Pregnancy-Induced High Plasma Levels of Soluble Endoglin in Mice Lead to Preeclampsia Symptoms and Placental Abnormalities

 , , and
, , and
Abstract
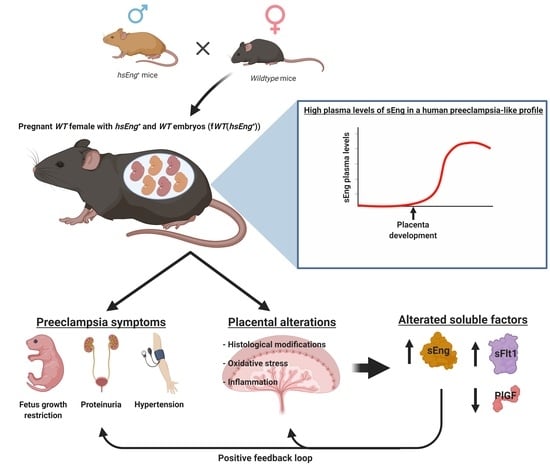
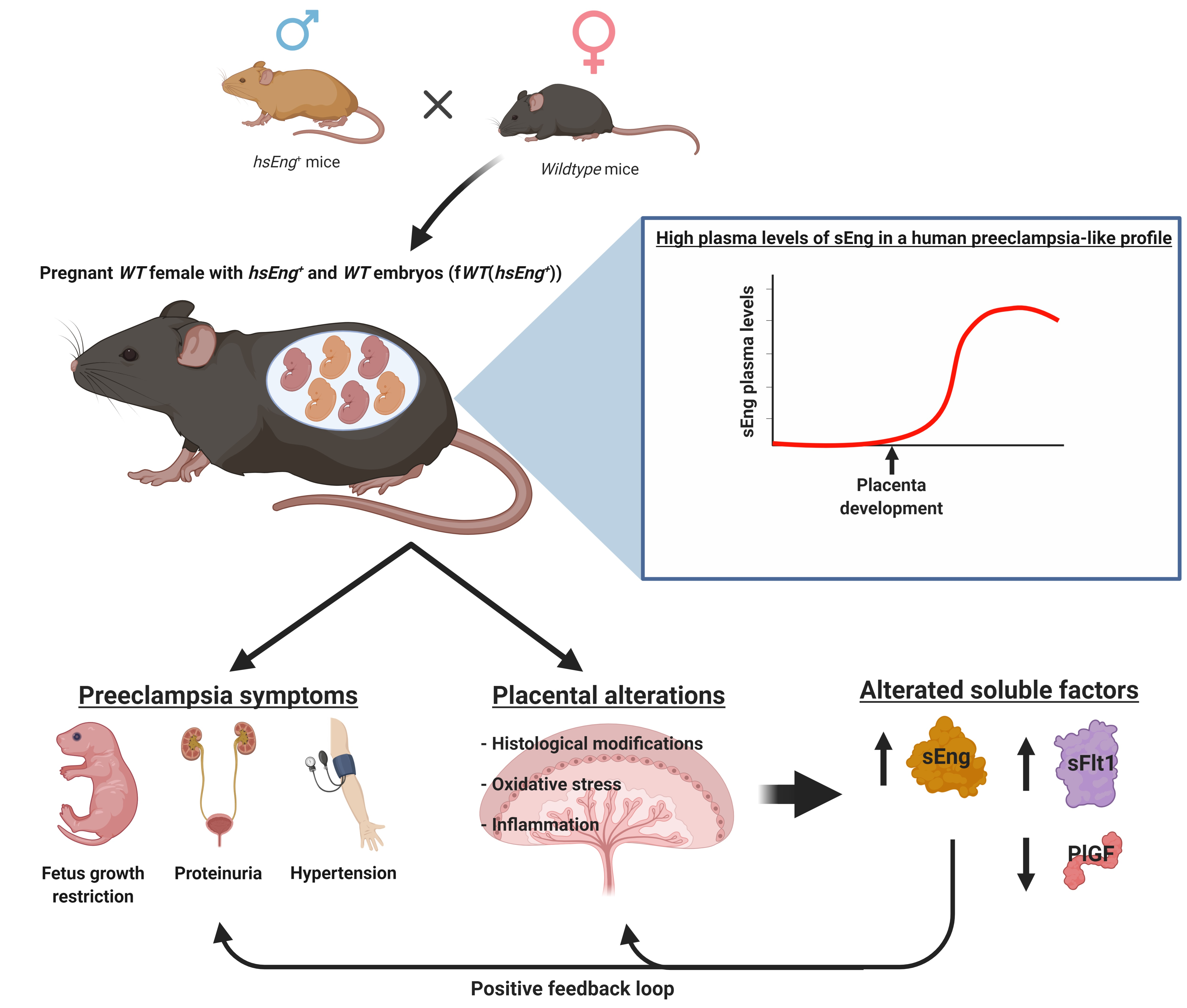
:
1. Introduction
2. Results
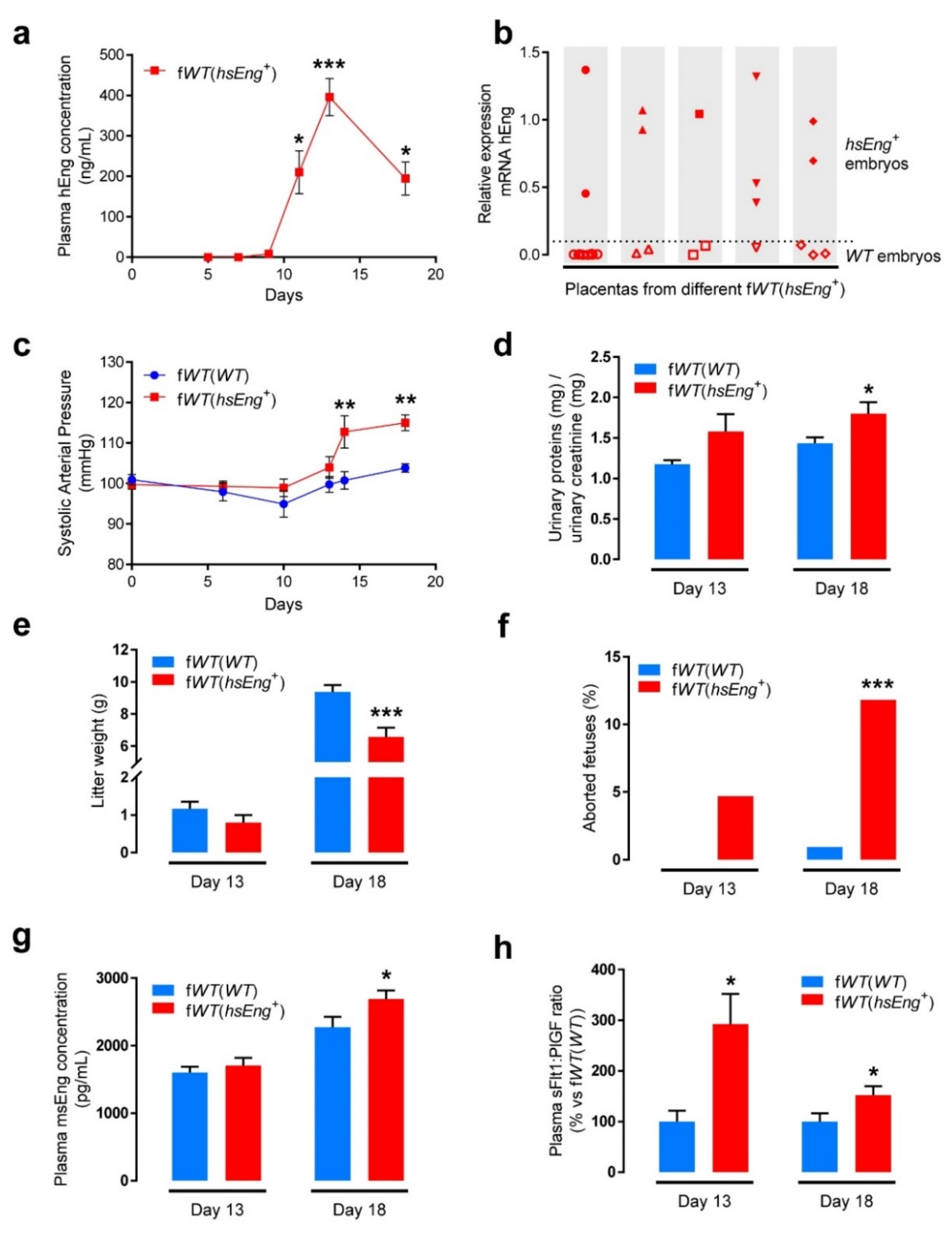
2.1. Wild Type Pregnant Females with hsEng+ Fetuses Show High Plasma Levels of Soluble Eng
2.2. Pregnant fWT(hsEng+) Mice Present Symptoms and Biomarkers of Preeclampsia
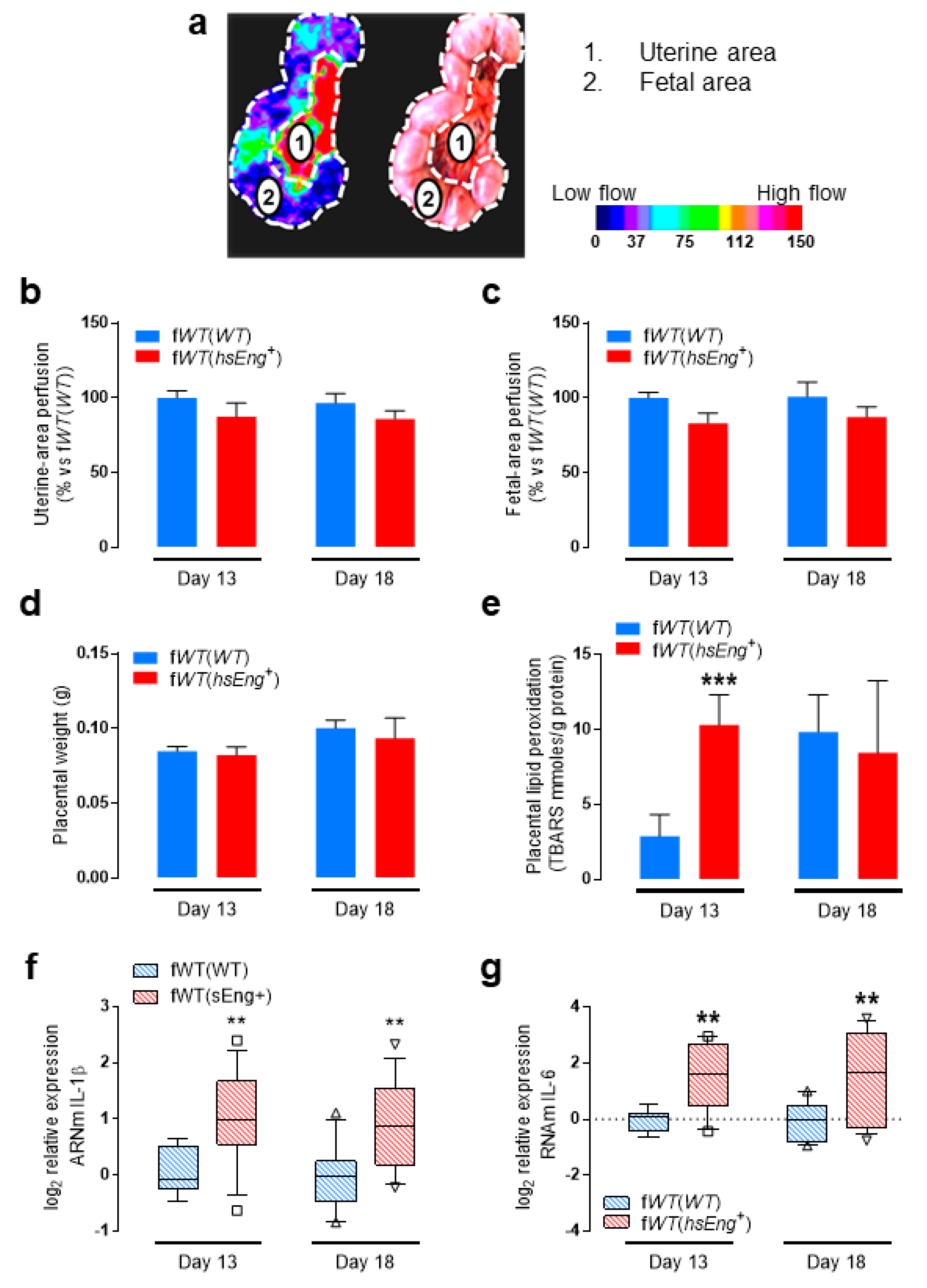
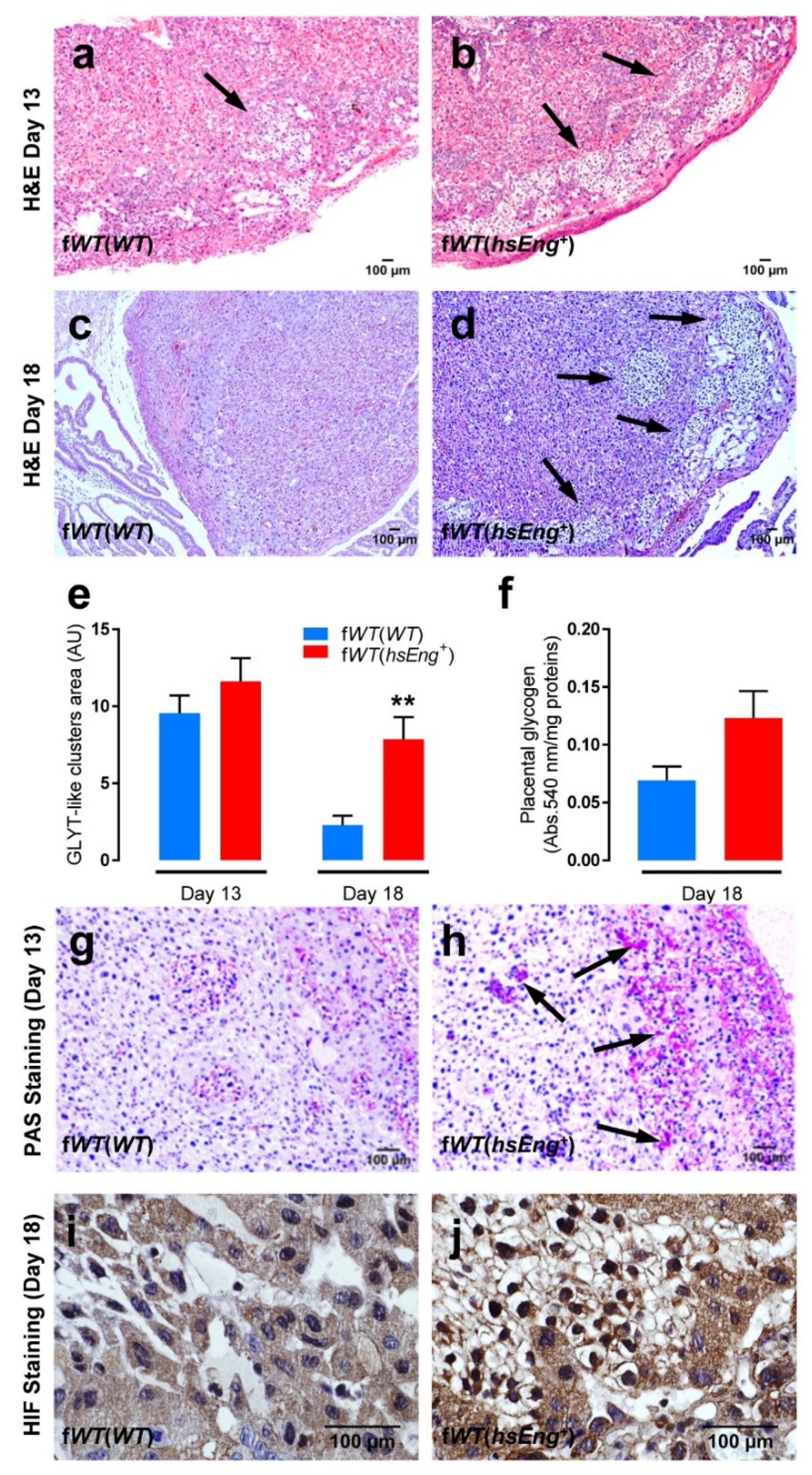
2.3. Presence of Soluble Endoglin Is Associated with Placental Alterations
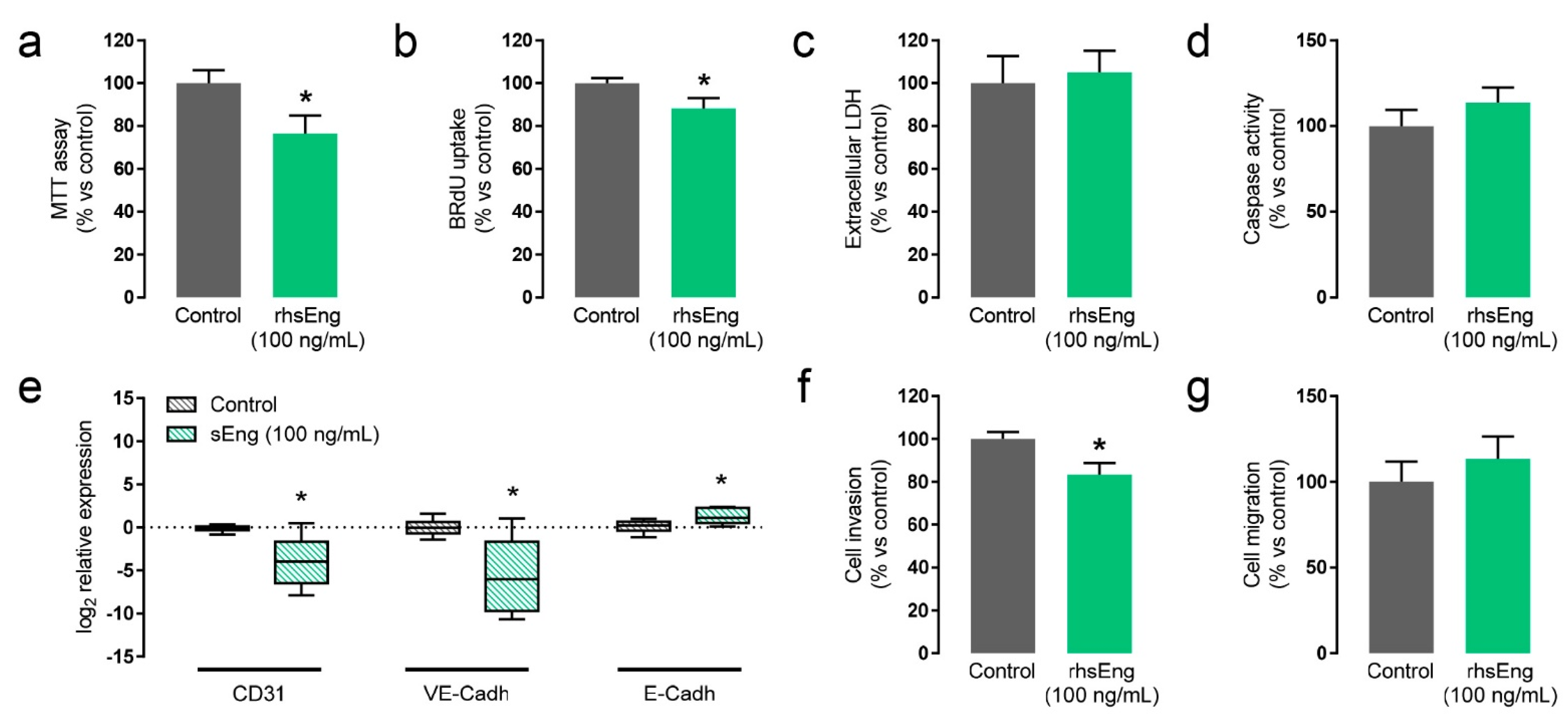
2.4. Soluble Endoglin Modifies Cytotrophoblast Cell Biology
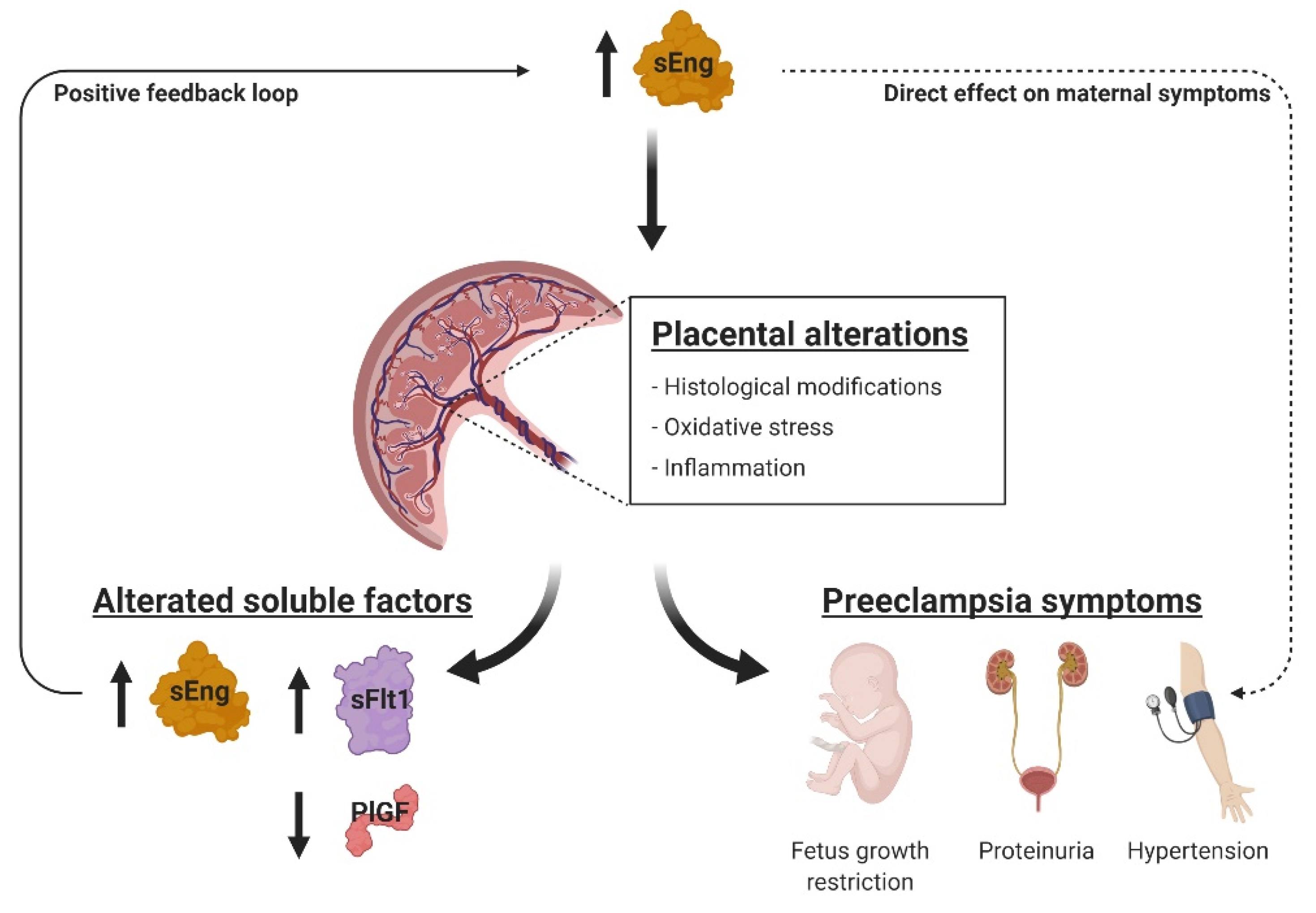
3. Discussion
4. Materials and Methods
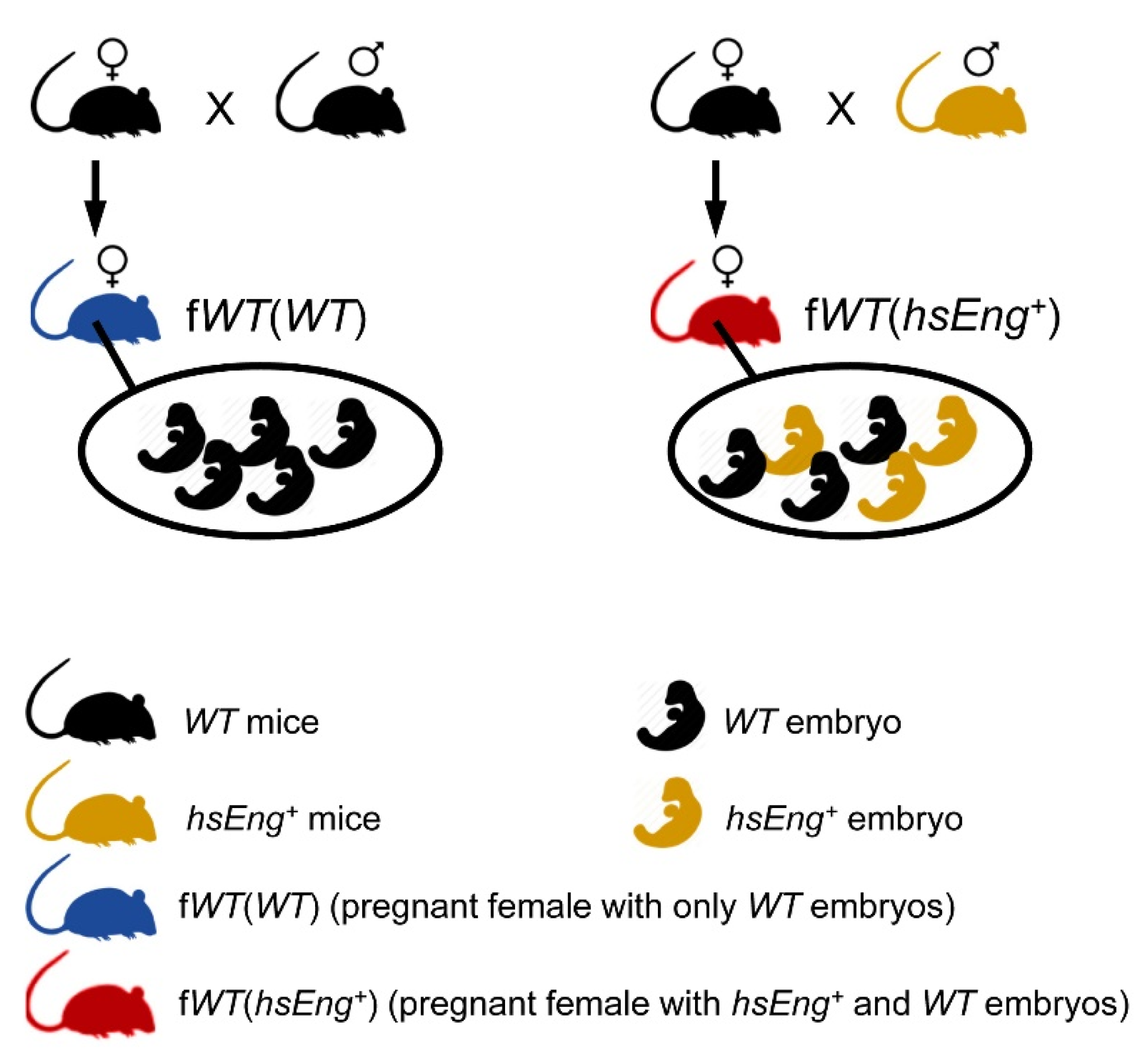
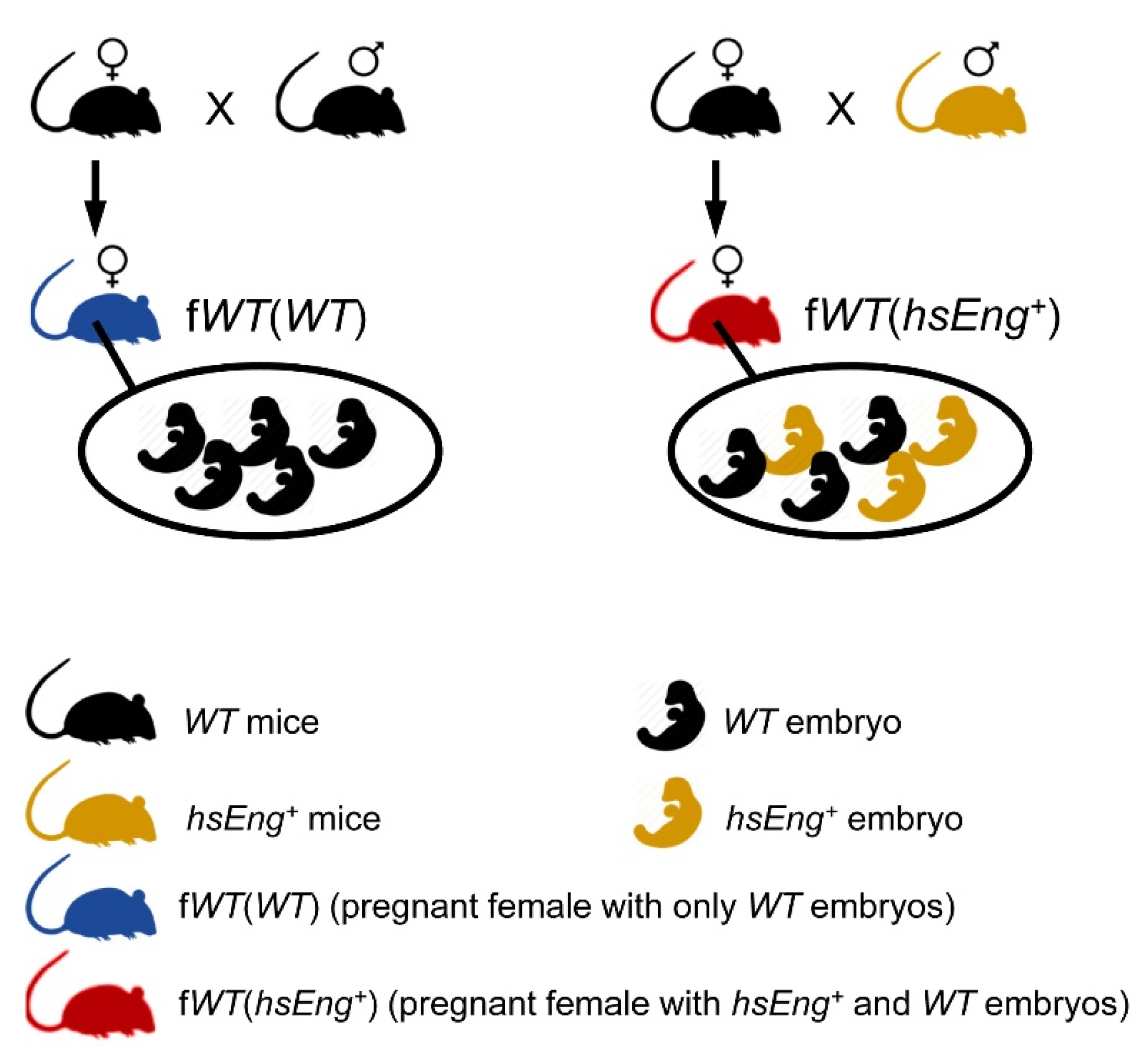
4.1. Animals and Study Design
4.2. Enzyme-Linked Immunosorbent Assays (ELISA)
4.3. Arterial Blood Pressure Measurement
4.4. Renal Function Measurement
4.5. Gene Expression Assays
4.6. Placental and Fetal Perfusion
4.7. Histological Studies
4.8. Placental Biochemical Determinations
4.9. Cell Culture and Placental Explants
4.10. Cell Proliferation and Apoptosis Assays
4.11. Transwell Migration and Invasion Assays
4.12. Data Presentation and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BMP4 | Bone morphogenetic protein 4 |
| CAMs | Cell adhesion molecules |
| BrdU | 5-Bromo-2-deoxyuridine |
| enCT | Endovascular cytotrophoblasts |
| Eng | Endoglin |
| EVCT | Extravillous cytotrophoblasts |
| FBS | Fetal bovine serum |
| fWT(hsEng+) | Pregnant WT females bearing both WT and hsEng+ fetuses |
| fWT(WT) | Pregnant WT females bearing only WT fetuses |
| GLYT | Glycogen-containing trophoblast |
| H&E | Hematoxylin and eosin |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| hsEng | Human soluble endoglin |
| IL | Interleukin |
| LDH | Lactate dehydrogenase |
| MMP | Matrix metalloprotease |
| msEng | Murine soluble endoglin |
| MTT | 3-[4.5-Dimethylthiazol-2-yl]-2.5-diphenyl tetrazolium bromide |
| PAS | Periodic acid-Schiff |
| PlGF | Placental growth factor |
| qRT-PCR | Quantitative reverse transcription polymerase chain reaction |
| rhsEng | Recombinant human soluble endoglin |
| ROS | Radical oxygen species |
| RUPP | Reduction of uterine perfusion |
| sEng | Soluble endoglin |
| SEM | Standard error of the mean |
| sFlt1 | Fms-like kinase 1 |
