013, 8, e77077. [Google Scholar] [CrossRef] [Green Version]
- Ryu, M.J.; Kim, S.J.; Choi, M.J.; Kim, Y.K.; Lee, M.J.; Lee, S.E.; Chung, H.K.; Jung, S.B.; Kim, H.-J.; Kim, K.S.; et al. Mitochondrial oxidative phosphorylation reserve is required for hormone- and PPARγ agonist-induced adipogenesis. Mol. Cells 2013, 35, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.G.; Yi, H.S.; Choi, M.J.; Ryu, M.J.; Jung, S.; Chung, H.K.; Chang, J.Y.; Kim, Y.K.; Lee, S.E.; Kim, H.W.; et al. ANGPTL6 expression is coupled with mitochondrial OXPHOS function to regulate adipose FGF21. J. Endocrinol. 2017, 233, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szkudelski, T.; Nogowski, L.; Szkudelska, K. Short-term regulation of adiponectin secretion in rat adipocytes. Physiol. Res. 2011, 60, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Yen, G.C. Effects of capsaicin on induction of apoptosis and inhibition of adipogenesis in 3T3-L1 cells. J. Agric. Food Chem. 2007, 55, 1730–1736. [Google Scholar] [CrossRef]
- Chevillotte, E.; Giralt, M.; Miroux, B.; Ricquier, D.; Villarroya, F. Uncoupling protein-2 controls adiponectin gene expression in adipose tissue through the modulation of reactive oxygen species production. Diabetes 2007, 56, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Stankov, M.; Lücke, T.; Das, A.; Schmidt, R.; Behrens, G.; HIV/AIDS, G.C.N. Relationship of mitochondrial DNA depletion and respiratory chain activity in preadipocytes treated with nucleoside reverse transcriptase inhibitors. Antivir. Ther. 2007, 12, 205–216. [Google Scholar]
- Stankov, M.V.; Schmidt, R.E.; Behrens, G.M.N. Zidovudine impairs adipogenic differentiation through inhibition of clonal expansion. Antimicrob. Agents Chemother. 2008, 52, 2882–2889. [Google Scholar] [CrossRef] [Green Version]
- Stankov, M.V.; Lücke, T.; Das, A.M.; Schmidt, R.E.; Behrens, G.M.N. Mitochondrial DNA depletion and respiratory chain activity in primary human subcutaneous adipocytes treated with nucleoside analogue reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2010, 54, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Hammond, E.; McKinnon, E.; Nolan, D. Human immunodeficiency virus treatment–induced adipose tissue pathology and lipoatrophy: Prevalence and metabolic consequences. Clin. Infect. Dis. 2010, 51, 591–599. [Google Scholar] [CrossRef]
- Llobet, L.; Toivonen, J.M.; Montoya, J.; Ruiz-Pesini, E.; López-Gallardo, E. Xenobiotics that affect oxidative phosphorylation alter differentiation of human adipose-derived stem cells at concentrations that are found in human blood. Dis. Model. Mech. 2015, 8, 1441–1455. [Google Scholar] [CrossRef] [Green Version]
- Dryden, M.S. Linezolid pharmacokinetics and pharmacodynamics in clinical treatment. J. Antimicrob. Chemother. 2011, 66, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, M.H.; Fleisher, M.S. Factors involved in the use of organic solvents as precipitating and drying agents of immune sera. J. Gen. Physiol. 1932, 16, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazier, T.P.; Gimble, J.M.; Kheterpal, I.; Rowan, B.G. Impact of low oxygen on the secretome of human adipose-derived stromal/stem cell primary cultures. Biochimie 2013, 95, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Reales-Calderon, J.A.; Corona, F.; Monteoliva, L.; Gil, C.; Martinez, J.L. Quantitative proteomics unravels that the post-transcriptional regulator Crc modulates the generation of vesicles and secreted virulence determinants of Pseudomonas aeruginosa. J. Proteom. 