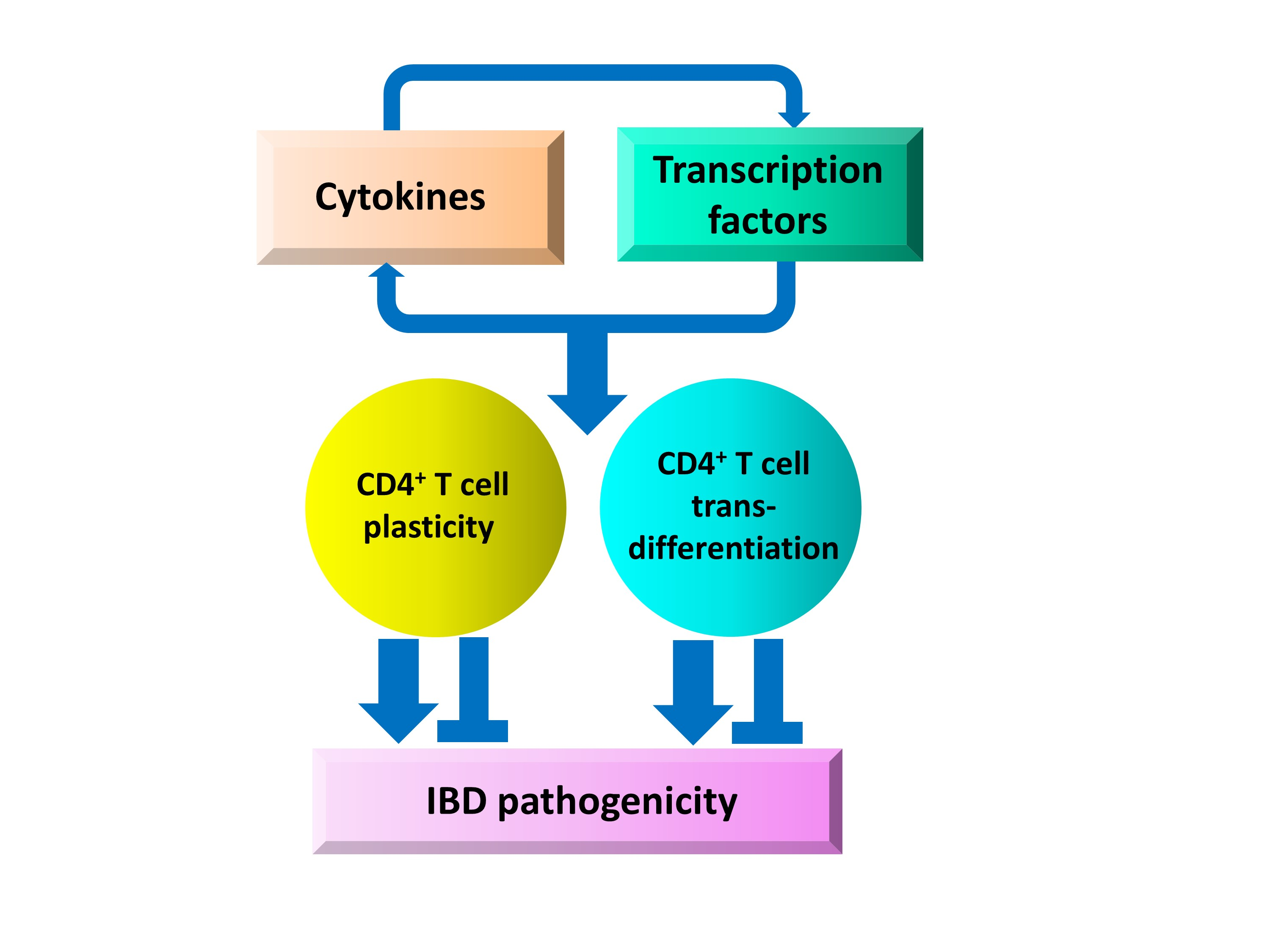
Interplay between Cytokine Circuitry and Transcriptional Regulation Shaping Helper T Cell Pathogenicity and Plasticity in Inflammatory Bowel Disease

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
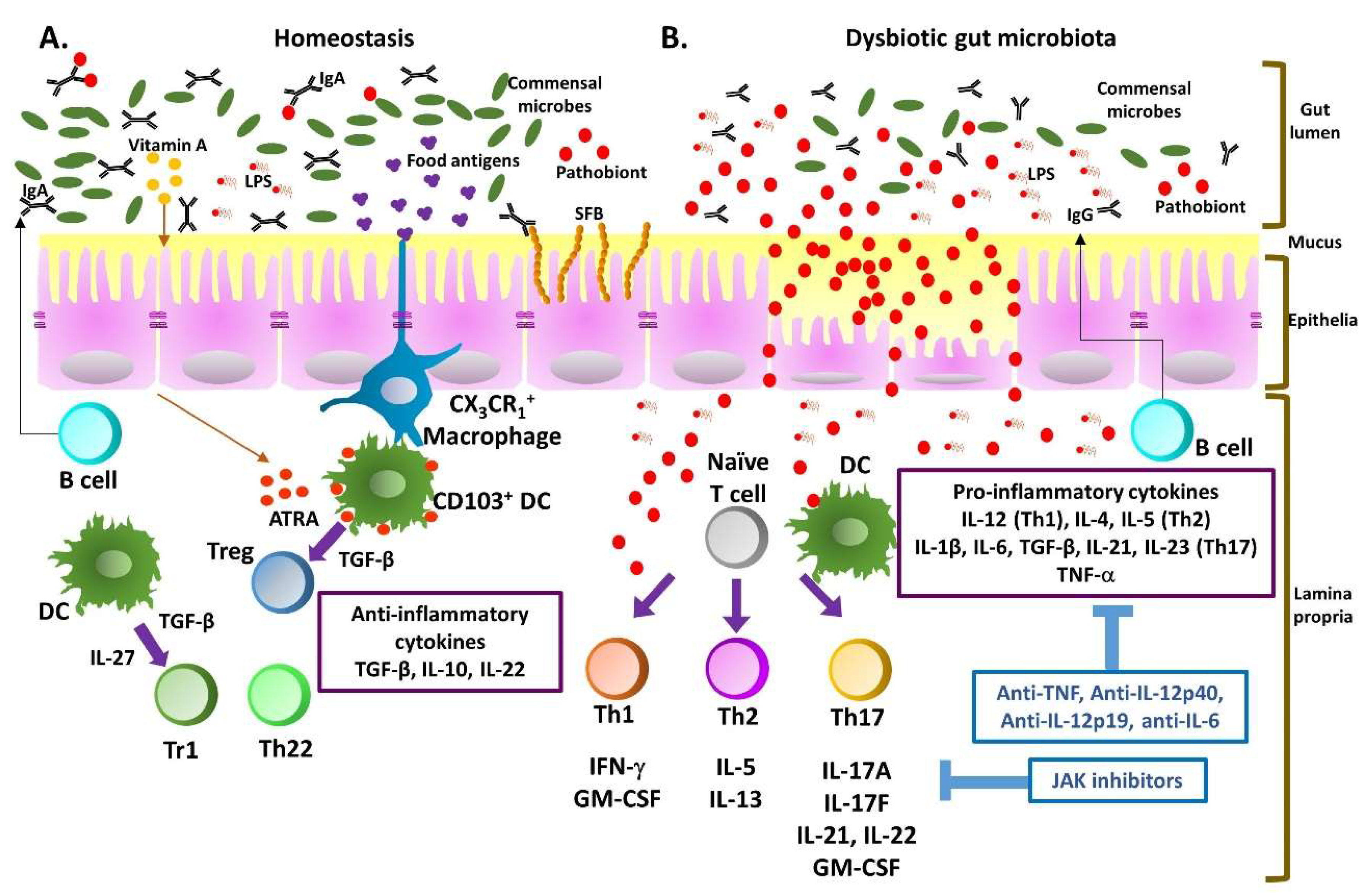
1. Introduction to Inflammatory Bowel Disease
1.1. Genetic Predisposition
1.2. CD4+ T Cells
1.3. Transcriptional Regulation
1.4. Cytokine-Mediated Regulation
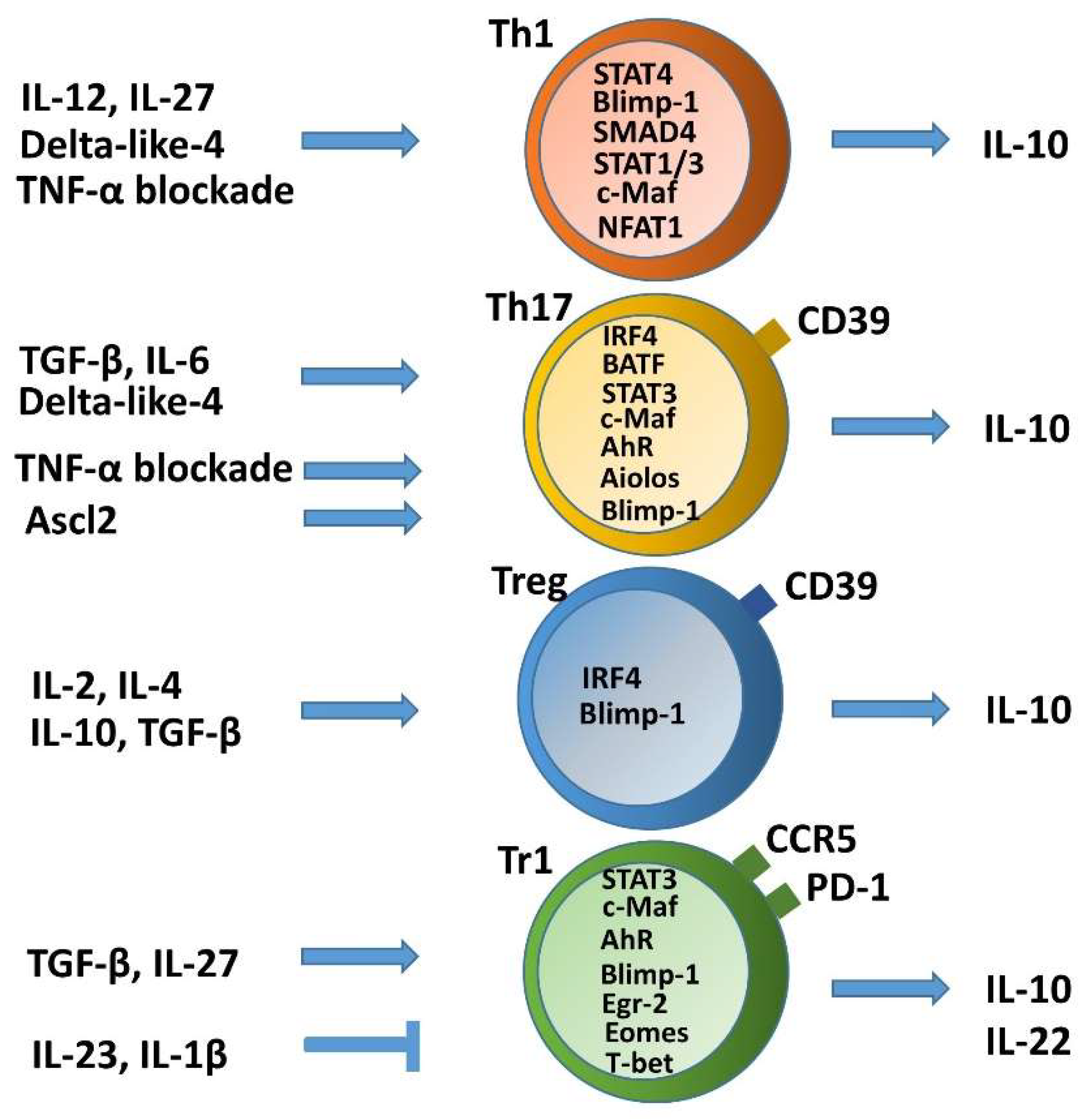
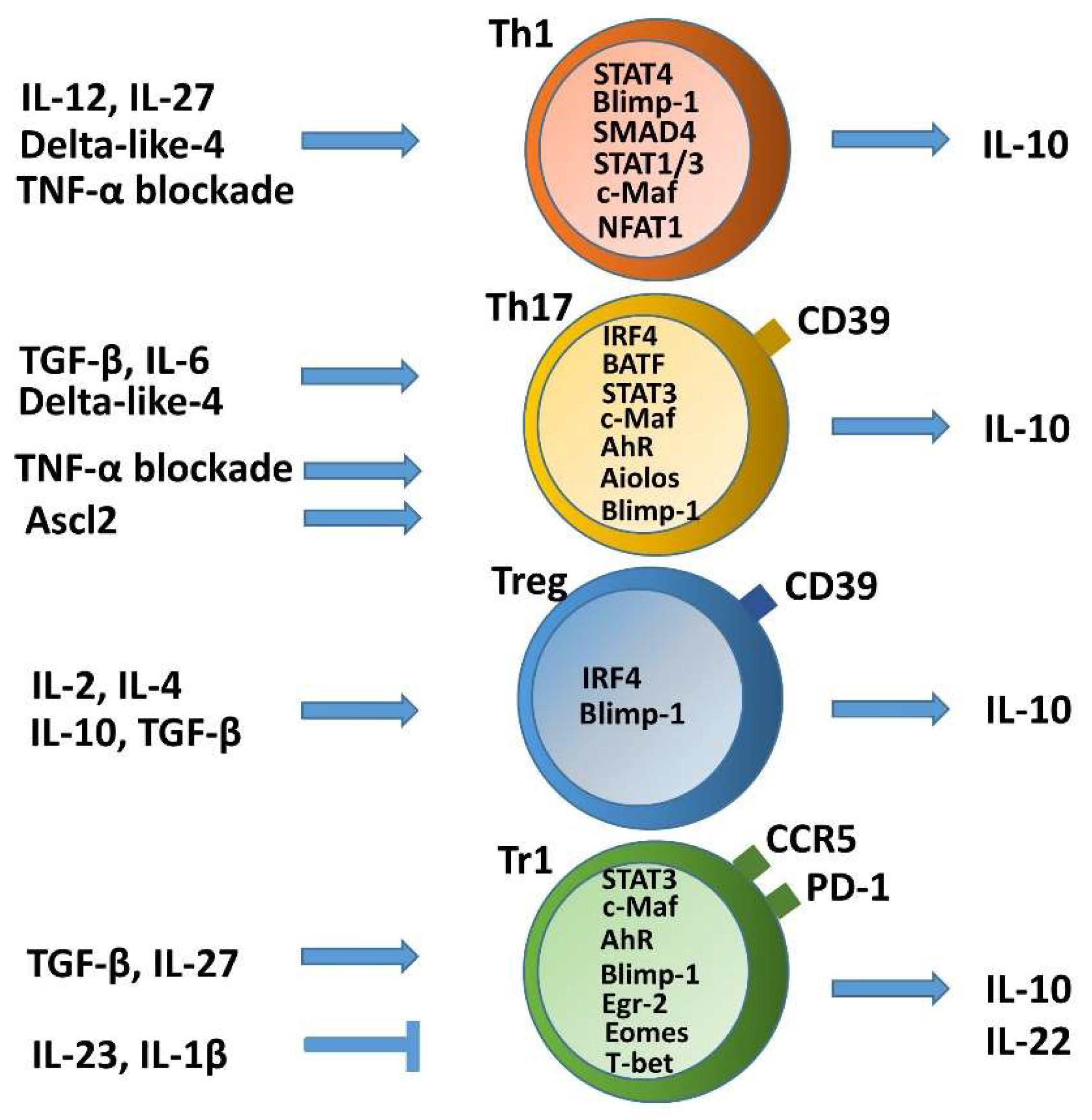
2. Self-Limitation of T Helper Cell Pathogenicity through IL-10
2.1. Non-Pathogenic or Anti-Inflammatory IL-10 Producing Th1 Cells and Plasticity toward Tr1 Cells
2.2. Non-Pathogenic or Anti-Inflammatory IL-10-Producing Th17 Cells
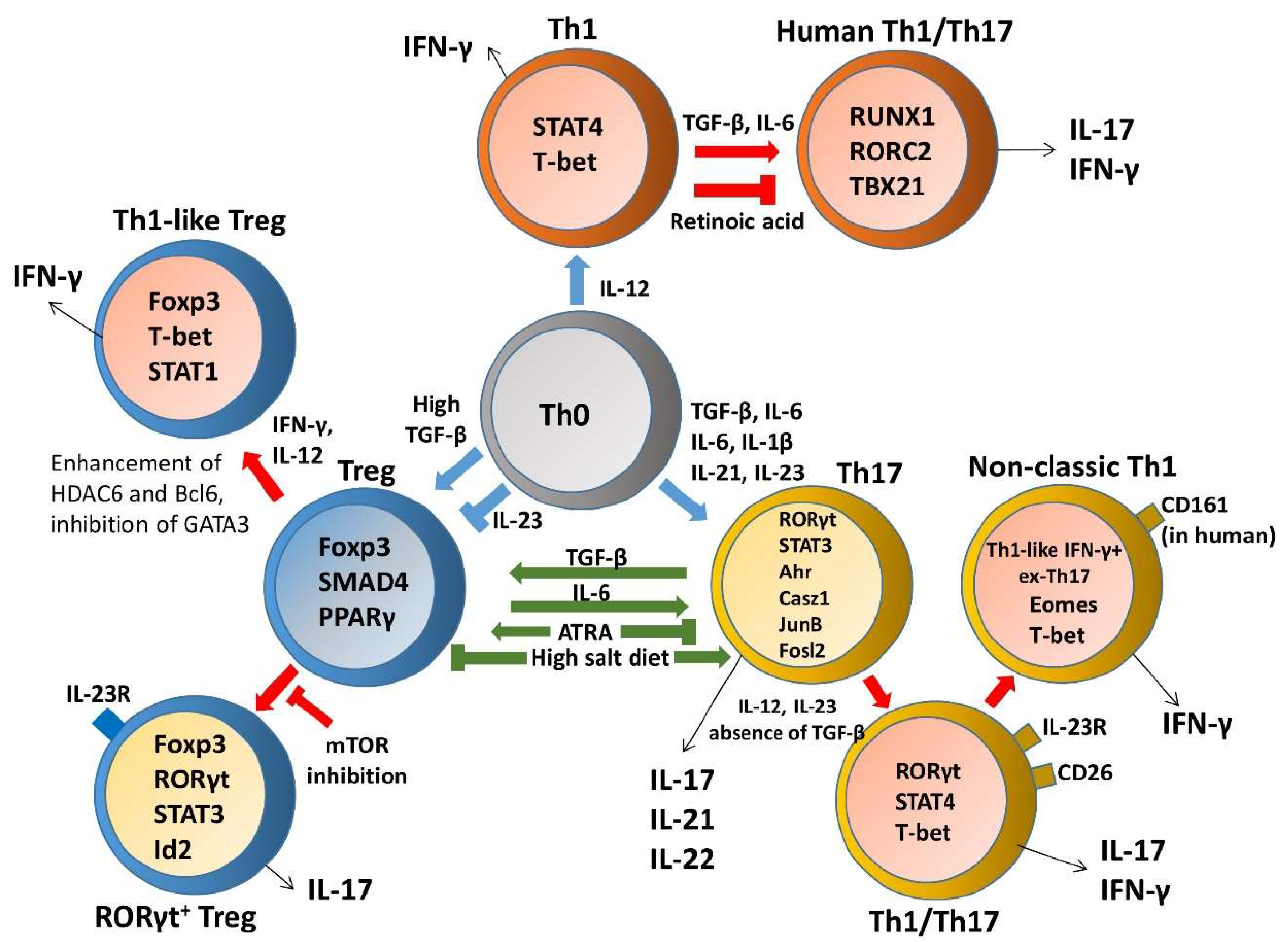
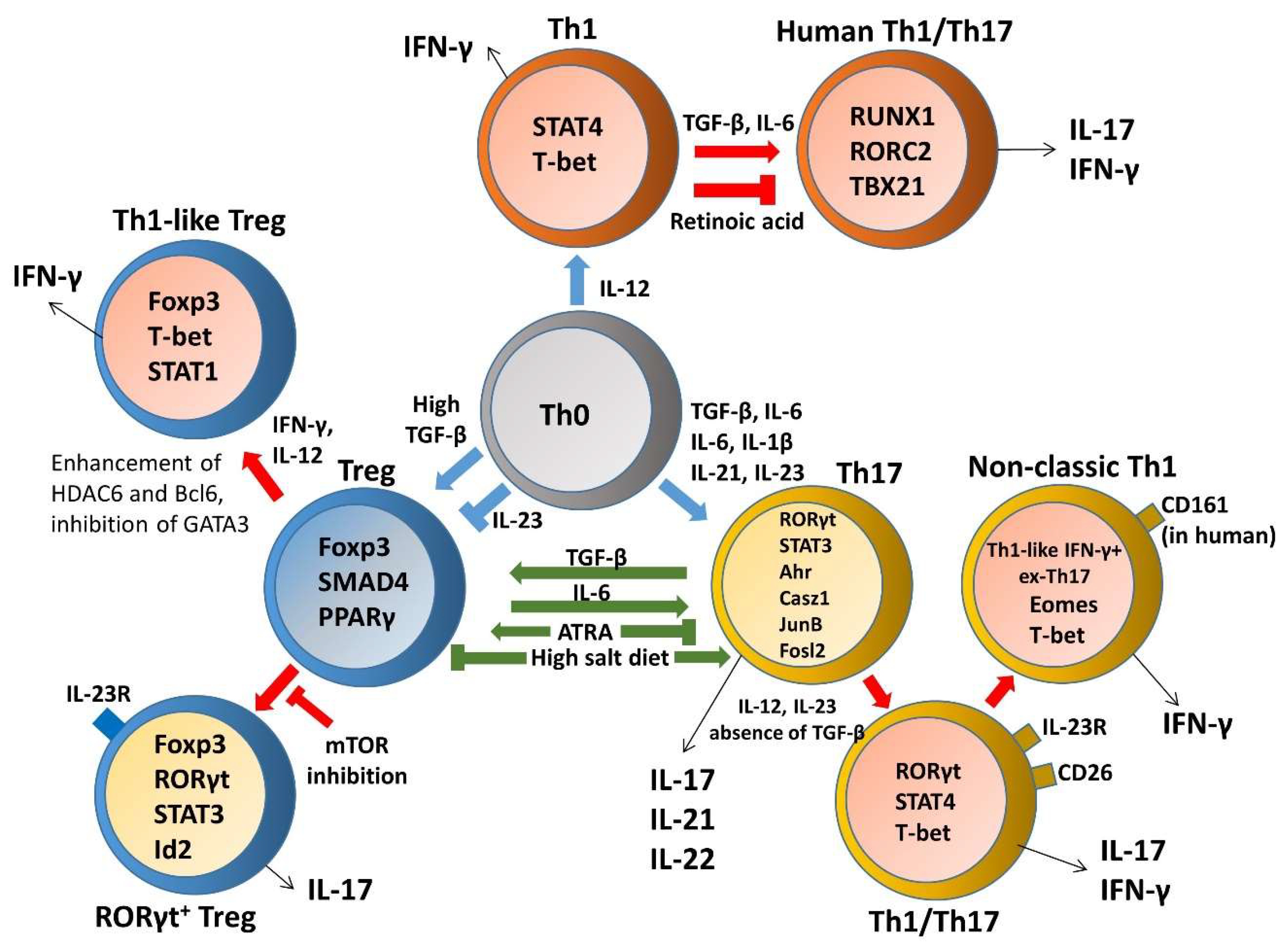
3. Heterogeneity and Plasticity of T Helper Cells in Inflammatory Bowel Disease (IBD)
3.1. Th1 Plasticity
3.2. Th17 and Treg Imbalance Paradigms
3.3. Th17 Plasticity
3.3.1. Th17–Th1 Plasticity and Transdifferentiation
3.3.2. Th17-Treg Plasticity
3.3.3. Th17-Tfh Plasticity
3.4. Treg Plasticity
3.4.1. Treg–Th17 Plasticity and Transdifferentiation
3.4.2. Treg-Th1 Plasticity and Transdifferentiation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef] [PubMed]
- De Vos, M.; De Keyser, F.; Cuvelier, C. The immunological basis of inflammatory bowel disease. Acta Gastroenterol. Belg. 1997, 60, 142–144. [Google Scholar] [PubMed]
- Kaistha, A.; Levine, J. Inflammatory bowel disease: The classic gastrointestinal autoimmune disease. Curr. Probl. Pediatr. Adolesc. Health Care 2014, 44, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Mirkov, M.U.; Verstockt, B.; Cleynen, I. Genetics of inflammatory bowel disease: Beyond NOD2. Lancet Gastroenterol. Hepatol. 2017, 2, 224–234. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Fang, M.; Jostins, L.; Umicevic Mirkov, M.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; Crins, F.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Fowler, S.A.; Ananthakrishnan, A.N.; Gardet, A.; Stevens, C.R.; Korzenik, J.R.; Sands, B.E.; Daly, M.J.; Xavier, R.J.; Yajnik, V. SMAD3 gene variant is a risk factor for recurrent surgery in patients with Crohn’s disease. J. Crohn’s Colitis 2014, 8, 845–851. [Google Scholar] [CrossRef]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Sventoraityte, J.; Nikolaus, S.; Mayr, G.; Domingues, F.S.; Albrecht, M.; Nothnagel, M.; Ellinghaus, D.; et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat. Genet. 2008, 40, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glocker, E.-O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schäffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiocchi, C. Susceptibility genes and overall pathogenesis of inflammatory bowel disease: Where do we stand? Dig. Dis. 2009, 27, 226–235. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Antov, A.; Flavell, R.A. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol. Med. 2009, 15, 199–207. [Google Scholar] [CrossRef]
- McAleer, J.P.; Kolls, J.K. Mechanisms controlling Th17 cytokine expression and host defense. J. Leukoc. Biol. 2011, 90, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Yousaf, W.; Giannella, R.; Shata, M.T. Th17 cells: Interactions with predisposing factors in the immunopathogenesis of inflammatory bowel disease. Expert Rev. Clin. Immunol. 2012, 8, 161–168. [Google Scholar] [CrossRef]
- Kanai, T.; Mikami, Y.; Sujino, T.; Hisamatsu, T.; Hibi, T. RORgammat-dependent IL-17A-producing cells in the pathogenesis of intestinal inflammation. Mucosal Immunol. 2012, 5, 240–247. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellavia, M.; Damiano, G.; Gioviale, M.C.; Palumbo, V.D.; Cacciabaudo, F.; Buscemi, G.; Lo Monte, A.I. Abnormal expansion of segmented filamentous bacteria in the gut: A role in pathogenesis of chronic inflammatory intestinal diseases? Rev. Med. Microbiol. 2011, 22, 45–47. [Google Scholar] [CrossRef]
- Li, J.; Ueno, A.; Fort Gasia, M.; Luider, J.; Wang, T.; Hirota, C.; Jijon, H.B.; Deane, M.; Tom, M.; Chan, R.; et al. Profiles of Lamina Propria T Helper Cell Subsets Discriminate Between Ulcerative Colitis and Crohn’s Disease. Inflamm. Bowel Dis. 2016, 22, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Verdier, J.; Begue, B.; Cerf-Bensussan, N.; Ruemmele, F.M. Compartmentalized expression of Th1 and Th17 cytokines in pediatric inflammatory bowel diseases. Inflamm. Bowel Dis. 2012, 18, 1260–1266. [Google Scholar] [CrossRef]
- Iboshi, Y.; Nakamura, K.; Ihara, E.; Iwasa, T.; Akiho, H.; Harada, N.; Nakamuta, M.; Takayanagi, R. Multigene analysis unveils distinctive expression profiles of helper T-cell-related genes in the intestinal mucosa that discriminate between ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Rovedatti, L.; Kudo, T.; Biancheri, P.; Sarra, M.; Knowles, C.H.; Rampton, D.S.; Corazza, G.R.; Monteleone, G.; Di Sabatino, A.; Macdonald, T.T. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut 2009, 58, 1629–1636. [Google Scholar] [CrossRef]
