PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects

 ,
,
Abstract
:1. Breast Cancer–Incidence and Risk Factors
2. Molecular Types of Breast Cancer
- ER-estrogen receptor status.
- PR-progesterone receptor status.
- Note that HR-represents the joint assessment of ER and PR status.
- 3.
- HER2-human epidermal growth factor receptor 2 status.
- HR+/HER2– corresponding to Luminal A subtype.
- HR+/HER2+ corresponding to Luminal B subtype.
- HR−/HER2+ corresponding to HER2 enriched subtype.
- HR−/HER2– corresponding to triple negative subtype.
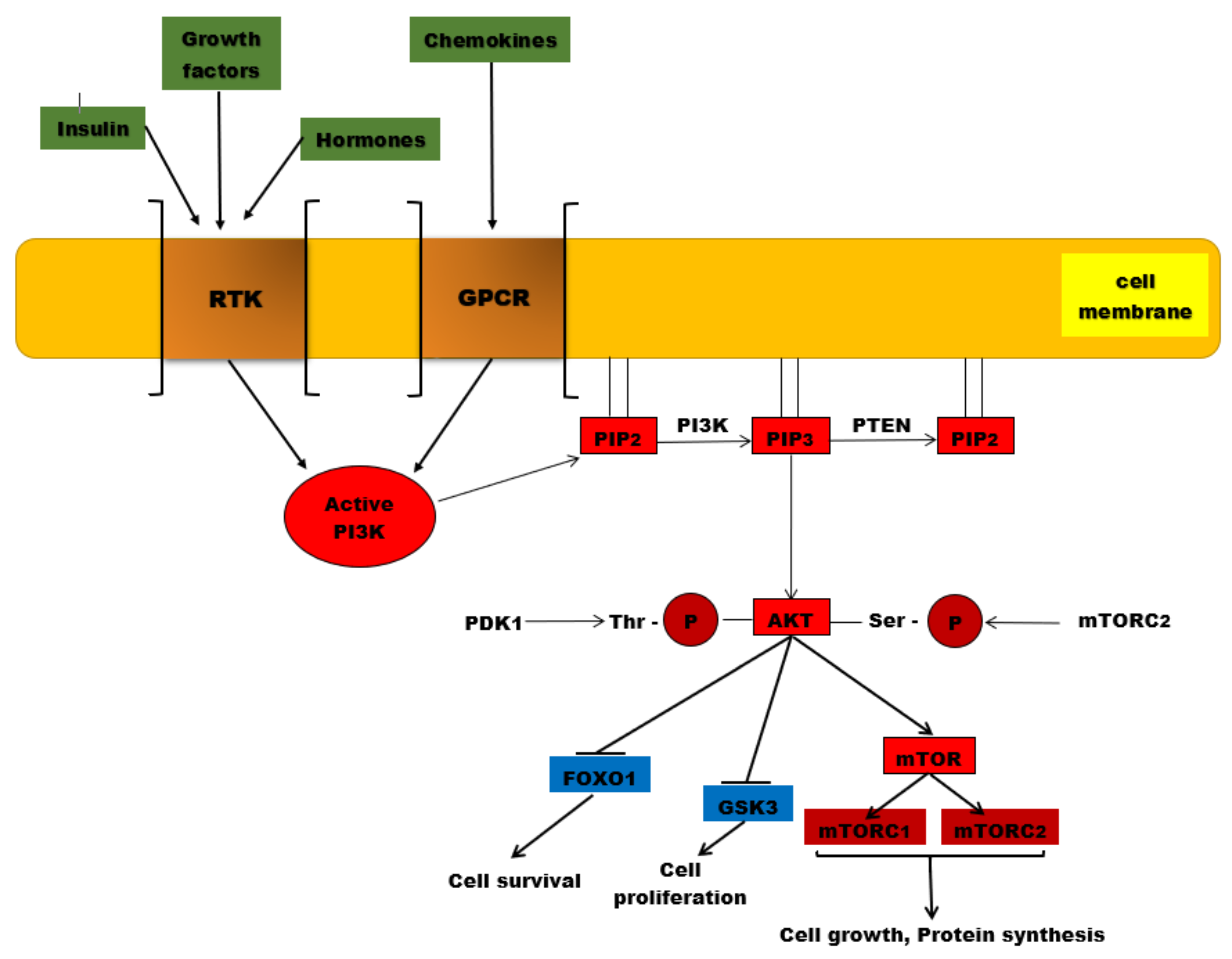
3. PI3K/AKT/mTOR Signaling Pathway
3.1. PI3K/AKT/mTOR Signaling Pathway Members
3.1.1. PI3K-Phosphatidylinositol 3-Kinase-(Phosphoinositide 3-Kinase)
3.1.2. AKT
3.1.3. mTOR
3.1.4. FoxO1
3.1.5. PTEN
4. PI3K/AKT/mTOR Mutations in Breast Cancer
