Postnatal Smoke Exposure Further Increases the Hepatic Nicotine Metabolism in Prenatally Smoke Exposed Male Offspring and Is Linked with Aberrant Cyp2a5 Methylation

 ,
,  ,
,
Abstract
:1. Introduction
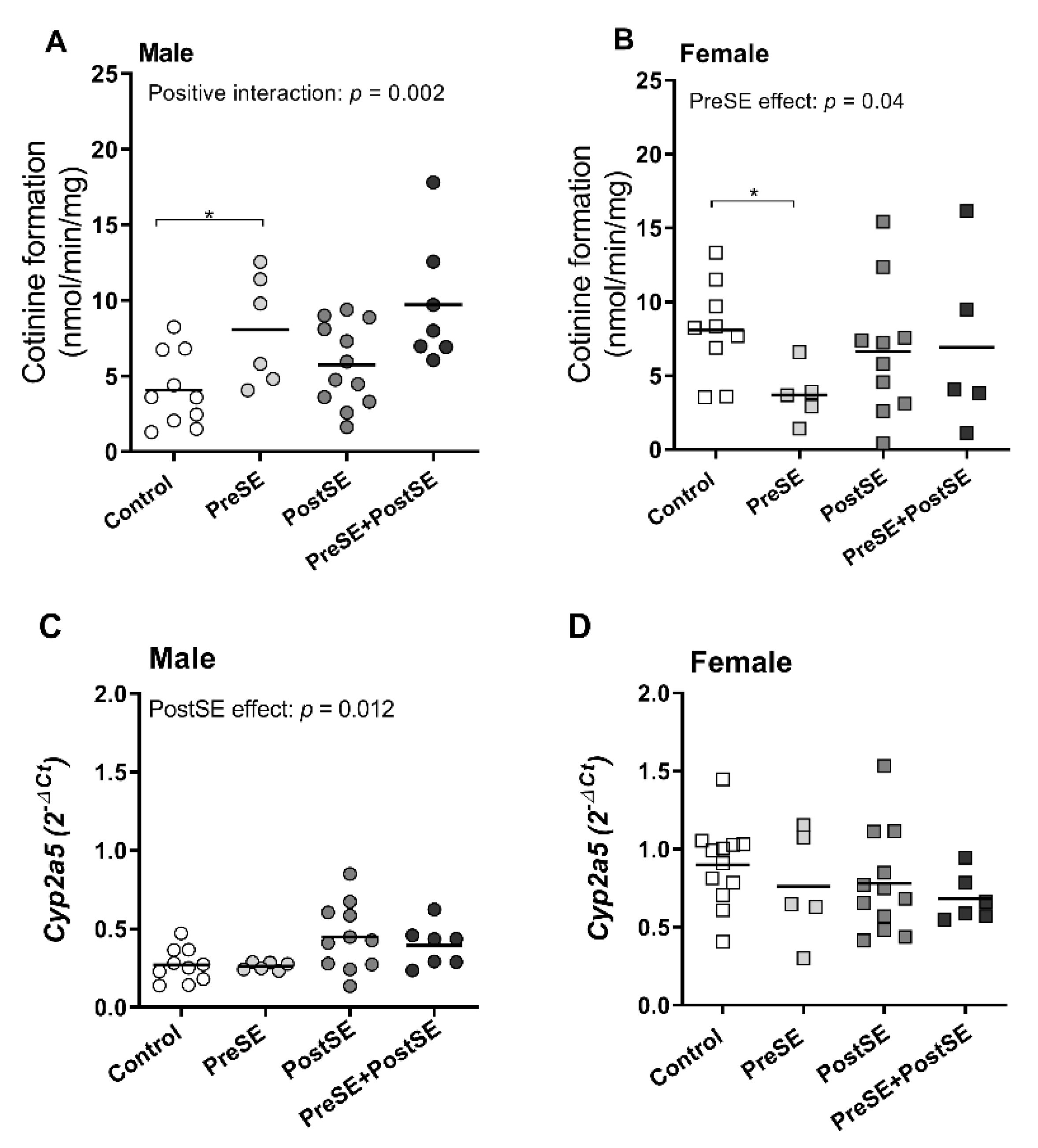
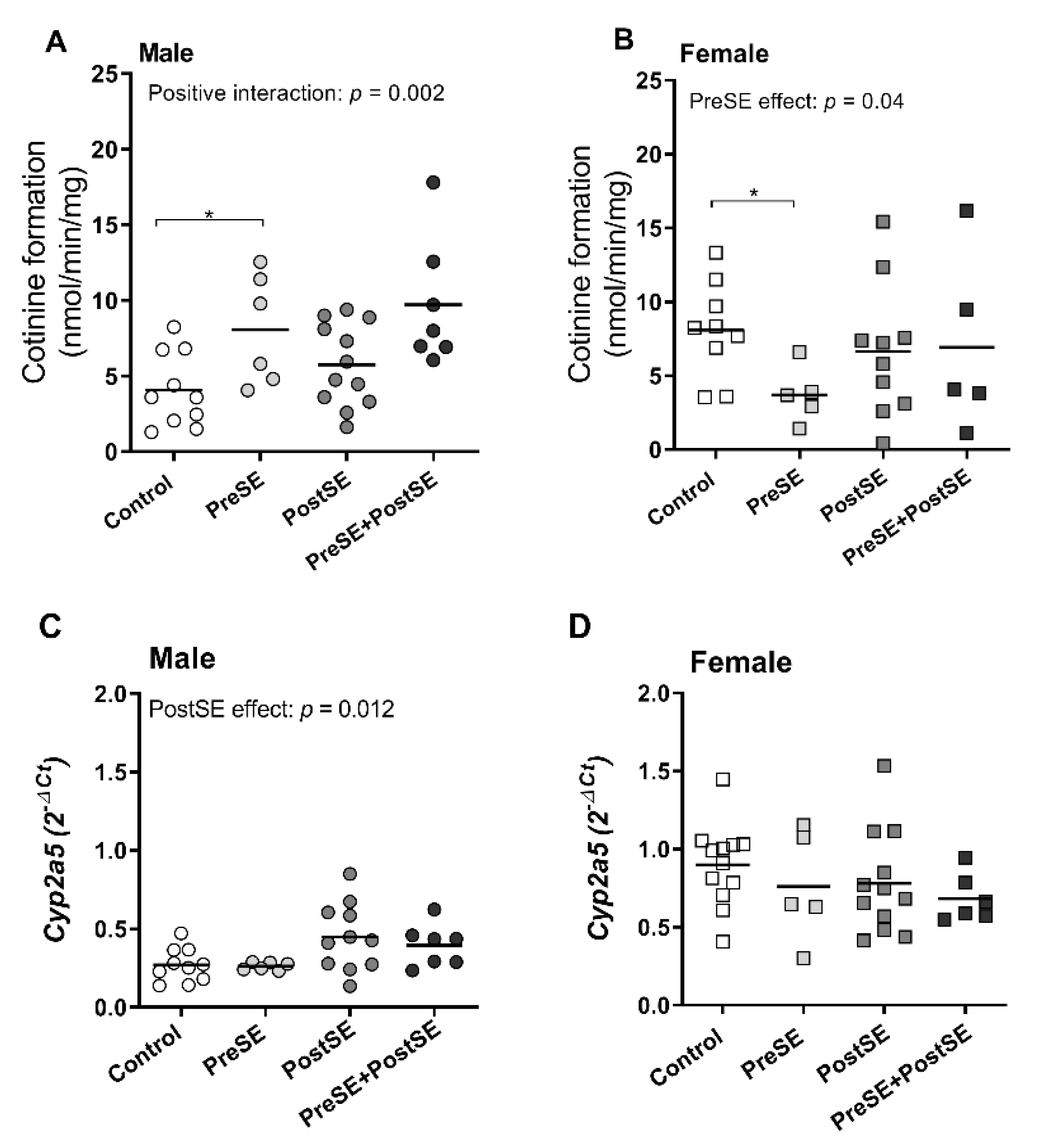
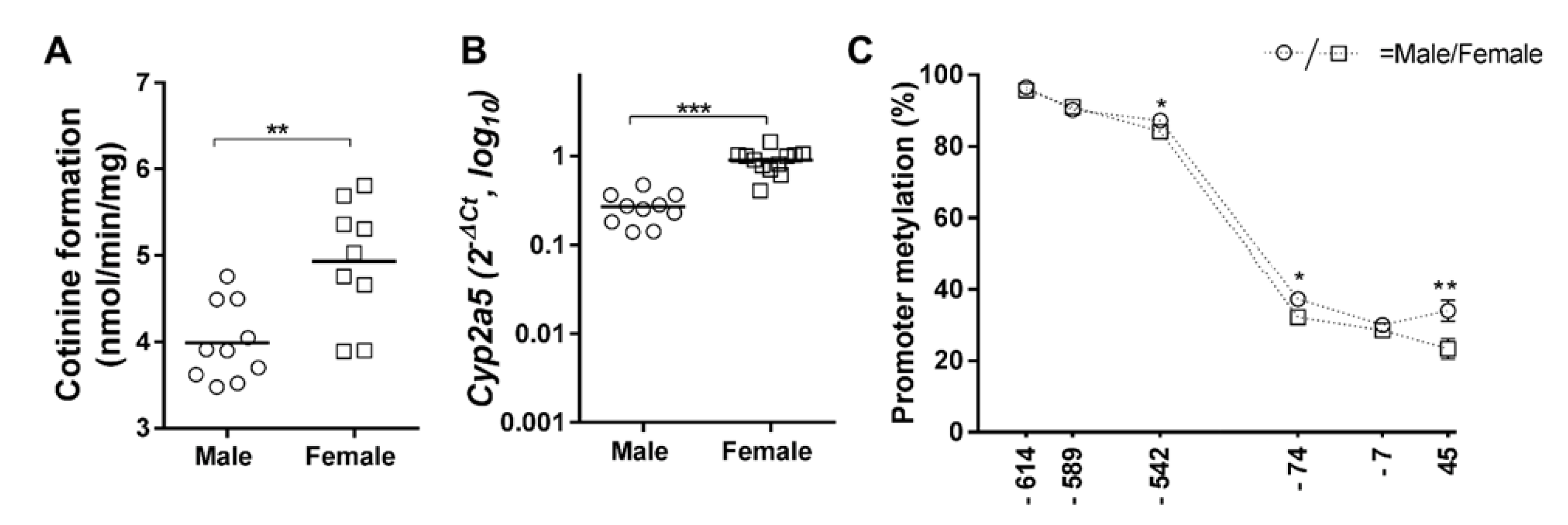
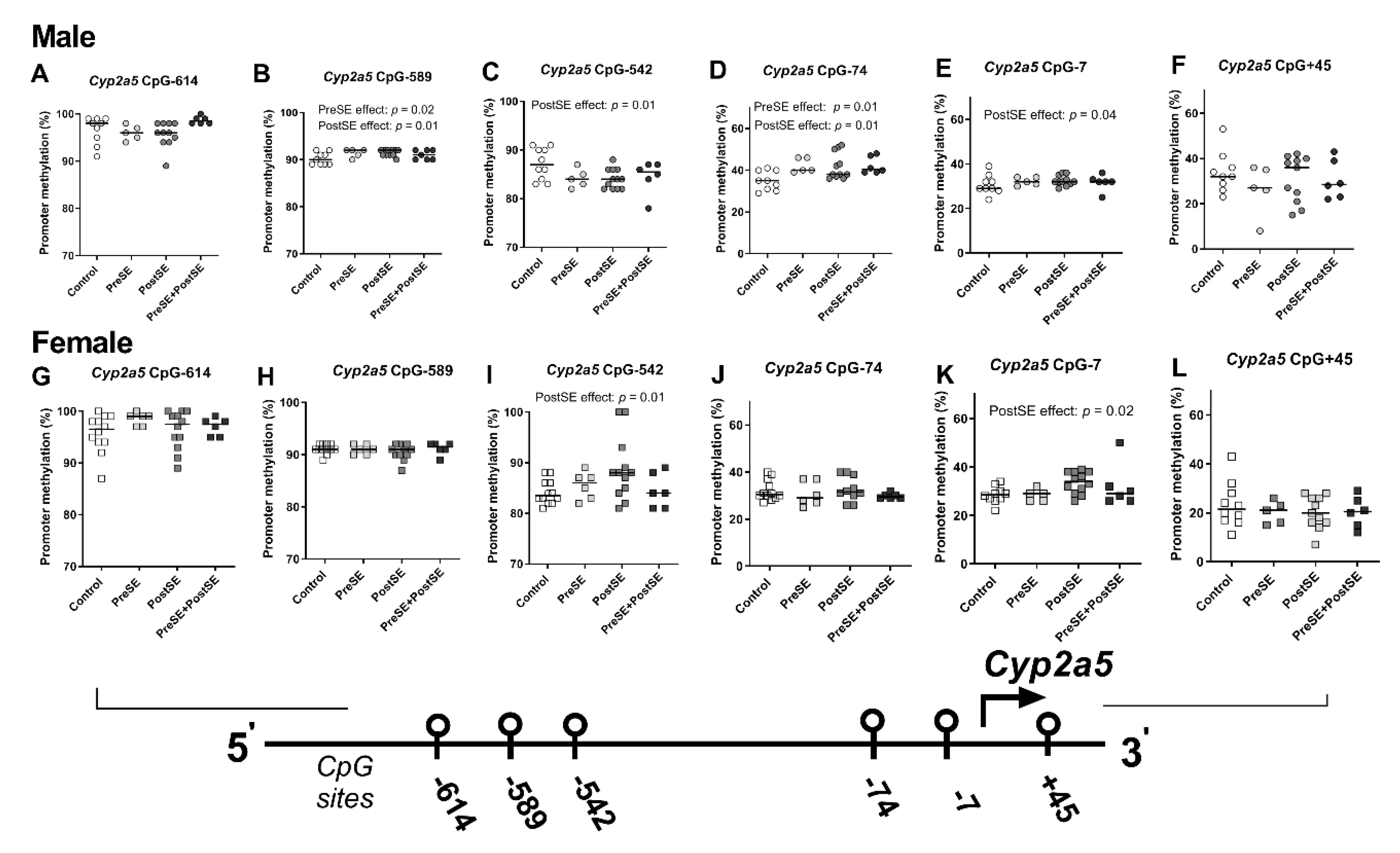
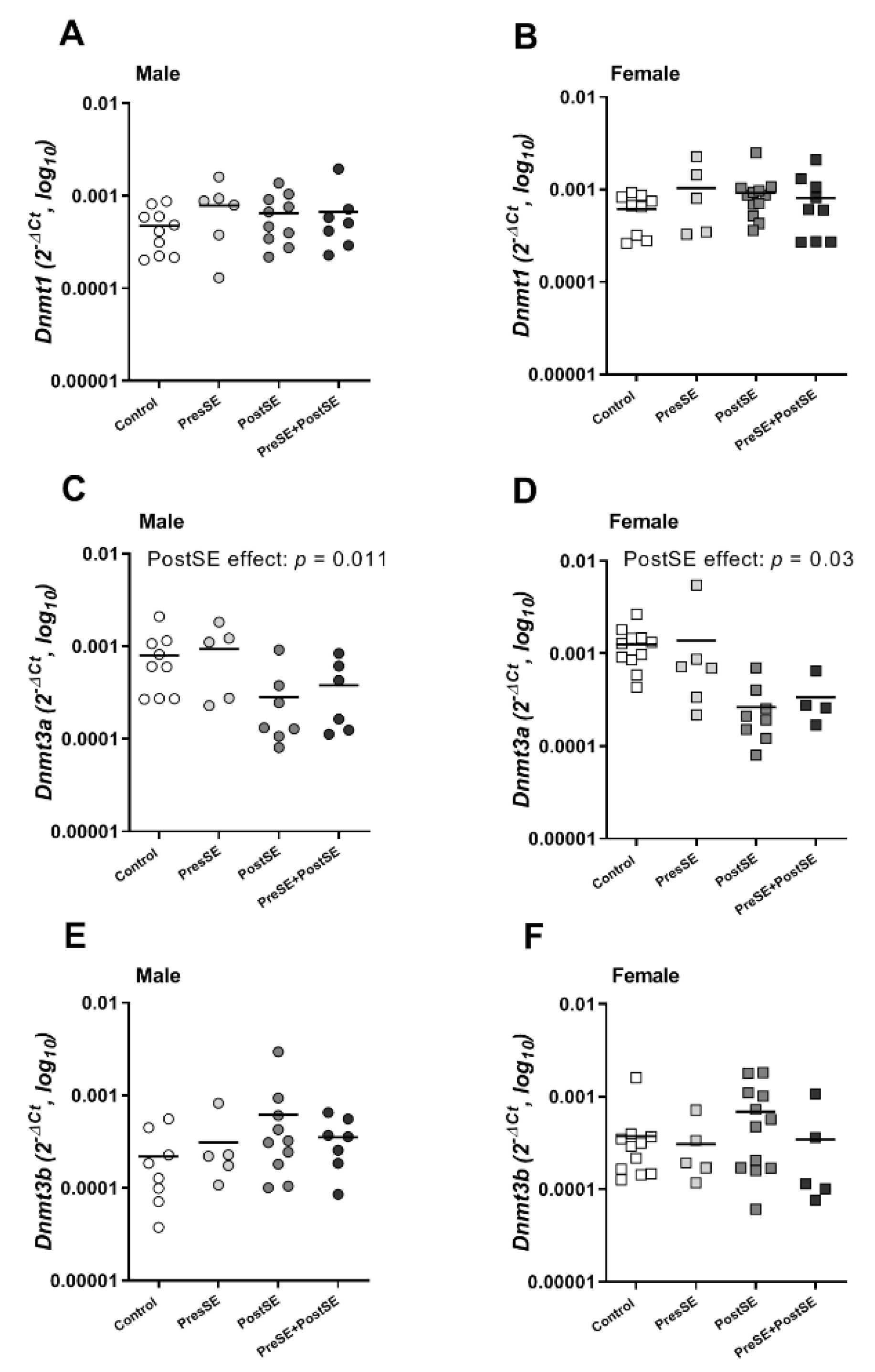
2. Results
3. Discussion
4. Materials and Methods
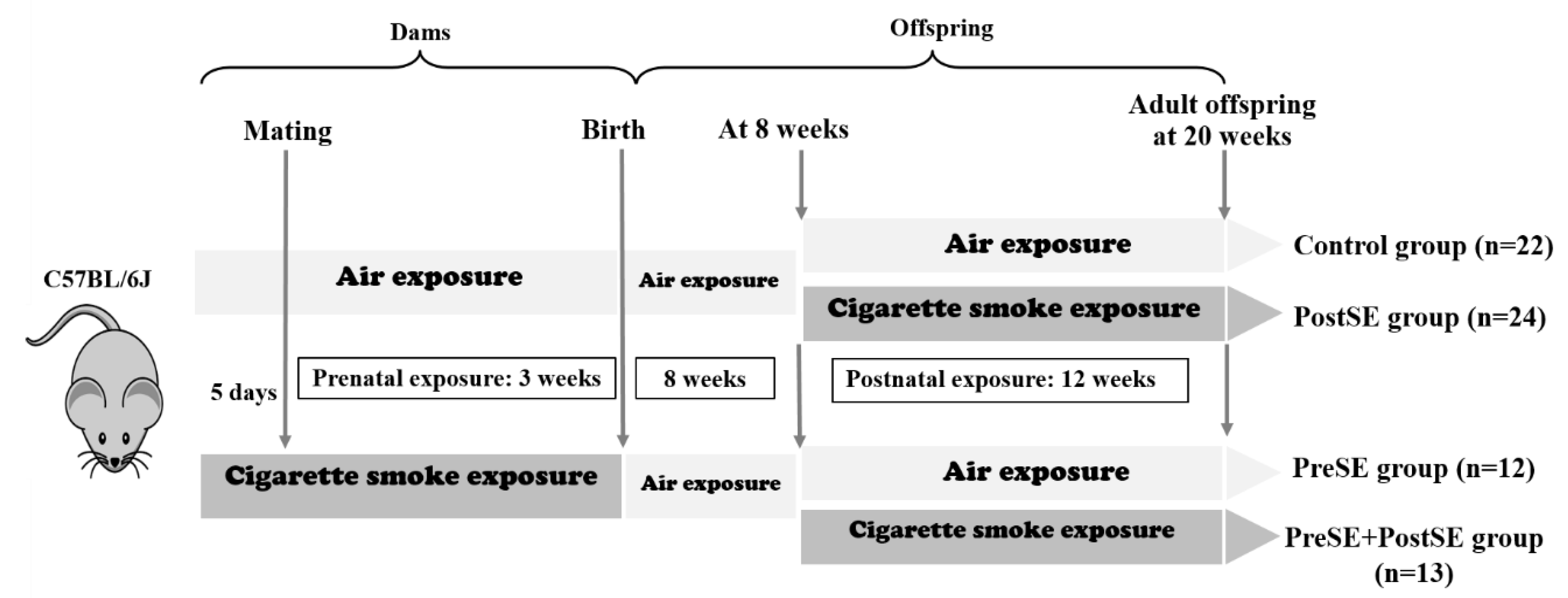
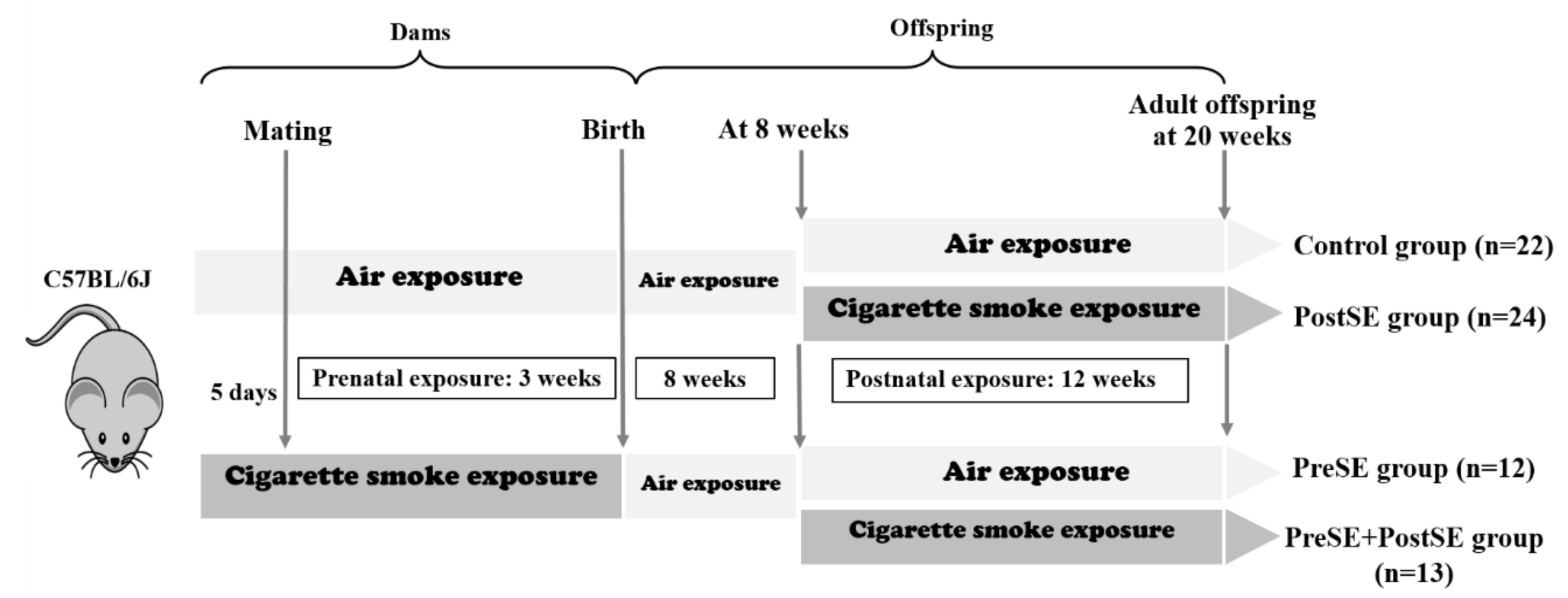
4.1. Animals & Smoke Exposure
4.2. mRNA Expression and Pyrosequencing-Based Bisulfite PCR Analysis
4.3. Isolation and Preparation of DNA of Liver Tissues for Bisulfite-Based Methylation Analysis
4.4. Microsome Preparation and In Vitro Assay for Nicotine Metabolism
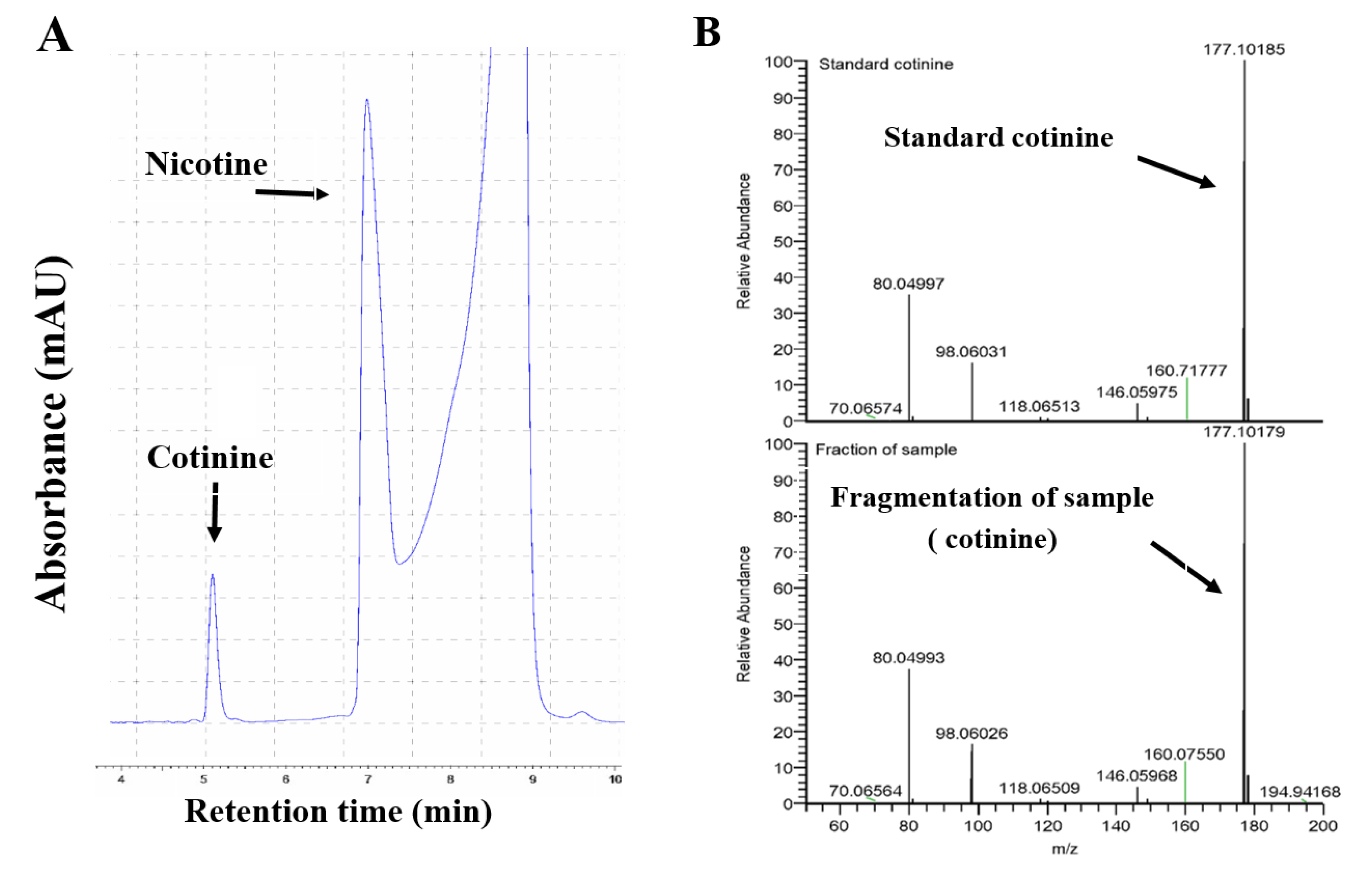
4.5. Nicotine Metabolite Analysis by HPLC and LCMS
4.6. Statistical Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PreSE | Prenatal smoke exposure |
| PostSE | Postnatal smoke exposure |
| CYP2A5 | Cytochrome P450 2A5 |
References
- Benowitz, N.L. Nicotine Addiction. N. Engl. J. Med. 2010, 362, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Villaça, Y.; Seidler, F.J.; Tate, C.A.; Cousins, M.M.; Slotkin, T.A. Prenatal Nicotine Exposure Alters the Response to Nicotine Administration in Adolescence: Effects on Cholinergic Systems During Exposure and Withdrawal. Neuropsychopharmacology 2004, 29, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandel, D.B.; Wu, P.; Davies, M. Maternal smoking during pregnancy and smoking by adolescent daughters. Am. J. Public Health 1994, 84, 1407–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, L.C. Maternal nicotine exposure increases nicotine preference in periadolescent male but not female C57Bl/6J mice. Nicotine Tob. Res. 2003, 5, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Siu, E.C.K.; Tyndale, R.F. Characterization and Comparison of Nicotine and Cotinine Metabolism in Vitro and in Vivo in DBA/2 and C57BL/6 Mice. Mol. Pharmacol. 2006, 71, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Messina, E.S.; Tyndale, R.F.; Sellers, E.M. A major role for CYP2A6 in nicotine C-oxidation by human liver microsomes. J. Pharmacol. Exp. Ther. 1997, 282, 1608–1614. [Google Scholar]