2015, 127, 352–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvonic, S.; Lefevre, M.; Kilroy, G.; Floyd, Z.E.; DeLany, J.P.; Kheterpal, I.; Gravois, A.; Dow, R.; White, A.; Wu, X.; et al. Secretome of primary cultures of human adipose-derived stem cells: Modulation of serpins by adipogenesis. Mol. Cell. Proteomics 2007, 6, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Llobet, L.; Bayona-Bafaluy, M.P.; Pacheu-Grau, D.; Torres-Pérez, E.; Arbones-Mainar, J.M.; Navarro, M.Á.; Gómez-Díaz, C.; Montoya, J.; López-Gallardo, E.; Ruiz-Pesini, E. Pharmacologic concentrations of linezolid modify oxidative phosphorylation function and adipocyte secretome. Redox Biol. 2017, 13, 244–254. [Google Scholar] [CrossRef]
- Passarelli, C.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. GSSG-mediated Complex I defect in isolated cardiac mitochondria. Int. J. Mol. Med. 2010, 25, 95–99. [Google Scholar]
- Meyer, J.G.; Garcia, T.Y.; Schilling, B.; Gibson, B.W.; Lamba, D.A. proteome and secretome dynamics of human retinal pigment epithelium in response to reactive oxygen species. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Boenzi, S.; Diodato, D. Biomarkers for mitochondrial energy metabolism diseases. Essays Biochem. 2018, 62, 443–454. [Google Scholar]
- Kajiyama, M.; Kawamura, I.; Fujita, A.; Hamamoto, K.; Nishi, Y.; Kitano, A.; Matsuda, I.; Ohtani, Y.; Miike, T. A case of mitochondrial encephalomyopathy with cardiomyopathy due to decreased complex I and IV activities. Brain Dev. 1989, 21, 369–373. [Google Scholar]
- Davis, R.L.; Liang, C.; Edema-Hildebrand, F.; Riley, C.; Needham, M.; Sue, C.M. Fibroblast growth factor 21 is a sensitive biomarker of mitochondrial disease. Neurology 2013, 81, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Kalko, S.G.; Paco, S.; Jou, C.; Rodríguez, M.A.; Meznaric, M.; Rogac, M.; Jekovec-Vrhovsek, M.; Sciacco, M.; Moggio, M.; Fagiolari, G.; et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genom. 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Ito, M.; Kojima, T.; Yatsuga, S.; Koga, Y.; Tanaka, M. GDF15 is a novel biomarker to evaluate efficacy of pyruvate therapy for mitochondrial diseases. Mitochondrion 2015, 20, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yatsuga, S.; Fujita, Y.; Ishii, A.; Fukumoto, Y.; Arahata, H.; Kakuma, T.; Kojima, T.; Ito, M.; Tanaka, M.; Saiki, R.; et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 2015, 78, 814–823. [Google Scholar] [CrossRef] [Green Version]
- Steele, H.E.; Horvath, R.; Lyon, J.J.; Chinnery, P.F. Monitoring clinical progression with mitochondrial disease biomarkers. Brain 2017, 140, 2530–2540. [Google Scholar] [CrossRef] [Green Version]
- Morovat, A.; Weerasinghe, G.; Nesbitt, V.; Hofer, M.; Agnew, T.; Quaghebeur, G.; Sergeant, K.; Fratter, C.; Guha, N.; Mirzazadeh, M.; et al. Use of FGF-21 as a biomarker of mitochondrial disease in clinical practice. J. Clin. Med. 2017, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.F.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Tyynismaa, H.; Peltola-Mjösund, K.; Wanrooij, S.; Lappalainen, I.; Ylikallio, E.; Jalanko, A.; Spelbrink, J.N.; Paetau, A.; Suomalainen, A. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17687–17692. [Google Scholar] [CrossRef] [Green Version]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-erkkilä, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjøsund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [Green Version]
