Pan-Genome Analyses of Geobacillus spp. Reveal Genetic Characteristics and Composting Potential


Abstract
:1. Introduction
2. Results
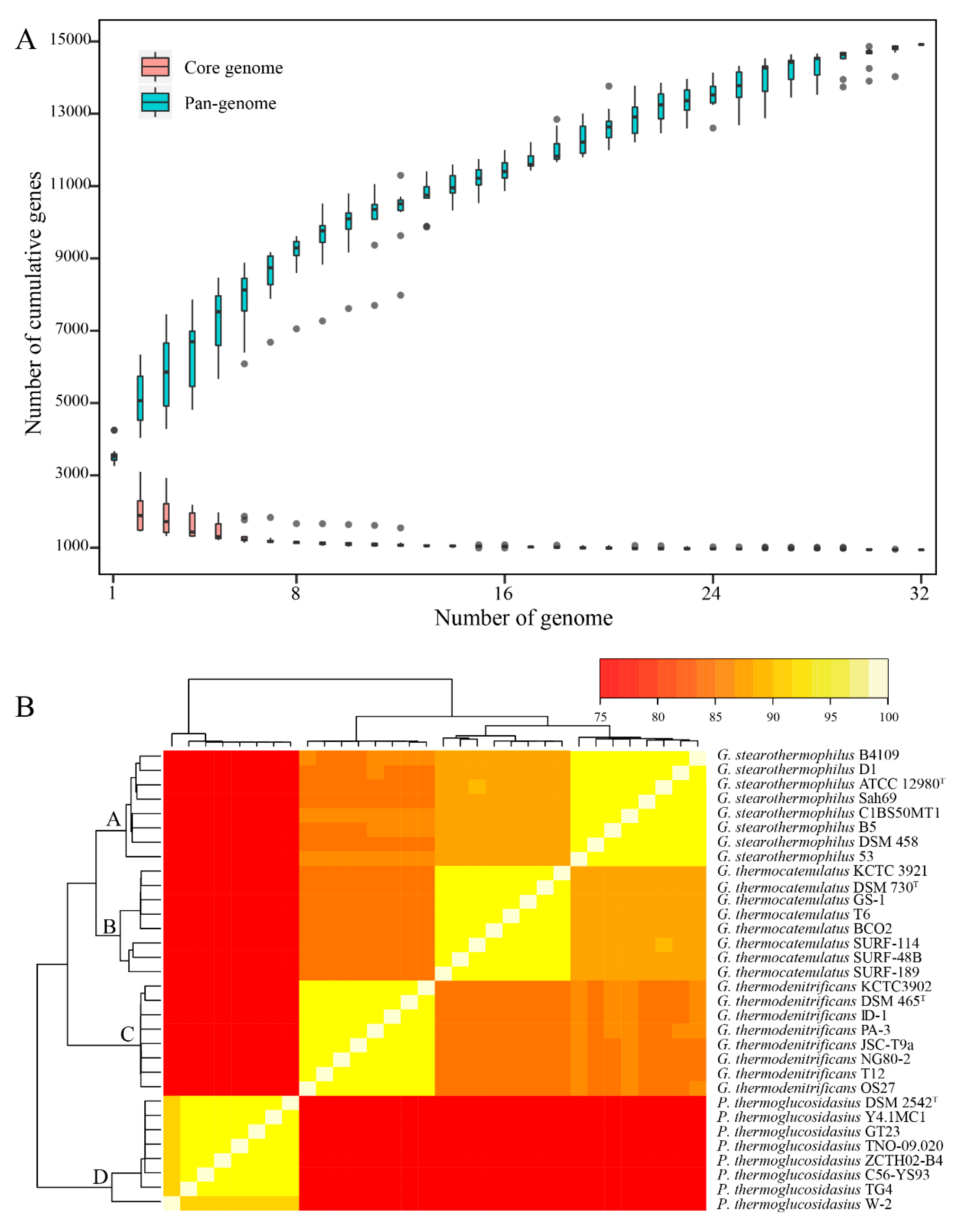
2.1. Pan- and Core Genomes Analysis of Different Geobacillus Species
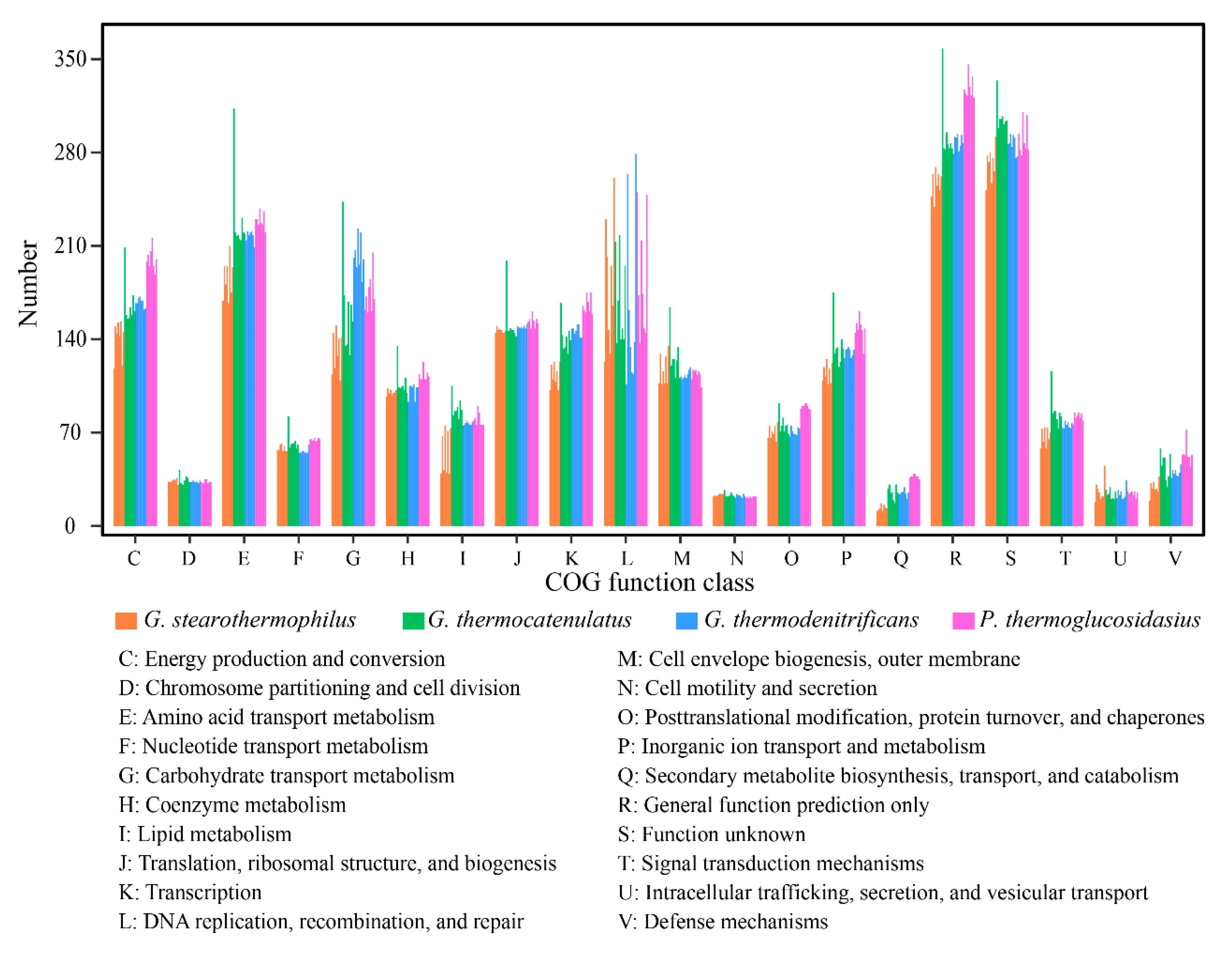
2.2. COG Distribution Between Different Geobacillus Species
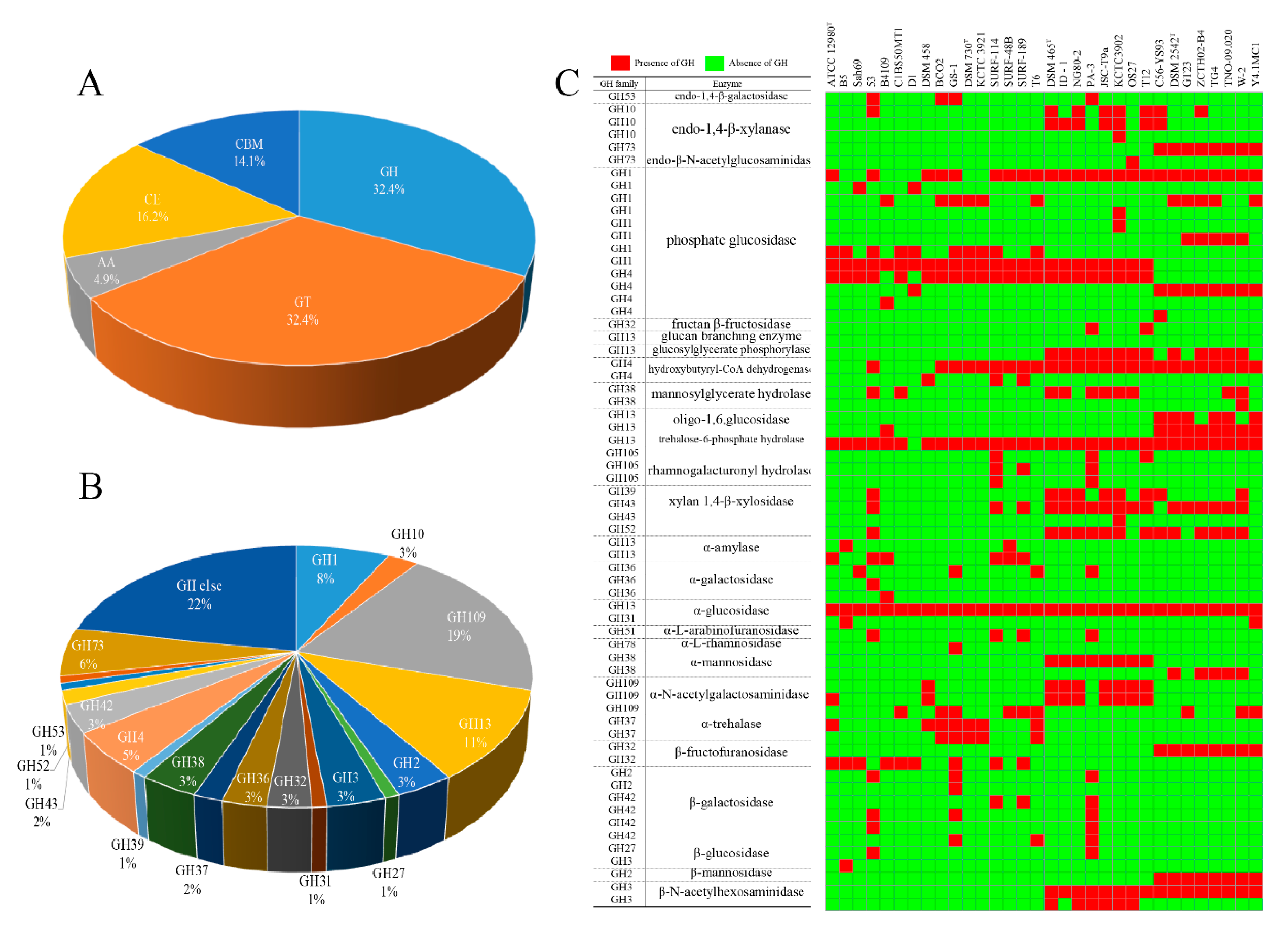
2.3. CAZyme Analysis of Different Geobacillus Species
2.4. Calculation of Pan- and Core Genomes of All Strains
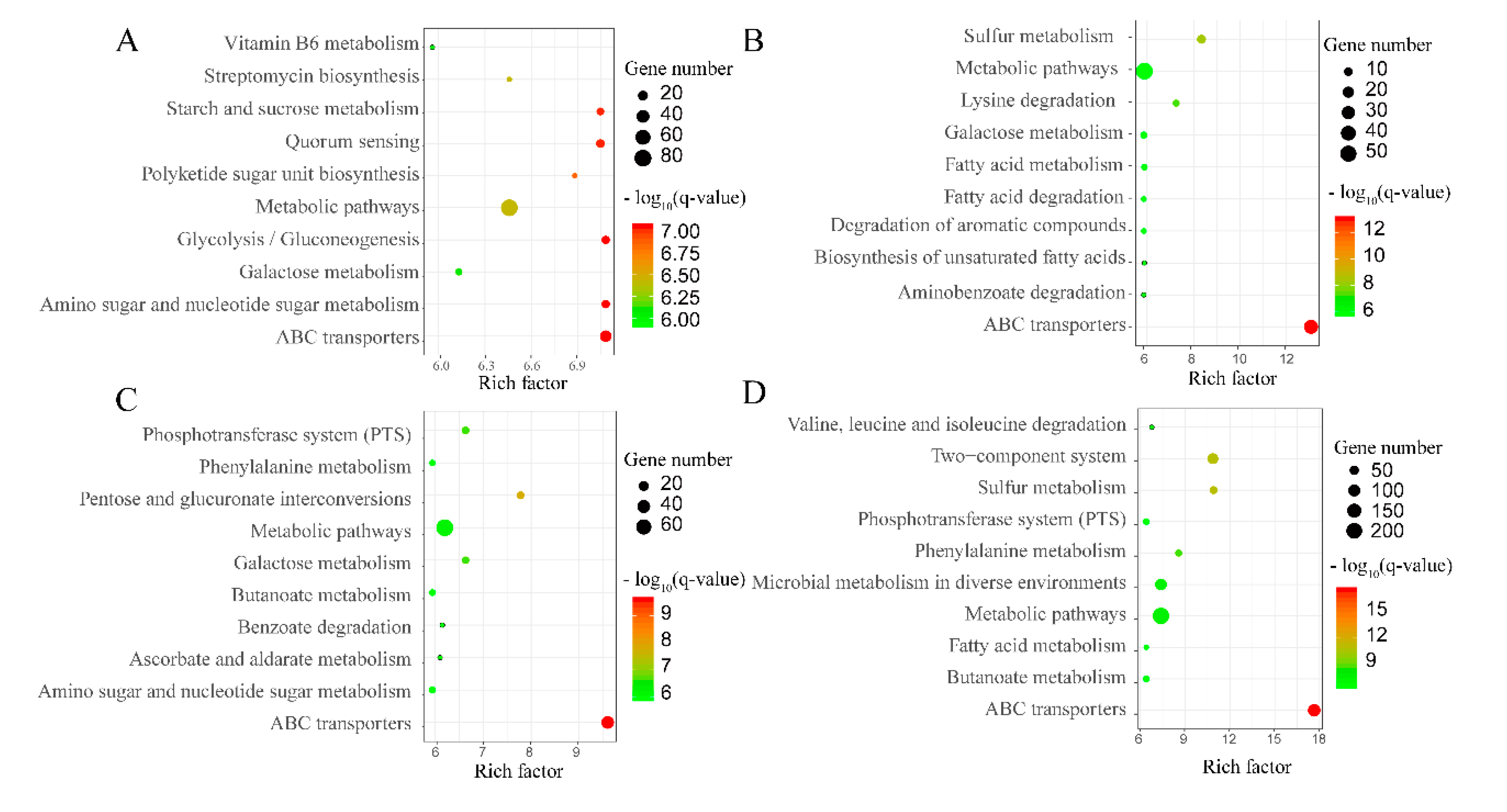
2.5. KEGG Analysis of Geobacillus Strains
2.6. Analyses of SNPs, Representative Genes, and HGT Events
3. Discussion
4. Materials and Methods
4.1. Genome Sequences
4.2. Calculation of Pan- and Core Genomes
4.3. Functional Classification of COGs and Identification of CAZymes
4.4. Clustering and GhostKOALA Analysis of the Pan-Genomes
4.5. Identification of Representative Genes and HGT Events
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| COG | Clusters of orthologous groups |
| CAZymes | Carbohydrate-active enzymes |
| HGT | Horizontal gene transfer |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ANI | Average nucleotide sequence identity |
| GT | Glycosyl transferase |
| GH | Glycoside hydrolase |
| AA | Auxiliary activity |
| CE | Carbohydrate esterase |
| CBM | Carbohydrate-binding module |
References
- Zeigler, D.R. The Geobacillus paradox: Why is a thermophilic bacterial genus so prevalent on a mesophilic planet? Microbiology 2014, 160, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studholme, D.J. Some (bacilli) like it hot: Genomics of Geobacillus species. Microb. Biotechnol. 2015, 8, 40–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, A.H.; Lisowska, B.K.; Leak, D.J. The genus Geobacillus and their biotechnological potential. In Advances in Applied Microbiology; Elsevier: Wallingford, UK, 2015; Volume 92, pp. 1–48. [Google Scholar]