- Crawford, E.L.; Weaver, D.A.; DeMuth, J.P.; Jackson, C.M.; Khuder, S.A.; Frampton, M.W.; Utell, M.J.; Thilly, W.G.; Willey, J.C. Measurement of cytochrome P450 2A6 and 2E1 gene expression in primary human bronchial epithelial cells. Carcinogenesis 1998, 19, 1867–1871. [Google Scholar] [CrossRef] [Green Version]
- Amos, C.I.; Spitz, M.R.; Cinciripini, P. Chipping away at the genetics of smoking behavior. Nat. Genet. 2010, 42, 366–368. [Google Scholar] [CrossRef]
- Wassenaar, C.A.; Dong, Q.; Wei, Q.; Amos, C.I.; Spitz, M.R.; Tyndale, R.F. Relationship Between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 Variation and Smoking Behaviors and Lung Cancer Risk. J. Natl. Cancer Inst. 2011, 103, 1342–1346. [Google Scholar] [CrossRef]
- Chenoweth, M.J.; O’Loughlin, J.; Sylvestre, M.-P.; Tyndale, R.F. CYP2A6 slow nicotine metabolism is associated with increased quitting by adolescent smokers. Pharm. Genom. 2013, 23, 232–235. [Google Scholar] [CrossRef] [Green Version]
- Bandiera, F.C.; Ross, K.C.; Taghavi, S.; Delucchi, K.; Tyndale, R.F.; Benowitz, N.L. Nicotine Dependence, Nicotine Metabolism, and the Extent of Compensation in Response to Reduced Nicotine Content Cigarettes. Nicotine Tob. Res. 2015, 17, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhuo, X.; Xie, F.; Kluetzman, K.; Shu, Y.-Z.; Humphreys, W.G.; Ding, X. Role of CYP2A5 in the Clearance of Nicotine and Cotinine: Insights from Studies on a Cyp2a5-null Mouse Model. J. Pharmacol. Exp. Ther. 2010, 332, 578–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Jia, K.; Zhou, X.; McCallum, S.E.; Hough, L.B.; Ding, X. Impact of Nicotine Metabolism on Nicotine’s Pharmacological Effects and Behavioral Responses: Insights from a Cyp2a(4/5)bgs-Null Mouse. J. Pharmacol. Exp. Ther. 2013, 347, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raunio, H.; Pokela, N.; Puhakainen, K.; Rahnasto, M.; Mauriala, T.; Auriola, S.; Juvonen, R.O. Nicotine metabolism and urinary elimination in mouse:in vitroandin vivo. Xenobiotica 2008, 38, 34–47. [Google Scholar] [CrossRef]