- Ahola-Erkkilä, S.; Carroll, C.J.; Peltola-Mjösund, K.; Tulkki, V.; Mattila, I.; Seppänen-Laakso, T.; Orešič, M.; Tyynismaa, H.; Suomalainen, A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 2010, 19, 1974–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, D.R.; Natarajan, T.G.; Jeong, S.Y.; Chen, C.; Park, S.Y.; Huang, H.; Ghosh, M.C.; Tong, W.H.; Haller, R.G.; Wu, C.; et al. Elevated FGF21 secretion, PGC-1α and ketogenic enzyme expression are hallmarks of iron-sulfur cluster depletion in human skeletal muscle. Hum. Mol. Genet. 2014, 23, 24–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtonen, J.M.; Forsström, S.; Bottani, E.; Viscomi, C.; Baris, O.R.; Isoniemi, H.; Höckerstedt, K.; Österlund, P.; Hurme, M.; Jylhävä, J.; et al. FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 2016, 87, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Chinnery, P.F. Mitochondrial DNA polymerase-γ and human disease. Hum. Mol. Genet. 2006, 15, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria (Nature Genetics (2001) 28 (223-231)). Nat. Genet. 2001, 29, 100. [Google Scholar]
- Nishino, I.; Spinazzola, A.; Hirano, M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999, 283, 689–692. [Google Scholar] [CrossRef]
- Baris, O.R.; Ederer, S.; Neuhaus, J.F.G.; von Kleist-Retzow, J.C.; Wunderlich, C.M.; Pal, M.; Wunderlich, F.T.; Peeva, V.; Zsurka, G.; Kunz, W.S.; et al. Mosaic deficiency in mitochondrial oxidative metabolism promotes cardiac arrhythmia during aging. Cell Metab. 2015, 21, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Nakada, K.; Hayashi, J.; Isobe, K. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000, 46, 829–837. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Jin, X.; Hsiao, E.C.; McGrath, S.A.; Esquela, A.F.; Koniaris, L.G. Growth differentiation factor-15/macrophage inhibitory cytokine-1 induction after kidney and lung injury. Shock 2005, 23, 543–548. [Google Scholar]
- Zimmers, T.A.; Jin, X.; Hsiao, E.C.; Perez, E.A.; Pierce, R.H.; Chavin, K.D.; Koniaris, L.G. Growth differentiation factor-15: Induction in liver injury through p53 and tumor necrosis factor-independent mechanisms. J. Surg. Res. 2006, 130, 45–51. [Google Scholar] [CrossRef]
- Strelau, J.; Böttner, M.; Lingor, P.; Suter-Crazzolara, C.; Galter, D.; Jaszai, J.; Sullivan, A.; Schober, A.; Krieglstein, K.; Unsicker, K. GDF-15/MIC-1 a novel member of the TGF-beta superfamily. J. Neural Transm. 2000, 60, 273–276. [Google Scholar]
- Unsicker, K.; Björn, S.; Krieglstein, K. The multiple facets of the TGF-B family cytokine growth/differentiation factor-15/ macrophage inhibitory cytokine-1. Cytokine Growth Facgor Rev. 2013, 24, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Povalko, N.; Inoue, E.; Nashiki, K.; Tanaka, M. Biomarkers and clinical rating scales for sodium pyruvate therapy in patients with mitochondrial disease. Mitochondrion 2019, 48, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Yubero, D.; Villarroya, J.; Henares, D.; Jou, C.; Rodríguez, M.A.; Ramos, F.; Nascimento, A.; Ortez, C.I.; Campistol, J.; et al. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS ONE 2016, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ribas, F.; Villarroya, J.; Elayne, H.; Giralt, M.; Villarroya, F. FGF21 expression and release in muscle cells: Involvement of MyoD and regulation by mitochondria-driven signalling. Biochem. J. 2014, 463, 191–199. [Google Scholar] [CrossRef]
- Koene, S.; De Laat, P.; Van Tienoven, D.H.; Vriens, D.; Brandt, A.M.; Sweep, F.C.G.J.; Rodenburg, R.J.T.; Donders, A.R.T.; Janssen, M.C.H.; Smeitink, J.A.M. Serum FGF21 levels in adult m.3243A>G carriers: Clinical implications. Neurology 2014, 83, 125–133. [Google Scholar] [CrossRef]
- Koene, S.; de Laat, P.; van Tienoven, D.H.; Weijers, G.; Vriens, D.; Sweep, F.C.; Timmermans, J.; Kapusta, L.; Janssen, M.C.; Smeitink, J.A. Serum GDF15 levels correlate to mitochondrial disease severity and myocardial strain, but not to disease progression in adult m.3243A>G carriers. JIMD Rep. 2015, 24, 69–81. [Google Scholar]