| SpT | Spongiotrophoblasts |
| VEGF | Vascular endothelial growth factor |
| WT | Wild type |
References
- Eiland, E.; Nzerue, C.; Faulkner, M. Preeclampsia 2012. J. Pregnancy 2012, 2012, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duley, L. The global impact of pre-eclampsia and eclampsia. Semin. Perinatol. 2009, 33, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.A.; Schlitt, J.M.; Jackson, D.L.; Schulz, L.C.; Schust, D.J. Preeclampsia: Multiple approaches for a multifactorial disease. Dis. Model. Mech. 2012, 5, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.M.; Escudero, C. The placenta in preeclampsia. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2012, 2, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piering, W.F.; Garancis, J.G.; Becker, C.G.; Beres, J.A.; Lemann, J. Preeclampsia related to a functioning extrauterine placenta: Report of a case and 25-year follow-up. Am. J. Kidney Dis. 1993, 21, 310–313. [Google Scholar] [CrossRef]
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 2007, 50, 1142–1147. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.S.; Gilbert, S.A.B.; Arany, M.; Granger, J.P. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension 2009, 53, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Hawinkels, L.J.A.C.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.M.; Ten Dijke, P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef] [Green Version]
- Gougos, A.; Letarte, M. Primary structure of endoglin, and RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar]
- Gougos, A.; St Jacques, S.; Greaves, A.; O’Connell, P.J.; d’Apice, A.J.; Bühring, H.J.; Bernabeu, C.; van Mourik, J.A.; Letarte, M. Identification of distinct epitopes of endoglin, an RGD-containing glycoprotein of endothelial cells, leukemic cells, and syncytiotrophoblasts. Int. Immunol. 1992, 4, 83–92. [Google Scholar] [CrossRef]
- Caniggia, I.; Taylor, C.V.; Ritchie, J.W.K.; Lye, S.J.; Letarte, M. Endoglin regulates trophoblast differentiation along the invasive pathway in human placental villous explants. Endocrinology 1997, 138, 4977–4988. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Novoa, J.M.; Bernabeu, C. The physiological role of endoglin in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H959–H974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaitu’u-Lino, T.J.; Palmer, K.R.; Whitehead, C.L.; Williams, E.; Lappas, M.; Tong, S. MMP-14 is expressed in preeclamptic placentas and mediates release of soluble endoglin. Am. J. Pathol. 