- Bartosiak-Jentys, J.; Hussein, A.H.; Lewis, C.J.; Leak, D.J. Modular system for assessment of glycosyl hydrolase secretion in Geobacillus thermoglucosidasius. Microbiology 2013, 159, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Okazaki, F.; Kondo, A.; Yoshida, K. Genome mining and motif modifications of glycoside hydrolase family 1 members encoded by Geobacillus kaustophilus HTA426 provide thermostable 6-phospho-β-glycosidase and β-fucosidase. Appl. Microbiol. Biotechnol. 2013, 97, 2929–2938. [Google Scholar] [CrossRef]
- Balan, A.; Ibrahim, D.; Abdul Rahim, R.; Ahmad Rashid, F.A. Purification and Characterization of a Thermostable Lipase from Geobacillus thermodenitrificans IBRL-nra. Enzym. Res. 2012, 2012, 987523. [Google Scholar] [CrossRef] [Green Version]
- Thebti, W.; Riahi, Y.; Belhadj, O. Purification and Characterization of a New Thermostable, Haloalkaline, Solvent Stable, and Detergent Compatible Serine Protease from Geobacillus toebii Strain LBT 77. Biomed. Res. Int. 2016, 2016, 9178962. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bi, L.; Liao, Y.; Lu, D.; Zhang, H.; Liao, X.; Liang, J.B.; Wu, Y. Influence and characteristics of Bacillus stearothermophilus in ammonia reduction during layer manure composting. Ecotoxicol Env. Saf 2019, 180, 80–87. [Google Scholar] [CrossRef]
- Brenner, D.J.; Staley, J.T.; Krieg, N.R. Classification of procaryotic organisms and the concept of bacterial speciation. In Bergey’s Manual «of Systematic Bacteriology»; Springer: Boston, MA, USA, 2005; pp. 27–32. [Google Scholar]