- Richmond, R.C.; Simpkin, A.J.; Woodward, G.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Smith, A.D.; Timpson, N.J.; Tilling, K.; et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: Findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum. Mol. Genet. 2015, 24, 2201–2217. [Google Scholar] [CrossRef] [Green Version]
- Maccani, J.Z.J.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Kelsey, K.T. Placental DNA methylation alterations associated with maternal tobacco smoking at theRUNX3gene are also associated with gestational age. Epigenomics 2013, 5, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Breton, C.V.; Siegmund, K.D.; Joubert, B.R.; Wang, X.; Qui, W.; Carey, V.; Nystad, W.; Håberg, S.E.; Ober, C.; Nicolae, D.; et al. Prenatal Tobacco Smoke Exposure Is Associated with Childhood DNA CpG Methylation. PLoS ONE 2014, 9, e99716. [Google Scholar] [CrossRef] [Green Version]
- Lkhagvadorj, K.; Meyer, K.F.; Verweij, L.P.; Kooistra, W.; Reinders-Luinge, M.; Dijkhuizen, H.W.; De Graaf, I.A.M.; Plösch, T.; Hylkema, M.N. Prenatal smoke exposure induces persistent Cyp2a5 methylation and increases nicotine metabolism in the liver of neonatal and adult male offspring. Epigenetics 2020, 15, 1370–1385. [Google Scholar] [CrossRef]
- Kandel, D.B.; Hu, M.-C.; Griesler, P.C.; Schaffran, C. On the development of nicotine dependence in adolescence. Drug Alcohol Depend. 2007, 91, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Kandel, D.B.; Udry, J.R. Prenatal effects of maternal smoking on daughters’ smoking: Nicotine or testosterone exposure? Am. J. Public Health 1999, 89, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.K.; Mwenifumbo, J.C.; Koudsi, N.A.; Okuyemi, K.S.; Ahluwalia, J.S.; Benowitz, N.L.; Tyndale, R.F. Association of Nicotine Metabolite Ratio and CYP2A6 Genotype With Smoking Cessation