- Kmiec, Z.; Cyman, M.; Slebioda, T.J. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv. Med. Sci. 2017, 62, 1–16. [Google Scholar] [CrossRef]
- Olsen, T.; Rismo, R.; Cui, G.; Goll, R.; Christiansen, I.; Florholmen, J. TH1 and TH17 interactions in untreated inflamed mucosa of inflammatory bowel disease, and their potential to mediate the inflammation. Cytokine 2011, 56, 633–640. [Google Scholar] [CrossRef]
- Gui, X.; Li, J.; Ueno, A.; Iacucci, M.; Qian, J.; Ghosh, S. Histopathological Features of Inflammatory Bowel Disease are Associated With Different CD4+ T Cell Subsets in Colonic Mucosal Lamina Propria. J. Crohns Colitis 2018, 12, 1448–1458. [Google Scholar] [CrossRef]
- Kanai, T.; Kawamura, T.; Dohi, T.; Makita, S.; Nemoto, Y.; Totsuka, T.; Watanabe, M. TH1/TH2-mediated colitis induced by adoptive transfer of CD4+CD45RBhigh T lymphocytes into nude mice. Inflamm. Bowel Dis. 2006, 12, 89–99. [Google Scholar] [CrossRef]
- He, Y.; Lin, L.J.; Zheng, C.Q.; Jin, Y.; Lin, Y. Cytokine expression and the role of Th17 cells in a mouse model of colitis. Mol. Med. Rep. 2012, 6, 1438–1442. [Google Scholar] [CrossRef] [Green Version]
- Tomasello, G.; Sinagra, E.; Raimondo, D.; Palumbo, V.D.; Puleio, R.; Cottone, M.; Damiani, P.; Traina, G.; Abruzzo, A.; Damiani, F.; et al. Validation of a modified model of TNBS-induced colitis in rats. How to induce a chemical colitis in rats. Acta Biomed. 2015, 86, 92–96. [Google Scholar]
- Alex, P.; Zachos, N.C.; Nguyen, T.; Gonzales, L.; Chen, T.E.; Conklin, L.S.; Centola, M.; Li, X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm. Bowel Dis. 2009, 15, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Allez, M.; Mayer, L. Regulatory T cells: Peace keepers in the gut. Inflamm. Bowel Dis. 2004, 10, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Eri, R.; Lyons, A.B.; Grimm, M.C.; Korner, H. CC Chemokine Ligand 20 and Its Cognate Receptor CCR6 in Mucosal T Cell Immunology and Inflammatory Bowel Disease: Odd Couple or Axis of Evil? Front. Immunol. 2013, 4, 194. [Google Scholar] [CrossRef] [Green Version]
- Igaki, K.; Nakamura, Y.; Komoike, Y.; Uga, K.; Shibata, A.; Ishimura, Y.; Yamasaki, M.; Tsukimi, Y.; Tsuchimori, N. Pharmacological Evaluation of TAK-828F, a Novel Orally Available RORgammat Inverse Agonist, on Murine Colitis Model. Inflammation 2019, 42, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Knosp, C.A.; Johnston, J.A. Regulation of CD4+ T-cell polarization by suppressor of cytokine signalling proteins. Immunology 2012, 135, 101–111. [Google Scholar] [CrossRef]
- Marafini, I.; Angelucci, E.; Pallone, F.; Monteleone, G. The IL-12/23/STAT Axis as a Therapeutic Target in Inflammatory Bowel Disease: Mechanisms and Evidence in Man. Dig. Dis. 2015, 33 (Suppl. 1), 113–119. [Google Scholar] [CrossRef] [Green Version]
- Schmetterer, K.G.; Pickl, W.F. The IL-10/STAT3 axis: Contributions to immune tolerance by thymus and peripherally derived regulatory T-cells. Eur. J. Immunol. 2017, 47, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Roychoudhuri, R.; Hirahara, K.; Mousavi, K.; Clever, D.; Klebanoff, C.A.; Bonelli, M.; Sciumè, G.; Zare, H.; Vahedi, G.; Dema, B.; et al. BACH2 represses effector programs to stabilize Treg-mediated immune homeostasis. Nature 2013, 498, 506–510. [Google Scholar] [CrossRef]
- Evans, C.M.; Jenner, R.G. Transcription factor interplay in T helper cell differentiation. Brief. Funct. Genom. 2013, 12, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoni, A.; Maggi, L.; Siracusa, F.; Ramazzotti, M.; Rossi, M.C.; Santarlasci, V.; Montaini, G.; Capone, M.; Rossettini, B.; De Palma, R.; et al. Eomes controls the development of Th17-derived (non-classic) Th1 cells during chronic inflammation. Eur. J. Immunol. 2019, 49, 79–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruarin, P.; Maglie, S.; De Simone, M.; Haringer, B.; Vasco, C.; Ranzani, V.; Bosotti, R.; Noddings, J.S.; Larghi, P.; Facciotti, F.; et al. Eomesodermin controls a unique differentiation program in human IL-10 and IFN-gamma coproducing regulatory T cells. Eur. J. Immunol. 2019, 49, 96–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessner, M.; Volk, B.A. Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT-PCR). Clin. Exp. Immunol. 1995, 101, 428–435. [Google Scholar] [CrossRef]
- Fantini, M.C.; Monteleone, G.; MacDonald, T.T. IL-21 comes of age as a regulator of effector T cells in the gut. Mucosal Immunol. 2008, 1, 110–115. [Google Scholar] [CrossRef]
- Hirahara, K.; Ghoreschi, K.; Laurence, A.; Yang, X.P.; Kanno, Y.; O’Shea, J.J. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010, 21, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, I.; Pallone, F.; Monteleone, G. Th17-related cytokines: New players in the control of chronic intestinal inflammation. BMC Med. 2011, 9, 122. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Munoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef] [Green Version]
- Mudter, J.; Neurath, M.F. Il-6 signaling in inflammatory bowel disease: Pathophysiological role and clinical relevance. Inflamm. Bowel Dis. 2007, 13, 1016–1023. [Google Scholar] [CrossRef]