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.-J.; Ludwig, W.; Kämpfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 2010, 60, 249–266. [Google Scholar] [CrossRef] [Green Version]
- Zeigler, D.R. Application of a recN sequence similarity analysis to the identification of species within the bacterial genus Geobacillus. Int. J. Syst. Evol. Microbiol. 2005, 55, 1171–1179. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Nazina, T.; Tourova, T.; Poltaraus, A.; Novikova, E.; Grigoryan, A.; Ivanova, A.; Lysenko, A.; Petrunyaka, V.; Osipov, G.; Belyaev, S. Taxonomic study of aerobic thermophilic bacilli: Descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int. J. Syst. Evol. Microbiol. 2001, 51, 433–446. [Google Scholar]
- Banat, I.M.; Marchant, R.; Rahman, T.J. Geobacillus debilis sp. nov., a novel obligately thermophilic bacterium isolated from a cool soil environment, and reassignment of Bacillus pallidus to Geobacillus pallidus comb. nov. Int. J. Syst. Evol. Microbiol. 2004, 54, 2197–2201. [Google Scholar] [CrossRef] [Green Version]
- Aliyu, H.; Lebre, P.; Blom, J.; Cowan, D.; De Maayer, P. Phylogenomic re-assessment of the thermophilic genus Geobacillus. Syst. Appl. Microbiol. 2016, 39, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Illeghems, K.; De Vuyst, L.; Weckx, S. Comparative genome analysis of the candidate functional starter culture strains Lactobacillus fermentum 222 and Lactobacillus plantarum 80 for controlled cocoa bean fermentation processes. BMC Genom. 2015, 16, 766. [Google Scholar] [CrossRef] [Green Version]