- Davis, R.L.; Liang, C.; Sue, C.M. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 2016, 86, 2010–2015. [Google Scholar] [CrossRef]
- Holmes, K.; Roberts, O.L.; Thomas, A.M.; Cross, M.J. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 2007, 19, 2003–2012. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Mourad, J.J.; Plouin, P.F.; Corvol, P.; Rötig, A.; Jeunemaitre, X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am. J. Hum. Genet. 2001, 69, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Kerlan, V.; Plouin, P.F.; Rötig, A.; Jeunemaitre, X. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Jung, H.J.; Jeong, S.H.; Kim, H.K.; Han, J.; Kwon, H.J. A mutation in the mitochondrial protein UQCRB promotes angiogenesis through the generation of mitochondrial reactive oxygen species. Biochem. Biophys. Res. Commun. 2014, 455, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Moon, H.-E.; Lee, M.-S.; Kim, S.-S. Loss of mitochondrial DNA enhances angiogenic and invasive potential of hepatoma cells. Oncol. Rep. 2010, 23, 779–786. [Google Scholar] [PubMed]
- Singh, B.; Schoeb, T.R.; Bajpai, P.; Slominski, A.; Singh, K.K. Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Koshikawa, N.; Iyozumi, A.; Gassmann, M.; Takenaga, K. Constitutive upregulation of hypoxia-inducible factor-1α mRNA occurring in highly metastatic lung carcinoma cells leads to vascular endothelial growth factor overexpression upon hypoxic exposure. Oncogene 2003, 22, 6717–6724. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamagucho, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Carrière, A.; Ebrahimian, T.G.; Dehez, S.; Augé, N.; Joffre, C.; André, M.; Arnal, S.; Duriez, M.; Barreau, C.; Arnaud, E.; et al. Preconditioning by mitochondrial reactive oxygen species improves the proangiogenic potential of adipose-derived cells-based therapy. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Barcia, C.; Bautista, V.; Sánchez-Bahillo, Á.; Fernández-Villalba, E.; Faucheux, B.; Poza, Y.; Poza, M.; Fernandez Barreiro, A.; Hirsch, E.C.; Herrero, M.T. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J. Neural Transm. 2005, 112, 1237–1248. [Google Scholar] [CrossRef]
- Bai, Y.; Zhu, X.; Chao, J.; Zhang, Y.; Qian, C.; Li, P.; Liu, D.; Han, B.; Zhao, L.; Zhang, J.; et al. Pericytes contribute to the disruption of the cerebral endothelial barrier via increasing VEGF expression: Implications for stroke. PLoS ONE 2015, 10, 1–20. [Google Scholar] [CrossRef]
- Longchamp, A.; Mirabella, T.; Arduini, A.; MacArthur, M.R.; Das, A.; Treviño-Villarreal, J.H.; Hine, C.; Ben-Sahra, I.; Knudsen, N.H.; Brace, L.E.; et al. Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell 2018, 173, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Satake, S.; Kuzuya, M.; Miura, H.; Asai, T.; Ramos, M.A.; Muraguchi, M.; Ohmoto, Y.; Iguchi, A. Up-regulation of vascular endothelial growth factor in response to glucose deprivation. Biol. Cell 1998, 90, 161–168. [Google Scholar] [CrossRef]
- Balogh, E.; Veale, D.J.; McGarry, T.; Orr, C.; Szekanecz, Z.; Ng, C.T.; Fearon, U.; Biniecka, M. Oxidative stress impairs energy metabolism in primary cells and synovial tissue of patients with rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 family cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, O.K.; Lora, M.; Cuillerier, A.; Shum-Tim, D.; Hamdy, R.; Burelle, Y.; Servant, M.J.; Stochaj, U.; Colmegna, I. Mitochondrial oxidative stress reduces the immunopotency of mesenchymal stromal cells in adults with coronary artery disease. Circ. Res. 2018, 122, 255–266. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Frink, M.; Kawasaki, T.; Thobe, B.M.; Choudhry, M.A.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Downregulation of TLR4-dependent ATP production is critical for estrogen-mediated immunoprotection in Kupffer cells following trauma-hemorrhage. J. Cell. Physiol. 2007, 211, 364–370. [Google Scholar] [CrossRef]