HER Receptors and Breast Cancers
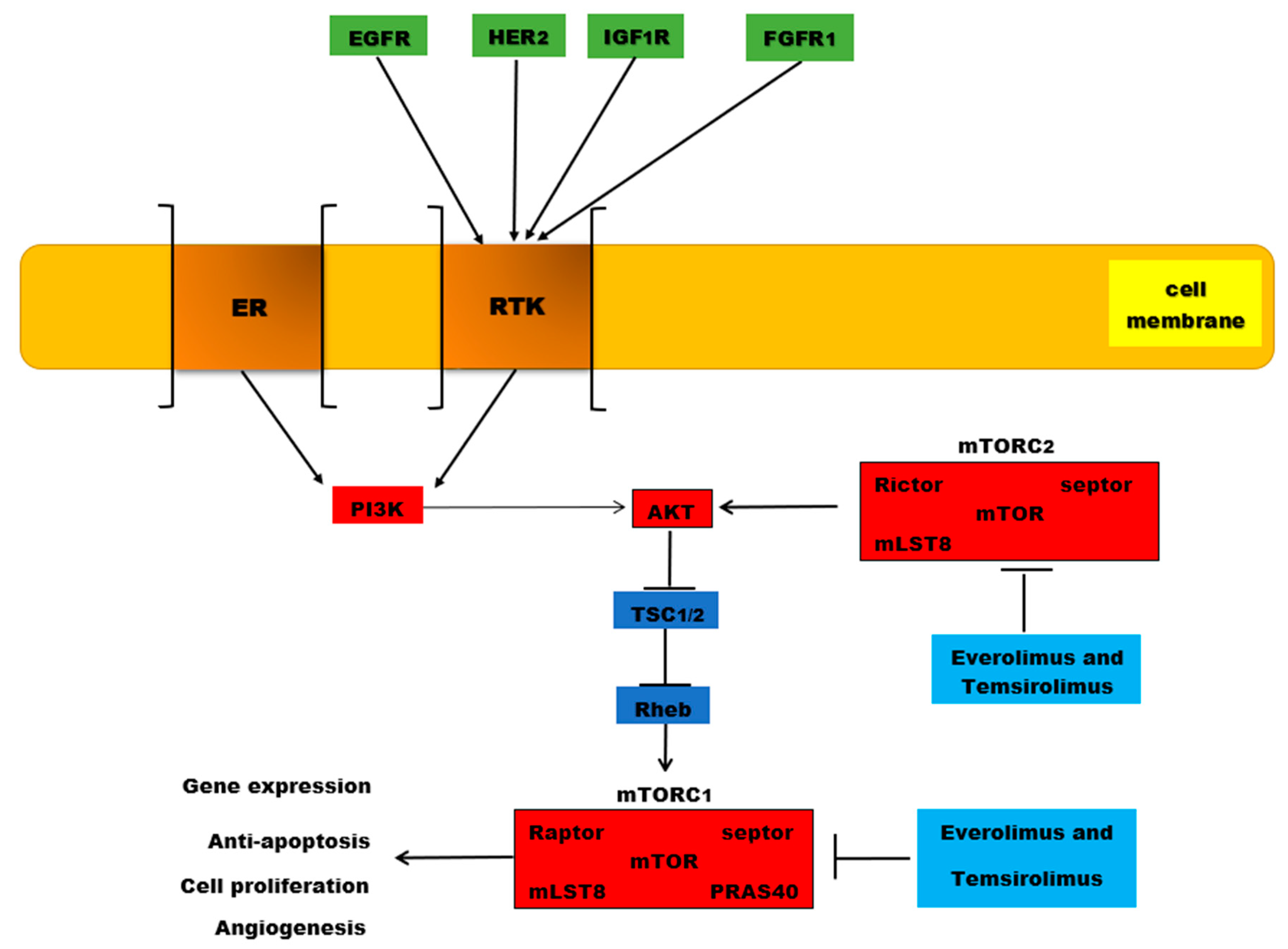
5. Mechanisms of Endocrine Resistance in Breast Cancer
6. mTOR Inhibitors: Everolimus and Temsirolimus
7. Conclusions
Funding
Conflicts of Interest
References
- Breast Cancer in Men—CDC Report 11 August 2020. Available online: www.cdc.gov/cancer/men (accessed on 6 October 2020).
- Sancho-Garnier, H.; Colonna, M. Épidémiologie des cancers du sein: Breast cancer epidemiology. Presse Med. 2019, 48, 1076–1084. [Google Scholar] [CrossRef]
- Graham, A.C. Breast Cancer Epidemiology and Risk Factors. Medscape Report 26 December 2019. Available online: https://emedicine.medscape.com/article/1697353-overview (accessed on 6 October 2020).
- Ferley, J.; Soerjomatarami, I.; Ervik, M.; Dikshit, R.; Eser, S. Cancer Incidence and Mortality Worldwide; IARC: Lyon, France, 2013.