2012, 180, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Valbuena-Diez, A.C.; Blanco, F.J.; Oujo, B.; Langa, C.; Gonzalez-Nunez, M.; Llano, E.; Pendas, A.M.; Diaz, M.; Castrillo, A.; Lopez-Novoa, J.M.; et al. Oxysterol-induced soluble endoglin release and its involvement in hypertension. Circulation 2012, 126, 2612–2624. [Google Scholar] [CrossRef] [Green Version]
- Kaitu’u-Lino, T.J.; Tuohey, L.; Ye, L.; Palmer, K.; Skubisz, M.; Tong, S. MT-MMPs in pre-eclamptic placenta: Relationship to soluble endoglin production. Placenta 2013, 34, 168–173. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Zhang, H.-Y.; Lu, S.; Jiang, L.-L.; Wu, J.; Yang, Y.-L.; Zhang, S.-A. MMP-14 aggravates onset of severe preeclampsia by mediating soluble endoglin release. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1209–1215. [Google Scholar]
- López-Novoa, J.M. Soluble endoglin is an accurate predictor and a pathogenic molecule in pre-eclampsia. Nephrol. Dial. Transplant. 2007. [Google Scholar] [CrossRef]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.-H.; Yuan, H.-T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Oujo, B.; Perez-Barriocanal, F.; Bernabeu, C.; Lopez-Novoa, J.M. Membrane and soluble forms of endoglin in preeclampsia. Curr. Mol. Med. 2013, 13, 1345–1357. [Google Scholar] [CrossRef]
- Rathouska, J.; Jezkova, K.; Nemeckova, I.; Nachtigal, P. Soluble endoglin, hypercholesterolemia and endothelial dysfunction. Atherosclerosis 2015, 243, 383–388. [Google Scholar] [CrossRef]
- Gallardo-Vara, E.; Blanco, F.J.; Roqué, M.; Friedman, S.L.; Suzuki, T.; Botella, L.M.; Bernabeu, C. Transcription factor KLF6 upregulates expression of metalloprotease MMP14 and subsequent release of soluble endoglin during vascular injury. Angiogenesis 2016, 19, 155–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P.; et al. MMP-12, secreted by pro-inflammatory macrophages, targets endoglin in human macrophages and endothelial cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Düwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabañas, C.; Lopez-Novoa, J.M.J.M.J.M.; Bernabeu, C.; et al. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, E.; Smadja, D.M.; Boscolo, E.; Langa, C.; Arevalo, M.A.; Pericacho, M.; Gamella-Pozuelo, L.; Kauskot, A.; Botella, L.M.; Gaussem, P.; et al. Endoglin regulates mural cell adhesion in the circulatory system. Cell. Mol. Life Sci. 2016, 73, 1715–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitverova, B.; Blazickova, K.; Najmanova, I.; Vicen, M.; Hyšpler, R.; Dolezelova, E.; Nemeckova, I.; Tebbens, J.D.; Bernabeu, C.; Pericacho, M.; et al. Soluble endoglin and hypercholesterolemia aggravate endothelial and vessel wall dysfunction in mouse aorta. Atherosclerosis 2018, 271. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Vara, E.; Gamella-Pozuelo, L.; Perez-Roque, L.; Bartha, J.L.; Garcia-Palmero, I.; Casal, J.I.; López-Novoa, J.M.; Pericacho, M.; Bernabeu, C. Potential role of circulating endoglin in hypertension via the upregulated expression of BMP4. Cells 2020, 9, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitverova, B.; Najmanova, I.; Vicen, M.; Tripska, K.; Cristina, I.; Sa, I.; Hyspler, R.; Pericacho, M.; Nachtigal, P. Long term effects of soluble endoglin and mild hypercholesterolemia in mice hearts. PLoS ONE 2020, 15, e0233725. [Google Scholar] [CrossRef]
- Than, N.G.; Romero, R.; Hillermann, R.; Cozzi, V.; Nie, G.; Huppertz, B. Prediction of preeclampsia—A workshop report. Placenta 2008, 29 (Suppl. A), S83–S85. [Google Scholar] [CrossRef] [Green Version]
- Hod, T.; Cerdeira, A.S.; Karumanchi, S.A. Molecular mechanisms of preeclampsia. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Grill, S.; Rusterholz, C.; Zanetti-Dällenbach, R.; Tercanli, S.; Holzgreve, W.; Hahn, S.; Lapaire, O. Potential markers of preeclampsi—A review. Reprod. Biol. Endocrinol. 2009, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-B.; Folger, J.K.; Rajput, S.K.; Smith, G.W. Temporal regulation of mRNAs for select bone morphogenetic proteins (BMP), BMP receptors and their associated SMAD proteins during bovine early embryonic development: Effects of exogenous BMP2 on embryo developmental progression. Reprod. Biol. Endocrinol. 2014, 12, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, A.; Sekizawa, A.; De Sanctis, P.; Purwosunu, Y.; Okai, T.; Cha, D.H.; Kang, J.H.; Vicenzi, C.; Tempesta, A.; Wibowo, N.; et al. Gene expression in chorionic villous samples at 11 weeks’ gestation from women destined to develop preeclampsia. Prenat. Diagn. 2008, 28, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Troisi, R.; Braekke, K.; Harsem, N.K.; Hyer, M.; Hoover, R.N.; Staff, A.C. Blood pressure augmentation and maternal circulating concentrations of angiogenic factors at delivery in preeclamptic and uncomplicated pregnancies. Am. J. Obstet. Gynecol. 2008, 199, 653.e1–653.