- Argemi, X.; Matelska, D.; Ginalski, K.; Riegel, P.; Hansmann, Y.; Bloom, J.; Pestel-Caron, M.; Dahyot, S.; Lebeurre, J.; Prevost, G. Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer. BMC Genom. 2018, 19, 621. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.; Nelson-Sathi, S.; Roettger, M.; Garg, S.; Hazkani-Covo, E.; Martin, W.F. Endosymbiotic gene transfer from prokaryotic pangenomes: Inherited chimerism in eukaryotes. Proc. Natl. Acad. Sci. USA 2015, 112, 10139–10146. [Google Scholar] [CrossRef] [Green Version]
- Bosi, E.; Monk, J.M.; Aziz, R.K.; Fondi, M.; Nizet, V.; Palsson, B. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 2016, 113, E3801–E3809. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.J.; Rocha, E.P. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011, 7, e1001284. [Google Scholar] [CrossRef]
- Bapteste, E.; Lopez, P.; Bouchard, F.; Baquero, F.; McInerney, J.O.; Burian, R.M. Evolutionary analyses of non-genealogical bonds produced by introgressive descent. Proc. Natl. Acad. Sci. USA 2012, 109, 18266–18272. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Li, M.; Guo, S.; Wang, W. Draft genome sequence of a thermophilic desulfurization bacterium, Geobacillus thermoglucosidasius strain W-2. Genome Announc. 2016, 4, e00793-16. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Song, Y.; Jeong, Y.; Cho, B.-K. Analysis of the core genome and pan-genome of autotrophic acetogenic bacteria. Front. Microbiol. 2016, 7, 1531. [Google Scholar] [CrossRef]
- Lukjancenko, O.; Wassenaar, T.M.; Ussery, D.W. Comparison of 61 sequenced Escherichia coli genomes. Microb. Ecol. 2010, 60, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Bezuidt, O.K.; Pierneef, R.; Gomri, A.M.; Adesioye, F.; Makhalanyane, T.P.; Kharroub, K.; Cowan, D.A. The Geobacillus Pan-Genome: Implications for the Evolution of the Genus. Front. Microbiol. 2016, 7, 723. [Google Scholar] [CrossRef]