- Croxford, A.L.; Kulig, P.; Becher, B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. 2014, 25, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.Y.; Bealgey, K.W.; Fang, Y.; Gong, Y.M.; Bao, S. Interleukin-23: Immunological roles and clinical implications. Int. J. Biochem. Cell Biol. 2009, 41, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J. The Interleukin-23/Interleukin-17 axis in intestinal inflammation. J. Intern. Med. 2008, 263, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Sarra, M.; Pallone, F.; Macdonald, T.T.; Monteleone, G. IL-23/IL-17 axis in IBD. Inflamm. Bowel Dis. 2010, 16, 1808–1813. [Google Scholar] [CrossRef] [PubMed]
- Ahern, P.P.; Izcue, A.; Maloy, K.J.; Powrie, F. The interleukin-23 Axis in Intestinal Inflammation. Immunol. Rev. 2008, 226, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Cho, J.H. IL-23 and autoimmunity: New insights into the pathogenesis of inflammatory bowel disease. Annu. Rev. Med. 2009, 60, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.J.; Kim, K.S.; Song, M.Y.; Seo, S.H.; Kim, S.J.; Yang, B.G.; Jang, M.H.; Sung, Y.C. Delivery of IL-12p40 ameliorates DSS-induced colitis by suppressing IL-17A expression and inflammation in the intestinal mucosa. Clin. Immunol. 2012, 144, 190–199. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Imamura, E.; Taguchi, K.; Sasaki-Iwaoka, H.; Kubo, S.; Furukawa, S.; Morokata, T. Anti-IL-23 receptor monoclonal antibody prevents CD4(+) T cell-mediated colitis in association with decreased systemic Th1 and Th17 responses. Eur. J. Pharmacol. 2018, 824, 163–169. [Google Scholar] [CrossRef]
- Shouval, D.S.; Konnikova, L.; Griffith, A.E.; Wall, S.M.; Biswas, A.; Werner, L.; Nunberg, M.; Kammermeier, J.; Goettel, J.A.; Anand, R.; et al. Enhanced TH17 Responses in Patients with IL10 Receptor Deficiency and Infantile-onset IBD. Inflamm. Bowel Dis. 2017, 23, 1950–1961. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Rennick, D.M.; Fort, M.M. Lessons from genetically engineered animal models. XII. IL-10-deficient (IL-10(-/-) mice and intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G829–G833. [Google Scholar] [CrossRef] [PubMed]
- Leach, M.W.; Davidson, N.J.; Fort, M.M.; Powrie, F.; Rennick, D.M. The Role of IL-10 in Inflammatory Bowel Disease: “Of Mice and Men”. Toxicol. Pathol. 1999, 27, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Kucharzik, T.; Stoll, R.; Lugering, N.; Domschke, W. Circulating antiinflammatory cytokine IL-10 in patients with inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 100, 452–456. [Google Scholar] [CrossRef]
- Schreiber, S.; Heinig, T.; Thiele, H.-G.; Raedler, A. Immunoregulatory Role of Interleukin 10 in Patients with Inflammatory Bowel Disease. Gastroenterology 1995, 108, 10. [Google Scholar] [CrossRef]
- Melgar, S.; Yeung, M.M.; Bas, A.; Forsberg, G.; Suhr, O.; Oberg, A.; Hammarstrom, S.; Danielsson, A.; Hammarstrom, M.L. Over-expression of interleukin 10 in mucosal T cells of patients with active ulcerative colitis. Clin. Exp. Immunol. 2003, 134, 127–137. [Google Scholar] [CrossRef]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson, W.R., Jr.; et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar] [CrossRef]
- Cope, A.; Le Friec, G.; Cardone, J.; Kemper, C. The Th1 life cycle: Molecular control of IFN-gamma to IL-10 switching. Trends Immunol. 2011, 32, 278–286. [Google Scholar] [CrossRef]
- Mizoguchi, A. Healing of intestinal inflammation by IL-22. Inflamm. Bowel Dis. 2012, 18, 1777–1784. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef]
- Leung, J.M.; Davenport, M.; Wolff, M.J.; Wiens, K.E.; Abidi, W.M.; Poles, M.A.; Cho, I.; Ullman, T.; Mayer, L.; Loke, P. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. 2014, 7, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ueno, A.; Iacucci, M.; Fort Gasia, M.; Jijon, H.B.; Panaccione, R.; Kaplan, G.G.; Beck, P.L.; Luider, J.; Barkema, H.W.; et al. Crossover Subsets of CD4(+) T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn’s Disease and Ulcerative Colitis. Dig. Dis. Sci. 2017, 62, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Cheroutre, H. The many face-lifts of CD4 T helper cells. Adv. Immunol. 2010, 107, 139–152. [Google Scholar] [PubMed]
- Zygmunt, B.; Veldhoen, M. T helper cell differentiation more than just cytokines. Adv. Immunol. 2011, 109, 159–196. [Google Scholar]