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purwosunu, Y.; Sekizawa, A.; Farina, A.; Wibowo, N.; Koide, K.; Okazaki, S.; Nakamura, M.; Okai, T. Evaluation of physiological alterations of the placenta through analysis of cell-free messenger ribonucleic acid concentrations of angiogenic factors. Am. J. Obstet. Gynecol. 2008, 198, 124.e1–124.e7. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Crovetto, F.; Figueras, F.; Triunfo, S.; Crispi, F.; Rodriguez-Sureda, V.; Peguero, A.; Dominguez, C.; Gratacos, E. Added value of angiogenic factors for the prediction of early and late preeclampsia in the first trimester of pregnancy. Fetal Diagn. Ther. 2014, 35, 258–266. [Google Scholar] [CrossRef]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Nemeckova, I.; Serwadczak, A.; Oujo, B.; Jezkova, K.; Rathouska, J.; Fikrova, P.; Varejckova, M.; Bernabeu, C.; Lopez-Novoa, J.M.; Chlopicki, S.; et al. High soluble endoglin levels do not induce endothelial dysfunction in mouse aorta. PLoS ONE 2015, 10, e0119665. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Chen, L.; Li, R. Values of serum sFlt-1, PLGF levels, and sFlt-1/PLGF ratio in diagnosis and prognosis evaluation of preeclamptic patients. Clin. Exp. Hypertens. 2020, 42, 601–607. [Google Scholar] [CrossRef]
- Whitley, G.S.J.; Cartwright, J.E. Cellular and molecular regulation of spiral artery remodelling: Lessons from the cardiovascular field. Placenta 2010, 31, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Damsky, C.H.; Fisher, S.J. Trophoblast pseudo-vasculogenesis: Faking it with endothelial adhesion receptors. Curr. Opin. Cell Biol. 1998, 10, 660–666. [Google Scholar] [CrossRef]
- Zhou, Y.; Damsky, C.H.; Fisher, S.J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Investig. 1997, 99, 2152–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, S.J. The placental problem: Linking abnormal cytotrophoblast differentiation to the maternal symptoms of preeclampsia. Reprod. Biol. Endocrinol. 2004, 2, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.E.; Moore Simas, T.A.; Bur, L.; Crawford, S.L.; Solitro, M.J.; Meyer, B.A. Soluble endoglin for the prediction of preeclampsia in a high risk cohort. Hypertens. Pregnancy 2010, 29, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Leaños-Miranda, A.; Navarro-Romero, C.S.; Sillas-Pardo, L.J.; Ramírez-Valenzuela, K.L.; Isordia-Salas, I.; Jiménez-Trejo, L.M. Soluble endoglin as a marker for preeclampsia, its severity, and the occurrence of adverse outcomes. Hypertension 2019, 74, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Hubel, C.A. The two stage model of preeclampsia: Variations on the theme. Placenta 2009, 30, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Sones, J.L.; Davisson, R.L. Preeclampsia, of mice and women. Physiol. Genom. 