Treatment in African-American Light Smokers. Clin. Pharmacol. Ther. 2009, 85, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, C.E.; Tyndale, R.; Patterson, F.; Wileyto, E.P.; Shields, P.G.; Pinto, A.; Benowitz, N. Nicotine metabolite ratio predicts efficacy of transdermal nicotine for smoking cessation. Clin. Pharmacol. Ther. 2006, 79, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Altea, S.; Fratta, W. Sex Differences in Drug Addiction: A Review of Animal and Human Studies. Women’s Health 2008, 4, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Yararbas, G.; Keser, A.; Kanit, L.; Pogun, S. Nicotine-induced conditioned place preference in rats: Sex differences and the role of mGluR5 receptors. Neuropharmacology 2010, 58, 374–382. [Google Scholar] [CrossRef]
- Tizabi, Y.; Popke, E.; Rahman, M.A.; Nespor, S.M.; Grunberg, N.E. Hyperactivity Induced by Prenatal Nicotine Exposure is Associated with an Increase in Cortical Nicotinic Receptors. Pharmacol. Biochem. Behav. 1997, 58, 141–146. [Google Scholar] [CrossRef]
- Guida, F.; Sandanger, T.M.; Castagné, R.; Campanella, G.; Polidoro, S.; Palli, D.; Krogh, V.; Tumino, R.; Sacerdote, C.; Panico, S.; et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet. 2015, 24, 2349–2359. [Google Scholar] [CrossRef]
- Markunas, C.A.; Xu, Z.; Harlid, S.; Wade, P.A.; Lie, R.T.; Taylor, J.A.; Wilcox, A.J. Identification of DNA Methylation Changes in Newborns Related to Maternal Smoking during Pregnancy. Environ. Health Perspect. 2014, 122, 1147–1153. [Google Scholar] [CrossRef]
- Lee, K.W.K.; Pausova, Z. Cigarette smoking and DNA methylation. Front. Genet. 2013, 4, 132. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: A systematic review of DNA methylation studies. Clin. Epigenetics 2015, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tsaprouni, L.G.; Yang, T.-P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Viñuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E.; et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Greene, R.; Pisano, M.M. Cigarette smoke induces proteasomal-mediated degradation of DNA methyltransferases and methyl CpG-/CpG domain-binding proteins in embryonic orofacial cells. Reprod. Toxicol. 