- Monteleone, I.; Sarra, M.; Pallone, F.; Monteleone, G. Th17-related cytokines in inflammatory bowel diseases: Friends or foes? Curr. Mol. Med. 2012, 12, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; Christensen, J.R.; Veldhoen, M.; Murphy, T.L.; Murphy, K.M.; O’Garra, A. Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity 2009, 31, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.D.; Helbig, C.; Tykocinski, L.; Kreher, S.; Koeck, J.; Niesner, U.; Radbruch, A. Expression of IL-10 in Th memory lymphocytes is conditional on IL-12 or IL-4, unless the IL-10 gene is imprinted by GATA-3. Eur. J. Immunol. 2007, 37, 807–817. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Qiu, G.; Lal, G.; Wu, Z.; Levy, D.E.; Ochando, J.C.; Bromberg, J.S.; Ding, Y. c-Maf regulates IL-10 expression during Th17 polarization. J. Immunol. 2009, 182, 6226–6236. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.; Heinrich, F.; Neumann, K.; Junghans, V.; Mashreghi, M.F.; Ahlers, J.; Janke, M.; Rudolph, C.; Mockel-Tenbrinck, N.; Kuhl, A.A.; et al. Role of Blimp-1 in programing Th effector cells into IL-10 producers. J. Exp. Med. 2014, 211, 1807–1819. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Kitani, A.; Fuss, I.; Nakamura, K.; Kumaki, F.; Usui, T.; Strober, W. Transforming growth factor (TGF)-beta1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-beta1-mediated fibrosis. J. Exp. Med. 2003, 198, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, C.; Blume, J.; Roy, U.; Teh, P.P.; Vasanthakumar, A.; Beller, A.; Liao, Y.; Heinrich, F.; Arenzana, T.L.; Hackney, J.A.; et al. c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat. Immunol. 2019, 20, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Pot, C.; Jin, H.; Awasthi, A.; Liu, S.M.; Lai, C.Y.; Madan, R.; Sharpe, A.H.; Karp, C.L.; Miaw, S.C.; Ho, I.C.; et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 2009, 183, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabryšová, L.; Alvarez-Martinez, M.; Luisier, R.; Cox, L.S.; Sodenkamp, J.; Hosking, C.; Pérez-Mazliah, D.; Whicher, C.; Kannan, Y.; Potempa, K.; et al. c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells. Nat. Immunol. 2018, 19, 497–507. [Google Scholar] [CrossRef]
- Martins, G.A.; Cimmino, L.; Shapiro-Shelef, M.; Szabolcs, M.; Herron, A.; Magnusdottir, E.; Calame, K. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat. Immunol. 2006, 7, 457–465. [Google Scholar] [CrossRef]
- Sun, J.; Dodd, H.; Moser, E.K.; Sharma, R.; Braciale, T.J. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by antiviral CTLs. Nat. Immunol. 2011, 12, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Fujio, K.; Okamura, T.; Yanai, A.; Sumitomo, S.; Shoda, H.; Tamura, T.; Yoshida, H.; Charnay, P.; Yamamoto, K. Egr-2 transcription factor is required for Blimp-1-mediated IL-10 production in IL-27-stimulated CD4+ T cells. Eur. J. Immunol. 2013, 43, 1063–1073. [Google Scholar] [CrossRef]
- Cretney, E.; Xin, A.; Shi, W.; Minnich, M.; Masson, F.; Miasari, M.; Belz, G.T.; Smyth, G.K.; Busslinger, M.; Nutt, S.L.; et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 2011, 12, 304–311. [Google Scholar] [CrossRef]
- Parish, I.A.; Marshall, H.D.; Staron, M.M.; Lang, P.A.; Brustle, A.; Chen, J.H.; Cui, W.; Tsui, Y.C.; Perry, C.; Laidlaw, B.J.; et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J. Clin. Investig. 2014, 124, 3455–3468. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Stumhofer, J.S.; Silver, J.S.; Laurence, A.; Porrett, P.M.; Harris, T.H.; Turka, L.A.; Ernst, M.; Saris, C.J.M.; O’Shea, J.J.; Hunter, C.A. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat. Immunol. 2007, 8, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Kastelein, R. Interleukin-27: Balancing protective and pathological immunity. Immunity 2012, 37, 960–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.; Hibbert, L.; Lieberman, L.; Wilson, E.; Mak, T.; Yoshida, H.; Kastelein, R.A.; Saris, C.; Hunter, C.A. The IL-27R (WSX-1) Is Required to Suppress T Cell Hyperactivity During Infection. Immunity 2003, 19, 10. [Google Scholar] [CrossRef] [Green Version]
- Rutz, S.; Janke, M.; Kassner, N.; Hohnstein, T.; Krueger, M.; Scheffold, A. Notch regulates IL-10 production by T helper 1 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3497–3502. [Google Scholar] [CrossRef] [Green Version]
- Kassner, N.; Krueger, M.; Yagita, H.; Dzionek, A.; Hutloff, A.; Kroczek, R.; Scheffold, A.; Rutz, S. Cutting edge: Plasmacytoid dendritic cells induce IL-10 production in T cells via the Delta-like-4/Notch axis. J. Immunol. 2010, 184, 550–554. [Google Scholar] [CrossRef]
- Zhang, P.; Lee, J.S.; Gartlan, K.H.; Schuster, I.S.; Comerford, I.; Varelias, A.; Ullah, M.A.; Vuckovic, S.; Koyama, M.; Kuns, R.D.; et al. Eomesodermin promotes the development of type 1 regulatory T (TR1) cells. Sci. Immunol. 2017, 2, eaah7152. [Google Scholar] [CrossRef] [Green Version]
- Boks, M.A.; Kager-Groenland, J.R.; van Ham, S.M.; ten Brinke, A. IL-10/IFNgamma co-expressing CD4(+) T cells induced by IL-10 DC display a regulatory gene profile and downmodulate T cell responses. Clin. Immunol. 2016, 162, 91–99. [Google Scholar] [CrossRef]
- Alfen, J.S.; Larghi, P.; Facciotti, F.; Gagliani, N.; Bosotti, R.; Paroni, M.; Maglie, S.; Gruarin, P.; Vasco, C.M.; Ranzani, V.; et al. Intestinal IFN-gamma-producing type 1 regulatory T cells coexpress CCR5 and programmed cell death protein 1 and downregulate IL-10 in the inflamed guts of patients with inflammatory bowel disease. J. Allergy Clin. Immunol. 2018, 142, 1537–1547. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.; Stahl, M.; Han, X.; Nazli, A.; MacDonald, K.N.; Wong, M.Q.; Tsai, K.; Dizzell, S.; Jacobson, K.; Bressler, B.; et al. Suppressive and Gut-Reparative Functions of Human Type 1 T Regulatory Cells. Gastroenterology 2019, 157, 1584–1598. [Google Scholar] [CrossRef] [Green Version]