- Bottacini, F.; Motherway, M.O.C.; Kuczynski, J.; O’Connell, K.J.; Serafini, F.; Duranti, S.; Milani, C.; Turroni, F.; Lugli, G.A.; Zomer, A. Comparative genomics of the Bifidobacterium breve taxon. BMC Genom. 2014, 15, 170. [Google Scholar] [CrossRef] [Green Version]
- Burgess, S.A.; Flint, S.H.; Lindsay, D.; Cox, M.P.; Biggs, P.J. Insights into the Geobacillus stearothermophilus species based on phylogenomic principles. BMC Microbiol. 2017, 17, 140. [Google Scholar] [CrossRef]
- Nanavati, D.M.; Thirangoon, K.; Noll, K.M. Several archaeal homologs of putative oligopeptide-binding proteins encoded by Thermotoga maritima bind sugars. Appl. Environ. Microbiol. 2006, 72, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Law, C.J.; Maloney, P.C.; Wang, D.-N. Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef] [Green Version]
- Deutscher, J.; Francke, C.; Postma, P.W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maayer, P.; Brumm, P.J.; Mead, D.A.; Cowan, D.A. Comparative analysis of the Geobacillus hemicellulose utilization locus reveals a highly variable target for improved hemicellulolysis. BMC Genom. 2014, 15, 836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graebin, N.; Schöffer, J.; Andrades, D.; Hertz, P.; Ayub, M.; Rodrigues, R. Immobilization of Glycoside Hydrolase Families GH1, GH13, and GH70: State of the Art and Perspectives. Molecules 2016, 21, 1074. [Google Scholar] [CrossRef]
- Cayuela, M.L.; Sánchez-Monedero, M.; Roig, A. Evaluation of two different aeration systems for composting two-phase olive mill wastes. Process Biochem. 2006, 41, 616–623. [Google Scholar] [CrossRef]