2015, 58, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauk, C.L.; Polyzos, A.; Kortubash, I.; Williams, A.; Kovalchuk, O.; Rowan-Carroll, A. Tandem repeat mutation, global DNA methylation, and regulation of DNA methyltransferases in cultured mouse embryonic fibroblast cells chronically exposed to chemicals with different modes of action. Environ. Mol. Mutagen. 2008, 49, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.F.; Verkaik-Schakel, R.N.; Timens, W.; Kobzik, L.; Plösch, T.; Hylkema, M.N. The fetal programming effect of prenatal smoking on Igf1r and Igf1 methylation is organ- and sex-specific. Epigenetics 2017, 12, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.F.; Krauss-Etschmann, S.; Kooistra, W.; Reinders-Luinge, M.; Timens, W.; Kobzik, L.; Plösch, T.; Hylkema, M.N. Prenatal exposure to tobacco smoke sex dependently influences methylation and mRNA levels of the Igf axis in lungs of mouse offspring. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L542–L555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Ryan, J.; Pereira, N.; Boughton, B.A.; Craig, J.; Saffery, R. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics 2013, 9, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.A.; Lesseur, C.; Conradt, E.; Lester, B.M.; Marsit, C.J. Global and gene-specific DNA methylation across multiple tissues in early infancy: Implications for children’s health research. FASEB J. 2014, 28, 2088–2097. [Google Scholar] [CrossRef] [Green Version]
- Holloway, A.C.; Kellenberger, L.D.; Petrik, J.J. Fetal and Neonatal Exposure to Nicotine Disrupts Ovarian Function and Fertility in Adult Female Rats. Endocrine 2006, 30, 213–216. [Google Scholar] [CrossRef]
- Lamba, V.; Lamba, J.; Yasuda, K.; Strom, S.C.; Davila, J.; Hancock, M.L.; Fackenthal, J.D.; Rogan, P.K.; Ring, B.; Wrighton, S.A.; et al. Hepatic CYP2B6 Expression: Gender and Ethnic Differences and Relationship to CYP2B6 Genotype and CAR (Constitutive Androstane Receptor) Expression. J. Pharmacol. Exp. Ther. 2003, 307, 906–922. [Google Scholar] [CrossRef]