- Vitale, A.; Strisciuglio, C.; Vitale, S.; Santopaolo, M.; Bruzzese, D.; Micillo, T.; Scarpato, E.; Miele, E.; Staiano, A.; Troncone, R.; et al. Increased frequency of regulatory T cells in pediatric inflammatory bowel disease at diagnosis: A compensative role? Pediatr. Res. 2019, 87, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Sasaoka, T.; Ito, M.; Yamashita, J.; Nakajima, K.; Tanaka, I.; Narita, M.; Hara, Y.; Hada, K.; Takahashi, M.; Ohno, Y.; et al. Treatment with IL-27 attenuates experimental colitis through the suppression of the development of IL-17-producing T helper cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G568–G576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, H.G.; Roostalu, U.; Walter, G.J.; Gullick, N.J.; Frederiksen, K.S.; Roberts, C.A.; Sumner, J.; Baeten, D.L.; Gerwien, J.G.; Cope, A.P.; et al. TNF-α blockade induces IL-10 expression in human CD4+ T cells. Nat. Commun. 2014, 5, 3199. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.A.; Durham, L.E.; Fleskens, V.; Evans, H.G.; Taams, L.S. TNF Blockade Maintains an IL-10(+) Phenotype in Human Effector CD4(+) and CD8(+) T Cells. Front. Immunol. 2017, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.H.; Lin, M.H.; Yeh, L.T.; Wang, Y.L.; Chien, M.W.; Lin, S.H.; Chang, D.M.; Sytwu, H.K. Targeting tumour necrosis factor receptor 1 assembly reverses Th17-mediated colitis through boosting a Th2 response. Gut 2015, 64, 765–775. [Google Scholar] [CrossRef]
- Sundrud, M.S.; Trivigno, C. Identity crisis of Th17 cells: Many forms, many functions, many questions. Semin. Immunol. 2013, 25, 263–272. [Google Scholar] [CrossRef]
- Nikoopour, E.; Schwartz, J.A.; Singh, B. Therapeutic benefits of regulating inflammation in autoimmunity. Inflamm. Allergy Drug Targets 2008, 7, 203–210. [Google Scholar] [CrossRef]
- Aschenbrenner, D.; Foglierini, M.; Jarrossay, D.; Hu, D.; Weiner, H.L.; Kuchroo, V.K.; Lanzavecchia, A.; Notarbartolo, S.; Sallusto, F. An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nat. Immunol. 2018, 19, 1126–1136. [Google Scholar] [CrossRef]
- Fernández, D.; Flores-Santibáñez, F.; Neira, J.; Osorio-Barrios, F.; Tejón, G.; Nuñez, S.; Hidalgo, Y.; Fuenzalida, M.J.; Meza, D.; Ureta, G.; et al. Purinergic Signaling as a Regulator of Th17 Cell Plasticity. PLoS ONE 2016, 11, e0157889. [Google Scholar] [CrossRef]
- Yi, Q.; Wang, J.; Song, Y.; Guo, Z.; Lei, S.; Yang, X.; Li, L.; Gao, C.; Zhou, Z. Ascl2 facilitates IL-10 production in Th17 cells to restrain their pathogenicity in inflammatory bowel disease. Biochem. Biophys. Res. Commun. 2019, 510, 435–441. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, J.; Chen, Y.; Zhou, Z.; Gao, C.; Guo, Z. Sauchinone ameliorates intestinal inflammation and promotes Th17 cell production of IL-10 via Blimp-1. Biochem. Biophys. Res. Commun. 2020, 522, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.T.; Gao, Y.; Lazarevic, V. Transcriptional regulation of CD4+ TH cells that mediate tissue inflammation. J. Leukoc. Biol. 2018, 104, 1069–1085. [Google Scholar] [CrossRef] [PubMed]
- Rowell, E.; Wilson, C.B. Programming perpetual T helper cell plasticity. Immunity 2009, 30, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.Y.; Watford, W.T.; et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the T helper 17 lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [Green Version]
- Yomogida, K.; Chou, Y.K.; Chu, C.Q. Superantigens induce IL-17 production from polarized Th1 clones. Cytokine 2013, 63, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Cohen, C.J.; Crome, S.Q.; MacDonald, K.G.; Dai, E.L.; Mager, D.L.; Levings, M.K. Human Th1 and Th17 cells exhibit epigenetic stability at signature cytokine and transcription factor loci. J. Immunol. 2011, 187, 5615–5626. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.C.; Esterhazy, D.; Sarde, A.; London, M.; Pullabhatla, V.; Osma-Garcia, I.; Al-Bader, R.; Ortiz, C.; Elgueta, R.; Arno, M.; et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015, 42, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.P.; Cao, A.T.; Feng, T.; Li, Q.; Zhang, W.; Yao, S.; Dann, S.M.; Elson, C.O.; Cong, Y. TGF-beta converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur. J. Immunol. 2015, 45, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, K.; Sadhukhan, S.; Han, S.-S.; Vyas, Y.M. SUMOylation-disrupting WAS mutation converts WASp from a transcriptional activator to a repressor of NF-κB response genes in T cells. Blood 2015, 126, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Caretto, D.; Katzman, S.D.; Villarino, A.V.; Gallo, E.; Abbas, A.K. Cutting edge: The Th1 response inhibits the generation of peripheral regulatory T cells. J. Immunol. 2010, 184, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Venigalla, R.K.; Guttikonda, P.J.; Eckstein, V.; Ho, A.D.; Sertel, S.; Lorenz, H.M.; Tretter, T. Identification of a human Th1-like IFNgamma-secreting Treg subtype deriving from effector T cells. J. Autoimmun. 2012, 39, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Ogino, H.; Nakamura, K.; Ihara, E.; Akiho, H.; Takayanagi, R. CD4+CD25+ regulatory T cells suppress Th17-responses in an experimental colitis model. Dig. Dis. Sci. 2011, 56, 376–386. [Google Scholar] [CrossRef]