2016, 48, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Doridot, L.; Passet, B.; Méhats, C.; Rigourd, V.; Barbaux, S.; Ducat, A.; Mondon, F.; Vilotte, M.; Castille, J.; Breuiller-Fouché, M.; et al. Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension 2013, 61, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Gatford, K.L.; Andraweera, P.H.; Roberts, C.T.; Care, A.S. Animal models of preeclampsia: Causes, consequences, and interventions. Hypertension 2020, 75, 1363–1381. [Google Scholar] [CrossRef]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [Green Version]
- Yinon, Y.; Nevo, O.; Xu, J.; Many, A.; Rolfo, A.; Todros, T.; Post, M.; Caniggia, I. Severe intrauterine growth restriction pregnancies have increased placental endoglin levels: Hypoxic regulation via transforming growth factor-β3. Am. J. Pathol. 2008, 172, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maria, F.J.; See, H.Y.C. Role of the placenta in pre-eclampsia. Am. J. Obstet. Gynecol. 1966, 94, 471–476. [Google Scholar] [CrossRef]
- Brosens, I.; Robertson, W.B.; Dixon, H.G. The physiological response of the vessels of the placental bed to normal pregnancy. J. Pathol. Bacteriol. 1967, 93, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Yung, H.-W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 2009, 30 (Suppl. A), S43–S48. [Google Scholar] [CrossRef] [Green Version]
- Pijnenborg, R.; Vercruysse, L.; Hanssens, M. The uterine spiral arteries in human pregnancy: Facts and controversies. Placenta 2006, 27, 939–958. [Google Scholar] [CrossRef]
- Bouillot, S.; Rampon, C.; Tillet, E.; Huber, P. Tracing the glycogen cells with protocadherin 12 during mouse placenta development. Placenta 2006, 27, 882–888. [Google Scholar] [CrossRef] [Green Version]
- Coan, P.M.; Conroy, N.; Burton, G.J.; Ferguson-Smith, A.C. Origin and characteristics of glycogen cells in the developing murine placenta. Dev. Dyn. 2006, 235, 3280–3294. [Google Scholar] [CrossRef]
- Rossant, J.; Cross, J.C. Placental development: Lessons from mouse mutants. Nat. Rev. Genet. 2001, 2, 538–548. [Google Scholar] [CrossRef]