- Brás, J.L.; Cartmell, A.; Carvalho, A.L.M.; Verzé, G.; Bayer, E.A.; Vazana, Y.; Correia, M.A.; Prates, J.A.; Ratnaparkhe, S.; Boraston, A.B.; et al. Structural insights into a unique cellulase fold and mechanism of cellulose hydrolysis. Proc. Nat. Acad. Sci. USA 2011, 108, 5237–5242. [Google Scholar]
- Sunna, A.; Antranikian, G. Xylanolytic enzymes from fungi and bacteria. Crit. Rev. Biotechnol. 1997, 17, 39–67. [Google Scholar] [CrossRef]
- Boraston, A.B.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. [Google Scholar] [CrossRef]
- Frantzen, C.A.; Kot, W.; Pedersen, T.B.; Ardö, Y.M.; Broadbent, J.R.; Neve, H.; Hansen, L.H.; Dal Bello, F.; Østlie, H.M.; Kleppen, H.P. Genomic characterization of dairy associated Leuconostoc species and diversity of Leuconostocs in undefined mixed mesophilic starter cultures. Front. Microbiol. 2017, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Mohr, T.; Aliyu, H.; Küchlin, R.; Zwick, M.; Cowan, D.; Neumann, A.; de Maayer, P. Comparative genomic analysis of Parageobacillus thermoglucosidasius strains with distinct hydrogenogenic capacities. BMC Genom. 2018, 19, 880. [Google Scholar] [CrossRef] [Green Version]
- Kapralou, S.; Fabbretti, A.; Garulli, C.; Gualerzi, C.O.; Pon, C.L.; Spurio, R. Characterization of Bacillus stearothermophilus infA and of its product IF1. Gene 2009, 428, 31–35. [Google Scholar] [CrossRef]
- Kuisiene, N.; Raugalas, J.; Chitavichius, D. Phylogenetic, inter, and intraspecific sequence analysis of spo0A gene of the genus Geobacillus. Curr. Microbiol. 2009, 58, 547. [Google Scholar] [CrossRef] [PubMed]
- Meintanis, C.; Chalkou, K.; Kormas, K.A.; Lymperopoulou, D.; Katsifas, E.; Hatzinikolaou, D.; Karagouni, A. Application of rpoB sequence similarity analysis, REP-PCR and BOX-PCR for the differentiation of species within the genus Geobacillus. Lett. Appl. Microbiol. 