- Koudsi, N.A.; Hoffmann, E.B.; Assadzadeh, A.; Tyndale, R.F. Hepatic CYP2A6 levels and nicotine metabolism: Impact of genetic, physiological, environmental, and epigenetic factors. Eur. J. Clin. Pharmacol. 2010, 66, 239–251. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Lessov-Schlaggar, C.N.; Swan, G.E.; Jacob, P. Female sex and oral contraceptive use accelerate nicotine metabolism. Clin. Pharmacol. Ther. 2006, 79, 480–488. [Google Scholar] [CrossRef]
- Torres, O.V.; Natividad, L.A.; Tejeda, H.A.; Van Weelden, S.A.; O’Dell, L.E. Female rats display dose-dependent differences to the rewarding and aversive effects of nicotine in an age-, hormone-, and sex-dependent manner. Psychopharmacology 2009, 206, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrod, S.B.; Booze, R.M.; Mactutus, C.F. Sex differences in nicotine levels following repeated intravenous injection in rats are attenuated by gonadectomy. Pharmacol. Biochem. Behav. 2007, 86, 32–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Bakar, A.; Hakkola, J.; Juvonen, R.; Rahnasto-Rilla, M.; Raunio, H.; Lang, M.A. Function and Regulation of the Cyp2a5/CYP2A6 Genes in Response to Toxic Insults in the Liver. Curr. Drug Metab. 2012, 14, 137–150. [Google Scholar] [CrossRef]
- Arpiainen, S.; Raffalli-Mathieu, F.; Lang, M.A.; Pelkonen, O.; Hakkola, J. Regulation of the Cyp2a5 gene involves an aryl hydrocarbon receptor-dependent pathway. Mol. Pharmacol. 2005, 67, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, E.C.K.; Wildenauer, D.B.; Tyndale, R.F. Nicotine self-administration in mice is associated with rates of nicotine inactivation by CYP2A5. Psychopharmacology 2006, 184, 401–408. [Google Scholar] [CrossRef]
- Dempsey, D.; Tutka, P.; Iii, P.J.P.; Allen, F.; Schoedel, K.; Tyndale, R.F.; Benowitz, N.L. Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin. Pharmacol. Ther. 2004, 76, 64–72. [Google Scholar] [CrossRef]