- Eastaff-Leung, N.; Mabarrack, N.; Barbour, A.; Cummins, A.; Barry, S. Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease. J. Clin. Immunol. 2010, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Xue, J. Expression of Treg/Th17 cells as well as related cytokines in patients with inflammatory bowel disease. Pak. J. Med. Sci. 2016, 32, 1164–1168. [Google Scholar] [CrossRef]
- Zhu, X.M.; Shi, Y.Z.; Cheng, M.; Wang, D.F.; Fan, J.F. Serum IL-6, IL-23 profile and Treg/Th17 peripheral cell populations in pediatric patients with inflammatory bowel disease. Pharmazie 2017, 72, 4. [Google Scholar]
- McNamee, E.N.; Masterson, J.C.; Veny, M.; Collins, C.B.; Jedlicka, P.; Byrne, F.R.; Ng, G.Y.; Rivera-Nieves, J. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohn’s-like murine ileitis. J. Leukoc. Biol. 2015, 97, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Bai, A.; Lu, N.; Guo, Y.; Liu, Z.; Chen, J.; Peng, Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J. Leukoc. Biol. 2009, 86, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Lu, C.; Chen, J.; Cui, G.; Wang, L.; Yu, T.; Yang, Y.; Wu, W.; Ding, Y.; Li, L.; et al. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget 2017, 8, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lecuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tian, M.; Li, W.; Hao, F. Preventative delivery of IL-35 by Lactococcus lactis ameliorates DSS-induced colitis in mice. Appl. Microbiol. Biotechnol. 2019, 103, 7931–7941. [Google Scholar] [CrossRef]
- Higashiyama, M.; Hokari, R.; Hozumi, H.; Kurihara, C.; Ueda, T.; Watanabe, C.; Tomita, K.; Nakamura, M.; Komoto, S.; Okada, Y.; et al. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J. Leukoc. Biol. 2012, 91, 901–909. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Zhong, W.; Di, C.; Lin, X.; Xia, Z. Heme oxygenase-1 ameliorates dextran sulfate sodium-induced acute murine colitis by regulating Th17/Treg cell balance. J. Biol. Chem. 2014, 289, 26847–26858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Chen, M.; Wang, Y.; Wang, Z.; Pei, Y.; Fan, R.; Liu, X.; Wang, L.; Zhou, J.; Zheng, S.; et al. mTOR Inhibition Attenuates Dextran Sulfate Sodium-Induced Colitis by Suppressing T Cell Proliferation and Balancing TH1/TH17/Treg Profile. PLoS ONE 2016, 11, e0154564. [Google Scholar] [CrossRef] [PubMed]
- Tosiek, M.J.; Fiette, L.; El Daker, S.; Eberl, G.; Freitas, A.A. IL-15-dependent balance between Foxp3 and RORgammat expression impacts inflammatory bowel disease. Nat. Commun. 2016, 7, 10888. [Google Scholar] [CrossRef] [Green Version]
- Diller, M.L.; Kudchadkar, R.R.; Delman, K.A.; Lawson, D.H.; Ford, M.L. Balancing Inflammation: The Link between Th17 and Regulatory T Cells. Mediat. Inflamm. 2016, 2016, 6309219. [Google Scholar] [CrossRef]
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414. [Google Scholar] [CrossRef]
- Basu, R.; Hatton, R.D.; Weaver, C.T. The Th17 family: Flexibility follows function. Immunol. Rev. 2013, 252, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Mukasa, R.; Hatton, R.D.; Weaver, C.T. Developmental plasticity of Th17 and Treg cells. Curr. Opin. Immunol. 2009, 21, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Jeffery, L.; Kobayashi, T.; Hibi, T.; Ghosh, S.; Jijon, H. Th17 plasticity and its relevance to inflammatory bowel disease. J. Autoimmun. 2018, 87, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Ghosh, A.; Hung, D.; Li, J.; Jijon, H. Th17 plasticity and its changes associated with inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 12283–12295. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Bot, A. The Th17 cell population and the immune homeostasis of the gastrointestinal tract. Int. Rev. Immunol. 2013, 32, 471–474. [Google Scholar] [CrossRef]
- Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Annunziato, F. Th17 plasticity: Pathophysiology and treatment of chronic inflammatory disorders. Curr. Opin. Pharmacol. 2014, 17, 12–16. [Google Scholar] [CrossRef]
- Kamali, A.N.; Noorbakhsh, S.M.; Hamedifar, H.; Jadidi-Niaragh, F.; Yazdani, R.; Bautista, J.M.; Azizi, G. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol. Immunol. 2019, 105, 107–115. [Google Scholar] [CrossRef]
- Globig, A.M.; Hennecke, N.; Martin, B.; Seidl, M.; Ruf, G.; Hasselblatt, P.; Thimme, R.; Bengsch, B. Comprehensive intestinal T helper cell profiling reveals specific accumulation of IFN-gamma+IL-17+coproducing CD4+ T cells in active inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 2321–2329. [Google Scholar] [CrossRef]