2008, 46, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Env. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Kaas, R.S.; Friis, C.; Ussery, D.W.; Aarestrup, F.M. Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genom. 2012, 13, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Rabiner, L.R. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 1989, 77, 257–286. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowicz, D.; Giaro, K.; Wróbel, B. TreeCmp: Comparison of trees in polynomial time. Evol. Bioinform. 2012, 8, EBO.S9657. [Google Scholar] [CrossRef]
- Estabrook, G.F.; McMorris, F.; Meacham, C.A. Comparison of undirected phylogenetic trees based on subtrees of four evolutionary units. Syst. Zool. 1985, 34, 193–200. [Google Scholar] [CrossRef]
- Bogdanowicz, D.; Giaro, K. Matching split distance for unrooted binary phylogenetic trees. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 150–160. [Google Scholar] [CrossRef]
- Song, W.; Wemheuer, B.; Zhang, S.; Steensen, K.; Thomas, T. MetaCHIP: Community-level horizontal gene transfer identification through the combination of best-match and phylogenetic approaches. Microbiome 2019, 7, 36. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Ngenome | Size (Mb) | Ngenes | Ncore | Nshell | Ncloud | Npan |
|---|---|---|---|---|---|---|---|
| - | 32 | 3.54 ± 0.35 | 3629 ± 356 | 940 | 5304 | 8496 | 14913 |
| G. stearothermophilus | 8 | 3.13 ± 0.33 | 3280 ± 281 | 1994 | 1925 | 1980 | 5899 |
| G. thermocatenulatus | 8 | 3.53 ± 0.11 | 3794 ± 360 | 2142 | 2333 | 2640 | 7115 |
| G. thermodenitrificans | 8 | 3.58 ± 0.13 | 3532 ± 153 | 2861 | 1101 | 1229 | 5191 |
| P. thermoglucosidasius | 8 | 3.92 ± 0.19 | 3912 ± 192 | 2659 | 1737 | 2352 | 6748 |
| Gene Numbers | n-Fold Extension | ||||