- Massadeh, A.M.; Gharaibeh, A.A.; Omari, K.W. A Single-Step Extraction Method for the Determination of Nicotine and Cotinine in Jordanian Smokers’ Blood and Urine Samples by RP-HPLC and GC-MS. J. Chromatogr. Sci. 2009, 47, 170–177. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
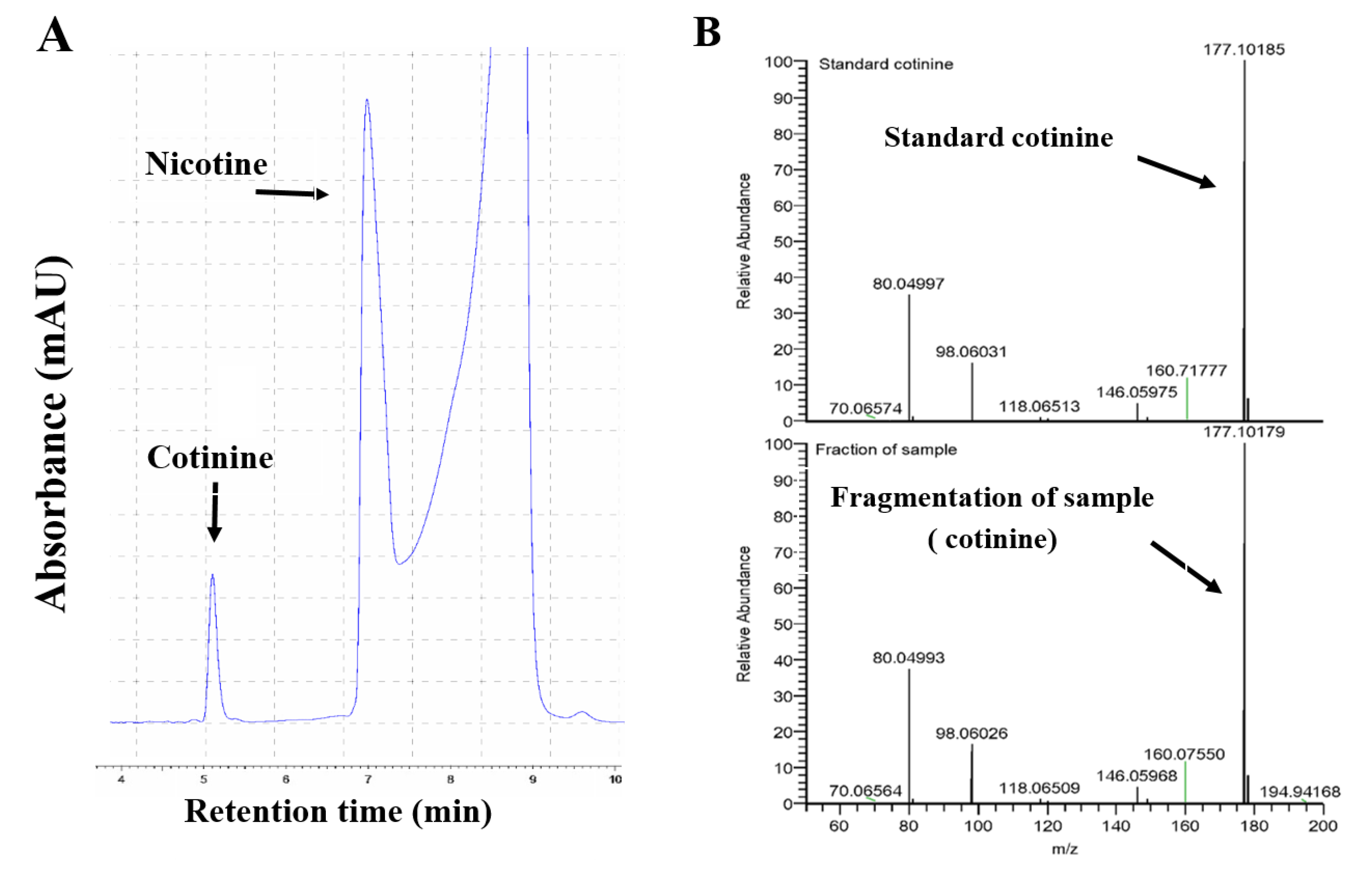{kind=link}
| Correlation of/with | All (Male and Female) | Male | Female | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyp2a5 (2-dCT) | All | All Control | All PreSE | All PostSE | All PreSE + PostSE | All | Control | PreSE | PostSE | PreSE + PostSE | All | Control | PreSE | PostSE | PreSE + PostSE | ||
| Cotinine formation (nmol/min/mg) | r | 0.10 | 0.67 | 0.07 | −0.03 | −0.61 | 0.28 | 0.21 | 0.26 | 0.33 | −0.25 | −0.04 | 0.47 | −0.40 | −0.19 | −0.63 | |
| p value | ns | 0.00 | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| Cyp2a5 promotor methylation (%) | CpG−614 | r | 0.20 | 0.02 | 0.83 | 0.39 | −0.31 | 0.06 | 0.38 | 0.20 | −0.25 | −0.06 | 0.44 | 0.12 | 0.82 | 0.76 | 0.53 |
| p value | ns | ns | 0.00 | ns | ns | ns | ns | ns | ns | ns | 0.00 | ns | ns | 0.00 | ns | ||
| CpG−589 | r | 0.27 | 0.38 | −0.10 | 0.23 | 0.48 | 0.31 | 0.03 | −0.11 | 0.08 | 0.9 | 0.55 | 0.29 | 0.32 | 0.93 | 0.22 | |
| p value | ns | ns | ns | ns | ns | ns | ns | ns | ns | 0.02 | 0.00 | ns | ns | 0.00 | ns | ||
| CpG−542 | r | −0.01 | −0.36 | 0.22 | 0.12 | 0.11 | −0.21 | 0.20 | −0.60 | −0.31 | −0.29 | 0.04 | 0.04 | −0.31 | −0.20 | 0.68 | |
| p value | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG−74 | r | −0.36 | −0.17 | −0.82 | −0.26 | −0.50 | 0.17 | 0.72 | 0.16 | −0.16 | −0.09 | 0.15 | 0.08 | −0.60 | 0.26 | 0.34 | |
| p value | 0.00 | ns | 0.00 | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG−7 | r | −0.20 | −0.33 | −0.85 | −0.03 | −0.32 | 0.19 | 0.35 | −0.67 | −0.03 | 0.03 | −0.35 | −0.67 | −0.63 | −0.39 | −0.46 | |
| p value | ns | ns | 0.00 | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ||
| CpG + 45 | r | −0.09 | −0.01 | −0.57 | 0.04 | −0.55 | 0.52 | 0.25 | −0.10 | 0.89 | −0.26 | 0.11 | 0.12 | 0.40 | 0.10 | −0.60 | |