- Calderon-Gomez, E.; Bassolas-Molina, H.; Mora-Buch, R.; Dotti, I.; Planell, N.; Esteller, M.; Gallego, M.; Marti, M.; Garcia-Martin, C.; Martinez-Torro, C.; et al. Commensal-Specific CD4(+) Cells From Patients With Crohn’s Disease Have a T-Helper 17 Inflammatory Profile. Gastroenterology 2016, 151, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Maggi, L.; Santarlasci, V.; Capone, M.; Rossi, M.C.; Querci, V.; Mazzoni, A.; Cimaz, R.; De Palma, R.; Liotta, F.; Maggi, E.; et al. Distinctive features of classic and nonclassic (Th17 derived) human Th1 cells. Eur. J. Immunol. 2012, 42, 3180–3188. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Defining the human T helper 17 cell phenotype. Trends Immunol. 2012, 33, 505–512. [Google Scholar] [CrossRef]
- Maggi, L.; Capone, M.; Giudici, F.; Santarlasci, V.; Querci, V.; Liotta, F.; Ficari, F.; Maggi, E.; Tonelli, F.; Annunziato, F.; et al. CD4+CD161+ T lymphocytes infiltrate Crohn’s disease-associated perianal fistulas and are reduced by anti-TNF-alpha local therapy. Int. Arch. Allergy Immunol. 2013, 161, 81–86. [Google Scholar] [CrossRef]
- Nurieva, R.; Yang, X.O.; Chung, Y.; Dong, C. Cutting edge: In vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J. Immunol. 2009, 182, 2565–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurschus, F.C.; Croxford, A.L.; Heinen, A.P.; Wortge, S.; Ielo, D.; Waisman, A. Genetic proof for the transient nature of the Th17 phenotype. Eur. J. Immunol. 2010, 40, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Prochazkova, J.; Pokorna, K.; Holan, V. IL-12 inhibits the TGF-beta-dependent T cell developmental programs and skews the TGF-beta-induced differentiation into a Th1-like direction. Immunobiology 2012, 217, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Mukasa, R.; Balasubramani, A.; Lee, Y.K.; Whitley, S.K.; Weaver, B.T.; Shibata, Y.; Crawford, G.E.; Hatton, R.D.; Weaver, C.T. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 2010, 32, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Bending, D.; Newland, S.; Krejci, A.; Phillips, J.M.; Bray, S.; Cooke, A. Epigenetic changes at Il12rb2 and Tbx21 in relation to plasticity behavior of Th17 cells. J. Immunol. 2011, 186, 3373–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taherian, M.; Razavi, A.R.; Izad, M.; Boghozian, R.; Namdari, H.; Ghayedi, M.; Rahimzadeh, P.; Bidad, K.; Salehi, E. The Role of interleukin-23 in Stability of in Vitro T helper-17 Cells. Iran. J. Allergy Asthma Immunol. 2014, 13, 6. [Google Scholar]
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066. [Google Scholar] [CrossRef] [Green Version]
- Morrison, P.J.; Bending, D.; Fouser, L.A.; Wright, J.F.; Stockinger, B.; Cooke, A.; Kullberg, M.C. Th17-cell plasticity in Helicobacter hepaticus-induced intestinal inflammation. Mucosal Immunol. 2013, 6, 1143–1156. [Google Scholar] [CrossRef]
- Brucklacher-Waldert, V.; Ferreira, C.; Innocentin, S.; Kamdar, S.; Withers, D.R.; Kullberg, M.C.; Veldhoen, M. Tbet or Continued RORgammat Expression Is Not Required for Th17-Associated Immunopathology. J. Immunol. 2016, 196, 4893–4904. [Google Scholar] [CrossRef] [Green Version]
- Bsat, M.; Chapuy, L.; Rubio, M.; Wassef, R.; Richard, C.; Schwenter, F.; Loungnarath, R.; Soucy, G.; Mehta, H.; Sarfati, M. Differential Pathogenic Th17 Profile in Mesenteric Lymph Nodes of Crohn’s Disease and Ulcerative Colitis Patients. Front. Immunol. 2019, 10, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, T.M.; Wheaton, J.D.; Houtz, G.M.; Ciofani, M. JunB promotes Th17 cell identity and restrains alternative CD4(+) T-cell programs during inflammation. Nat. Commun. 2017, 8, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskaran, N.; Liu, Z.; Saravanamuthu, S.S.; Yan, C.; Hu, Y.; Dong, L.; Zelenka, P.; Zheng, L.; Bletsos, V.; Harris, R.; et al. Identification of Casz1 as a Regulatory Protein Controlling T Helper Cell Differentiation, Inflammation, and Immunity. Front. Immunol. 2018, 9, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmaus, P.W.F.; Chen, X.; Lim, S.A.; Herrada, A.A.; Nguyen, T.M.; Xu, B.; Dhungana, Y.; Rankin, S.; Chen, W.; Rosencrance, C.; et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 2019, 565, 101–105. [Google Scholar] [CrossRef]
- Galvez, J. Role of Th17 Cells in the Pathogenesis of Human IBD. ISRN Inflamm. 2014, 2014, 928461. [Google Scholar] [CrossRef] [Green Version]
- Hoechst, B.; Gamrekelashvili, J.; Manns, M.P.; Greten, T.F.; Korangy, F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 2011, 117, 6532–6541. [Google Scholar] [CrossRef]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkhurst, C.N.; Muratet, M.; et al. A validated regulatory network for Th17 cell specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Carbo, A.; Hontecillas, R.; Kronsteiner, B.; Viladomiu, M.; Pedragosa, M.; Lu, P.; Philipson, C.W.; Hoops, S.; Marathe, M.; Eubank, S.; et al. Systems modeling of molecular mechanisms controlling cytokine-driven CD4+ T cell differentiation and phenotype plasticity. PLoS Comput. Biol. 2013, 9, e1003027. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Turner, J.E.; Villa, M.; Duarte, J.H.; Demengeot, J.; Steinmetz, O.M.; Stockinger, B. Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat. Immunol. 2013, 14, 372–379. [Google Scholar] [CrossRef]