- Imanishi, H.; Takibuchi, G.; Kobayashi, T.; Ishikawa, K.; Nakada, K.; Mori, M.; Kikkawa, Y.; Takenaga, K.; Toyama-Sorimachi, N.; Hayashi, J.I. Specific mtDNA mutations in mouse carcinoma cells suppress their tumor formation via activation of the host innate immune system. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.V.; Sharief, F.S.; Chan, S.S.L.; Copeland, W.C.; Naviaux, R.K. Molecular diagnosis of Alpers syndrome. J. Hepatol. 2006, 45, 108–116. [Google Scholar] [CrossRef]
- Hasselmann, O.; Blau, N.; Ramaekers, V.T.; Quadros, E.V.; Sequeira, J.M.; Weissert, M. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol. Genet. Metab. 2010, 99, 58–61. [Google Scholar] [CrossRef]
- Semba, R.D.; Sun, K.; Egan, J.M.; Crasto, C.; Carlson, O.D.; Ferrucci, L. Relationship of serum fibroblast growth factor 21 with abnormal glucose metabolism and insulin resistance: The Baltimore longitudinal study of aging. J. Clin. Endocrinol. Metab. 2012, 97, 1375–1382. [Google Scholar] [CrossRef]
- Chow, W.S.; Xu, A.; Woo, Y.C.; Tso, A.W.K.; Cheung, S.C.W.; Fong, C.H.Y.; Tse, H.F.; Chau, M.T.; Cheung, B.M.Y.; Lam, K.S.L. Serum fibroblast growth factor-21 levels are associated with carotid atherosclerosis independent of established cardiovascular risk factors. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2454–2459. [Google Scholar] [CrossRef] [Green Version]
- Lovadi, E.; Csereklyei, M.; Merkli, H.; FüLöp, K.; Sebők, Á.; Karcagi, V.; Komoly, S.; Pál, E. Elevated FGF 21 in myotonic dystrophy type 1 and mitochondrial diseases. Muscle Nerve 2017, 55, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Margáin, A.; Pohlmann, A.; Ryan, P.; Schierwagen, R.; Chi-Cervera, L.A.; Jansen, C.; Mendez-Guerrero, O.; Flores-García, N.C.; Lehmann, J.; Torre, A.; et al. Fibroblast growth factor 21 is an early predictor of acute-on-chronic liver failure in critically Ill cirrhotic patients. Liver Transplant. 2018, 24, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, L.; Magnusson, M.K.; Mavroudis, G.; Polster, A.; Jonefjäll, B.; Törnblom, H.; Sundin, J.; Simrén, M.; Strid, H.; Öhman, L. Systemic inflammatory protein profiles distinguish irritable bowel syndrome (IBS) and ulcerative colitis, irrespective of inflammation or IBS-like symptoms. Inflamm. Bowel Dis. 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Novotny, D.; Malina, P.; Krumpholcova, P.; Tozzi, I. Total adiponectin, adipocyte fatty acid binding protein, fibriblast growth factor 21 and proinflamatory marker levels during the early satge of acute pancretaitis. Clin. Lab. 2015, 61, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Boulogne, M.; Sadoune, M.; Launay, J.M.; Baudet, M.; Cohen-Solal, A.; Logeart, D. Inflammation versus mechanical stretch biomarkers over time in acutely decompensated heart failure with reduced ejection fraction. Int. J. Cardiol. 2017, 226, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Choi, Y.K.; Min, B.K.; Park, K.S.; Seong, K.; Lee, I.K.; Kim, J.G. Relationship between hepcidin and GDF15 in anemic patients with type 2 diabetes without overt renal impairment. Diabetes Res. Clin. Pract. 2015, 109, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Lukaszyk, E.; Lukaszyk, M.; Koc-Zorawska, E.; Bodzenta-Lukaszyk, A.; Malyszko, J. GDF-15, iron, and inflammation in early chronic kidney disease among elderly patients. Int. Urol. Nephrol. 2016, 48, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Burýšek, L.; Houštěk, J. β-Adrenergic stimulation of interleukin-1α and interleukin-6 expression in mouse brown adipocytes. FEBS Lett. 1997, 411, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Verdeguer, F.; Soustek, M.S.; Hatting, M.; Blättler, S.M.; McDonald, D.; Barrow, J.J.; Puigserver, P. Brown Adipose YY1 Deficiency Activates Expression of Secreted Proteins Linked to Energy Expenditure and Prevents Diet-Induced Obesity. Mol. Cell. Biol. 2015, 36, 184–196. [Google Scholar] [CrossRef] [Green Version]