|---|---|---|---|---|---|
| Pan | Softcore | Core | Softcore-Pan | Core-Pan | |
| Brite mapping | |||||
| Orthologs and modules | 2846 | 1625 | 1419 | 1.8 | 2.0 |
| Protein families: metabolism | 1342 | 798 | 698 | 1.7 | 1.9 |
| Protein families: genetic information processing | 516 | 399 | 372 | 1.3 | 1.4 |
| Protein families: signaling and cellular processing | 539 | 228 | 190 | 2.4 | 2.8 |
| Total | 5243 | 3050 | 2679 | 1.7 | 2.0 |
| Pathway reconstruction | |||||
| Metabolism | 2958 | 1850 | 1666 | 1.6 | 1.8 |
| Genetic Information Processing | 198 | 180 | 167 | 1.1 | 1.2 |
| Environmental Information Processing | 278 | 98 | 79 | 2.8 | 3.5 |
| Cellular Processes | 156 | 102 | 78 | 1.5 | 2.0 |
| Organismal Systems | 61 | 30 | 27 | 2.0 | 2.3 |
| Human Diseases | 88 | 49 | 47 | 1.8 | 1.9 |
| Total | 3739 | 2309 | 2064 | 1.6 | 1.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhu, H.; Kong, Z.; Li, T.; Ma, L.; Liu, D.; Shen, Q. Pan-Genome Analyses of Geobacillus spp. Reveal Genetic Characteristics and Composting Potential. Int. J. Mol. Sci. 2020, 21, 3393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093393
Wang M, Zhu H, Kong Z, Li T, Ma L, Liu D, Shen Q. Pan-Genome Analyses of Geobacillus spp. Reveal Genetic Characteristics and Composting Potential. International Journal of Molecular Sciences. 2020; 21(9):3393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093393
Chicago/Turabian StyleWang, Mengmeng, Han Zhu, Zhijian Kong, Tuo Li, Lei Ma, Dongyang Liu, and Qirong Shen. 2020. "Pan-Genome Analyses of Geobacillus spp. Reveal Genetic Characteristics and Composting Potential" International Journal of Molecular Sciences 21, no. 9: 3393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093393