- Hori, S. Lineage Stability and Phenotypic Plasticity of Foxp3⁺ Regulatory T Cells. Immunol. Rev. 2014, 259, 13. [Google Scholar] [CrossRef]
- Komatsu, N.; Mariotti-Ferrandiz, M.E.; Wang, Y.; Malissen, B.; Waldmann, H.; Hori, S. Heterogeneity of natural Foxp3+ T cells: A committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. USA 2009, 106, 1903–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, M.; Ruffini, F.; Bergami, A.; Garzetti, L.; Borsellino, G.; Battistini, L.; Martino, G.; Furlan, R. IL-17- and IFN-gamma-secreting Foxp3+ T cells infiltrate the target tissue in experimental autoimmunity. J. Immunol. 2010, 185, 7467–7473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulahad, W.H.; Boots, A.M.; Kallenberg, C.G. FoxP3+ CD4+ T cells in systemic autoimmune diseases: The delicate balance between true regulatory T cells and effector Th-17 cells. Rheumatology (Oxf.) 2011, 50, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Kitani, A.; Xu, L. Regulatory T cells and the induction of IL-17. Mucosal Immunol. 2008, 1 (Suppl. 1), S43–S46. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, A.; Jijon, H.; Chan, R.; Ford, K.; Hirota, C.; Kaplan, G.G.; Beck, P.L.; Iacucci, M.; Fort Gasia, M.; Barkema, H.W.; et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm. Bowel Dis. 2013, 19, 2522–2534. [Google Scholar] [CrossRef]
- Yang, J.; Xu, L. Elevated IL-23R Expression and Foxp3+Rorgt+ Cells in Intestinal Mucosa During Acute and Chronic Colitis. Med. Sci. Monit. 2016, 22, 2785–2792. [Google Scholar] [CrossRef] [Green Version]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORgammat(+) Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Yurchenko, E.; Shio, M.T.; Huang, T.C.; Da Silva Martins, M.; Szyf, M.; Levings, M.K.; Olivier, M.; Piccirillo, C.A. Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS ONE 2012, 7, e35572. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.M.; Sharma, G.; Verma, R.; Byun, S.; Rudra, D.; Im, S.H. Inflammation-induced Id2 promotes plasticity in regulatory T cells. Nat. Commun. 2018, 9, 4736. [Google Scholar] [CrossRef]
- Zheng, J.; Liu, Y.; Qin, G.; Lam, K.T.; Guan, J.; Xiang, Z.; Lewis, D.B.; Lau, Y.L.; Tu, W. Generation of human Th1-like regulatory CD4+ T cells by an intrinsic IFN-gamma-and T-bet-dependent pathway. Eur. J. Immunol. 2011, 41, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Di Giovangiulio, M.; Rizzo, A.; Franze, E.; Caprioli, F.; Facciotti, F.; Onali, S.; Favale, A.; Stolfi, C.; Fehling, H.J.; Monteleone, G.; et al. Tbet Expression in Regulatory T Cells Is Required to Initiate Th1-Mediated Colitis. Front. Immunol. 2019, 10, 2158. [Google Scholar] [CrossRef] [PubMed]
- Kitz, A.; Dominguez-Villar, M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell. Mol. Life Sci. 2017, 74, 4059–4075. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Cao, A.T.; Weaver, C.T.; Elson, C.O.; Cong, Y. Interleukin-12 converts Foxp3+ regulatory T cells to interferon-gamma-producing Foxp3+ T cells that inhibit colitis. Gastroenterology 2011, 140, 2031–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landman, S.; Cruijsen, M.; Urbano, P.C.M.; Huls, G.; van Erp, P.E.J.; van Rijssen, E.; Joosten, I.; Koenen, H. DNA Methyltransferase Inhibition Promotes Th1 Polarization in Human CD4(+)CD25(high) FOXP3(+) Regulatory T Cells but Does Not Affect Their Suppressive Capacity. J. Immunol. Res. 2018, 2018, 4973964. [Google Scholar] [CrossRef] [Green Version]
- Sarmento, O.F.; Svingen, P.A.; Xiong, Y.; Sun, Z.; Bamidele, A.O.; Mathison, A.J.; Smyrk, T.C.; Nair, A.A.; Gonzalez, M.M.; Sagstetter, M.R.; et al. The Role of the Histone Methyltransferase Enhancer of Zeste Homolog 2 (EZH2) in the Pathobiological Mechanisms Underlying Inflammatory Bowel Disease (IBD). J. Biol. Chem. 2017, 292, 706–722. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Yang, W.Y.; Nanayakkara, G.K.; Shao, Y.; Yang, F.; Hu, W.; Choi, E.T.; Wang, H.; Yang, X. GATA3, HDAC6, and BCL6 Regulate FOXP3+ Treg Plasticity and Determine Treg Conversion into Either Novel Antigen-Presenting Cell-Like Treg or Th1-Treg. Front. Immunol. 2018, 9, 45. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, S.-H.; Chien, M.-W.; Hsu, C.-Y.; Liu, Y.-W.; Sytwu, H.-K. Interplay between Cytokine Circuitry and Transcriptional Regulation Shaping Helper T Cell Pathogenicity and Plasticity in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 3379. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093379
Fu S-H, Chien M-W, Hsu C-Y, Liu Y-W, Sytwu H-K. Interplay between Cytokine Circuitry and Transcriptional Regulation Shaping Helper T Cell Pathogenicity and Plasticity in Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2020; 21(9):3379. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093379
Chicago/Turabian StyleFu, Shin-Huei, Ming-Wei Chien, Chao-Yuan Hsu, Yu-Wen Liu, and Huey-Kang Sytwu. 2020. "Interplay between Cytokine Circuitry and Transcriptional Regulation Shaping Helper T Cell Pathogenicity and Plasticity in Inflammatory Bowel Disease" International Journal of Molecular Sciences 21, no. 9: 3379. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093379