- Campderrós, L.; Moure, R.; Cairó, M.; Gavaldà-Navarro, A.; Quesada-López, T.; Cereijo, R.; Giralt, M.; Villarroya, J.; Villarroya, F. Brown adipocytes secrete GDF15 in response to thermogenic activation. Obesity 2019, 27, 1606–1616. [Google Scholar] [CrossRef]
- Bal, N.C.; Maurya, S.K.; Pani, S.; Sethy, C.; Banerjee, A.; Das, S.; Patnaik, S.; Kundu, C.N. Mild cold induced thermogenesis: Are BAT and skeletal muscle synergistic partners? Biosci. Rep. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, M.A.; Noc, M.; Lang, I.; Holzer, M.; Clemmensen, P.; Jensen, U.; Metzler, B.; Erlinge, D. Proteomics in hypothermia as adjunctive therapy in patients with ST-segment elevation myocardial infarction: A CHILL-MI substudy. Ther. Hypothermia Temp. Manag. 2017, 7, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Oxholm, A.; Oxholm, P.; Avnstorp, C.; Bendtzen, K. Keratinocyte-expression of interleukin-6 but not of tumour necrosis factor-alpha is increased in the allergic and the irritant patch test reaction. Acta Derm Venereol. 1991, 71, 93–98. [Google Scholar] [PubMed]
- Guéniche, A.; Viac, J.; Lizard, G.; Charveron, M.; Schmitt, D. Effect of nickel on the activation state of normal human keratinocytes through interleukin 1 and intercellular adhesion molecule 1 expression. Br. J. Dermatol. 1994, 131, 250–256. [Google Scholar] [CrossRef]
- Lisby, S.; Müller, K.M.; Jongeneel, C.V.; Saurat, J.H.; Hauser, C. Nickel and skin irritants up-regulate tumor necrosis factor-α mRNA in keratinocytes by different but potentially synergistic mechanisms. Int. Immunol. 1995, 7, 343–352. [Google Scholar] [CrossRef]
- Gazel, A.; Rosdy, M.; Tornier, C.; De Fraissinette, A.D.B.; Blumenberg, M. Transcriptional profiling defines the effects of nickel in human epidermal keratinocytes. J. Cell. Physiol. 2008, 217, 686–692. [Google Scholar] [CrossRef]
- Muraresku, C.C.; McCormick, E.M.; Falk, M.J. Mitochondrial disease: Advances in clinical diagnosis, management, therapeutic development, and preventative strategies. Curr. Genet. Med. Rep. 2018, 6, 62–72. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido-Pérez, N.; Vela-Sebastián, A.; López-Gallardo, E.; Emperador, S.; Iglesias, E.; Meade, P.; Jiménez-Mallebrera, C.; Montoya, J.; Bayona-Bafaluy, M.P.; Ruiz-Pesini, E. Oxidative Phosphorylation Dysfunction Modifies the Cell Secretome. Int. J. Mol. Sci. 2020, 21, 3374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093374
Garrido-Pérez N, Vela-Sebastián A, López-Gallardo E, Emperador S, Iglesias E, Meade P, Jiménez-Mallebrera C, Montoya J, Bayona-Bafaluy MP, Ruiz-Pesini E. Oxidative Phosphorylation Dysfunction Modifies the Cell Secretome. International Journal of Molecular Sciences. 2020; 21(9):3374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093374
Chicago/Turabian StyleGarrido-Pérez, Nuria, Ana Vela-Sebastián, Ester López-Gallardo, Sonia Emperador, Eldris Iglesias, Patricia Meade, Cecilia Jiménez-Mallebrera, Julio Montoya, M. Pilar Bayona-Bafaluy, and Eduardo Ruiz-Pesini. 2020. "Oxidative Phosphorylation Dysfunction Modifies the Cell Secretome" International Journal of Molecular Sciences 21, no. 9: 3374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093374