- Wiemers, D.O.; Shao, L.J.; Ain, R.; Dai, G.; Soares, M.J. The mouse prolactin gene family locus. Endocrinology 2003, 144, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Adamson, S.L.; Lu, Y.; Whiteley, K.J.; Holmyard, D.; Hemberger, M.; Pfarrer, C.; Cross, J.C. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev. Biol. 2002, 250, 358–373. [Google Scholar] [CrossRef]
- Coan, P.M.; Ferguson-Smith, A.C.; Burton, G.J. Developmental dynamics of the definitive mouse placenta assessed by stereology. Biol. Reprod. 2004, 70, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, L. The placental imprintome and imprinted gene function in the trophoblast glycogen cell lineage. Reprod. Biomed. Online 2012, 25, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Chamorro-Jorganes, A.; Grande, M.T.; Herranz, B.; Jerkic, M.; Griera, M.; Gonzalez-Nuñez, M.; Santos, E.; Rodriguez-Puyol, D.; Lopez-Novoa, J.M.; Rodriguez-Puyol, M.; et al. Targeted genomic disruption of h-ras induces hypotension through a NO-cGMP-PKG pathway-dependent mechanism. Hypertension 2010, 56, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Nunez, M.; Riolobos, A.S.; Castellano, O.; Fuentes-Calvo, I.; de los Angeles Sevilla, M.; Oujo, B.; Pericacho, M.; Cruz-Gonzalez, I.; Perez-Barriocanal, F.; ten Dijke, P.; et al. Heterozygous disruption of activin receptor-like kinase 1 is associated with increased arterial pressure in mice. Dis. Model. Mech. 2015, 8, 1427–1439. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Félix, J.M.; Pérez-Roque, L.; Núñez-Gómez, E.; Oujo, B.; Arévalo, M.; Ruiz-Remolina, L.; Cuesta, C.; Langa, C.; Pérez-Barriocanal, F.; Bernabeu, C.; et al. Overexpression of the short endoglin isoform reduces renal fibrosis and inflammation after unilateral ureteral obstruction. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 1801–1814. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Prieto-García, L.; Vicente-Vicente, L.; Blanco-Gozalo, V.; Hidalgo-Thomas, O.; García-Macías, M.C.; Kurtz, A.; Layton, A.T.; Sanz, A.B.; Morales, A.I.; Martínez-Salgado, C.; et al. Pathophysiological mechanisms underlying a rat model of triple whammy acute kidney injury. Lab. Investig. 2020, 100, 1455–1464. [Google Scholar] [CrossRef]
- Munoz-Felix, J.M.; Fuentes-Calvo, I.; Cuesta, C.; Eleno, N.; Crespo, P.; Lopez-Novoa, J.M.; Martinez-Salgado, C. Absence of K-Ras reduces proliferation and migration but increases extracellular matrix synthesis in fibroblasts. J. Cell. Physiol. 2016. [Google Scholar] [CrossRef]