| p value | ns | ns | ns | ns | ns | 0.00 | ns | ns | 0.00 | ns | ns | ns | ns | ns | ns | ||
| Gene | Targeted CpG-Sites | Sequence 5′-3′ |
|---|---|---|
| Cyp2a5 | CpG-614 to −542 | For: TTTGTGTTTGTTTTGAGTGTTGGGATTA Rev: CCCCATCCACAACCATTCTT Seq: GTTGGGATTATAGGTTTATATTA Sequence to analyze: TTATATTYGATTTTTGGGAGTTTTTTAATGAAGAGGATTTTGAATTTAAGGATGYGAGAA GTGGAGATTT TAGGGTTATYGG |
| CpG-74 to +45 | For: AGTGGATAGTTTGGAGGTGAAAT Rev: ACAACTTTCCTAAAAACTTTCTCTACTTC Seq1: GGAGGTGAAATAGTTGTATAATTAA Seq2: TAGTTATTATTGTTTGTTTATTAT Sequence to analyze: S1: GATTAAAGTTYGTTTTTTTGTTTTTGGATGTATAAAAGTAAGTTAATT S2: TATYGTTATTATGTTGATTTTAGGATTTTTTTTGGTGGTTGTAGTGGTTTTTTTTAGYGTTTTGGTTTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lkhagvadorj, K.; Zeng, Z.; Meyer, K.F.; Verweij, L.P.; Kooistra, W.; Reinders-Luinge, M.; Dijkhuizen, H.W.; de Graaf, I.A.M.; Plösch, T.; Hylkema, M.N. Postnatal Smoke Exposure Further Increases the Hepatic Nicotine Metabolism in Prenatally Smoke Exposed Male Offspring and Is Linked with Aberrant Cyp2a5 Methylation. Int. J. Mol. Sci. 2021, 22, 164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010164
Lkhagvadorj K, Zeng Z, Meyer KF, Verweij LP, Kooistra W, Reinders-Luinge M, Dijkhuizen HW, de Graaf IAM, Plösch T, Hylkema MN. Postnatal Smoke Exposure Further Increases the Hepatic Nicotine Metabolism in Prenatally Smoke Exposed Male Offspring and Is Linked with Aberrant Cyp2a5 Methylation. International Journal of Molecular Sciences. 2021; 22(1):164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010164
Chicago/Turabian StyleLkhagvadorj, Khosbayar, Zhijun Zeng, Karolin F. Meyer, Laura P. Verweij, Wierd Kooistra, Marjan Reinders-Luinge, Henk W. Dijkhuizen, Inge A. M. de Graaf, Torsten Plösch, and Machteld N. Hylkema. 2021. "Postnatal Smoke Exposure Further Increases the Hepatic Nicotine Metabolism in Prenatally Smoke Exposed Male Offspring and Is Linked with Aberrant Cyp2a5 Methylation" International Journal of Molecular Sciences 22, no. 1: 164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010164