- Sancho-Martínez, S.M.; Prieto-García, L.; Prieto, M.; Fuentes-Calvo, I.; López-Novoa, J.M.; Morales, A.I.; Martínez-Salgado, C.; López-Hernández, F.J. N-acetylcysteine transforms necrosis into apoptosis and affords tailored protection from cisplatin cytotoxicity. Toxicol. Appl. Pharmacol. 2018, 349, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Letamendía, A.; Lastres, P.; Almendro, N.; Raab, U.; Bühring, H.-J.; Kumar, S.; Bernabéu, C. Endoglin, a component of the TGF-β receptor system, is a differentiation marker of human choriocarcinoma cells. Int. J. Cancer 1998, 76, 541–546. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Protein) | qPCR Primers | Organism |
|---|---|---|
| ENG (endoglin) | Forward-AGGTGCTTCTGGTCCTCAGT Reverse-CCACTCAAGGATCTGGGTCT | Human |
| Il1b (Interleukin-1β) | Forward-GCCTGTGTTTTCCTCCTTGC Reverse-TGCTGCCTAATGTCCCCTTG | Mouse |
| Il6 (Interleukin-6) | Forward-TCCAGTTGCCTTCTTGGGAC Reverse-AGTCTCCTCTCCGGACTTGT | Mouse |
| Actb (β-actin) | Forward-TCTACAAATGTGGCTGAGGACT Reverse-GAGGGACTTCCTGTAACCACTT | Mouse |
| CADH1 (E-cadherin) | Forward-CAAGCTATCCTTGCACCTCAG Reverse-GCATCAGAGAACTCCTATCTT | Human |
| CADH5 (VE-cadherin) | Forward-TGGTCACCCATGCATCTTCC Reverse-CCATGACGAAGGGTGAGCTT | Human |
| PECAM1 (CD31) | Forward-TGCCGTGGAAAGCAGATAC Reverse-GGAGCAGGGCAGGTTCATAA | Human |
| GAPDH | Forward-CAATGACCCCTTCATTGACC Reverse-GACAAGCTTCCCGTTCTCAG | Human |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Roque, L.; Núñez-Gómez, E.; Rodríguez-Barbero, A.; Bernabéu, C.; López-Novoa, J.M.; Pericacho, M. Pregnancy-Induced High Plasma Levels of Soluble Endoglin in Mice Lead to Preeclampsia Symptoms and Placental Abnormalities. Int. J. Mol. Sci. 2021, 22, 165. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010165
Pérez-Roque L, Núñez-Gómez E, Rodríguez-Barbero A, Bernabéu C, López-Novoa JM, Pericacho M. Pregnancy-Induced High Plasma Levels of Soluble Endoglin in Mice Lead to Preeclampsia Symptoms and Placental Abnormalities. International Journal of Molecular Sciences. 2021; 22(1):165. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010165
Chicago/Turabian StylePérez-Roque, Lucía, Elena Núñez-Gómez, Alicia Rodríguez-Barbero, Carmelo Bernabéu, José M. López-Novoa, and Miguel Pericacho. 2021. "Pregnancy-Induced High Plasma Levels of Soluble Endoglin in Mice Lead to Preeclampsia Symptoms and Placental Abnormalities" International Journal of Molecular Sciences 22, no. 1: 165. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010165