- National Cancer Institute Surveillance. Epidemiology and End Results Programme (SEER)—Cancer Stat Facts: Female Breast Cancer. 2020. Available online: http://seer.cancer.gov/statfacts/html/breast.html (accessed on 7 October 2020).
- DeSantis, C.E.; Miller, K.D.; Sauer, A.G.; Siegel, R.L. Cancer Statistics for African Americans, 2019. CA Cancer J. Clin. 2017, 69, 211–233. [Google Scholar] [CrossRef] [Green Version]
- Surveillance, Epidemiology, and End Results (SEER) Program. SEER*Stat Database: Mortality-All COD, Aggregated with State, Total U.S. (1990–2017) <Early release with Vintage 2017 Katrina/Rita Population Adjustment>; National Cancer Institute, Division of Cancer Control and Population Sciences, Surveillance Research Program: North Bethesda, MD, USA, 2019; Underlying mortality data provided the by National Center for Health Statistics.
- DeSantis, C.E.; Ma, J.; Goding, S.A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017: Racial disparity in mortality by state. CA Cancer. J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, C.E.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer. J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoncheh, M.; Mohammadian-Hafshejani, A.; Salehiniya, H. Incidence and mortality of breast cancer and their relationship to development in Asia. Asian Pac. J. Cancer Prev. 2015, 16, 6081–6087. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, L.; Ross, R.K. Endogenous hormones and breast cancer risk. Epidemiol. Rev. 1993, 15, 48–65. [Google Scholar] [CrossRef]
- Wu, A.H.; Stanczyk, F.Z.; Seow, A.; Lee, H.P.; Yu, M.C. Soy intake and other lifestyle determinants of serum estrogen levels among postmenopausal Chinese women in Singapore. Cancer Epidemiol. Biomark. Prev. 2002, 11, 844–851. [Google Scholar]
- Thakur, P.; Seam, R.K.; Gupta, M.K.; Gupta, M.; Sharma, M.; Fotedar, V. Breast cancer risk factor evaluation in a Western Himalayan state: A case-control study and comparison with the Western World. South Asian J. Cancer 2017, 6, 106–109. [Google Scholar]
- Colditz, G.A.; Rosner, B. Cumulative risk of breast cancer to age 70 years according to risk factor status: Data from the Nurses’ Health Study. Am. J. Epidemiol. 2000, 152, 950–964. [Google Scholar] [CrossRef] [Green Version]
- Giordano, S.H.; Buzdar, A.U.; Hortobagyi, G.N. Breast Cancer in Men. Ann. Intern. Med. 2002, 137, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Meo, S.A.; Suraya, F.; Jamil, B.; Al Rouq, F.; Meo, A.S.; Sattar, K.; Javed, M.; Alasiri, S.A. Association of ABO and Rh blood groups with breast cancer. Saudi J. Biol. Sci. 2017, 24, 1609–1613. [Google Scholar] [CrossRef] [PubMed]
- National Center for Health Statistics. SEER Cancer Statistics Review, 1973–1999; National Cancer Institute: Bethesda, MD, USA, 1998.
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding, S.A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Lilienfeld, A.M. The relationship of cancer of the female breast to artificial menopause and marital status. Cancer 1956, 9, 927–934. [Google Scholar] [CrossRef]
- Dai, Q.; Liu, B.; Du, Y. Meta-analysis of the risk factors of breast cancer concerning reproductive factors and oral contraceptive use. Front. Med. China 2009, 3, 452–458. [Google Scholar] [CrossRef]
- Ma, H.; Henderson, K.D.; Sullivan-Halley, J.; Duan, L.; Marshall, S.F.; Ursin, G.; Horn-Ross, P.L.; Largent, J.; Deapen, D.M.; Lacey, J.V., Jr. Pregnancy-related factors and the risk of breast carcinoma in situ and invasive breast cancer among postmenopausal women in the California Teachers Study cohort. Breast Cancer Res. 2010, 12, R35. [Google Scholar] [CrossRef] [Green Version]
- Balekouzou, A.; Yin, P.; Pamatika, C.M.; Bekolo, C.E.; Nambei, S.W.; Djeintote, M.; Kota, K.; Mossoro-Kpinde, C.D.; Shu, C.; Yin, M.; et al. Reproductive risk factors associated with breast cancer in women in Bangui: A case-control study. BMC. Women’s Health 2017, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Rosner, B.; Colditz, G.A.; Willett, W.C. Reproductive risk factors in a prospective study of breast cancer: The Nurses’ Health Study. Am. J. Epidemiol. 1994, 139, 819–835. [Google Scholar] [CrossRef] [Green Version]
- Rosner, B.; Colditz, G.A.; Martínez, M.E.; Giovannucci, E.L.; Stampfer, M.J.; Hunter, D.J.; Speizer, F.E.; Wing, A.; Willett, W.C. Nurses’ health study: Log-incidence mathematical model of breast cancer incidence. J. Natl. Cancer Inst. 1996, 88, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Mahouri, K.; Zahedani, M.D.; Zare, S. Breast cancer risk factors in south of Islamic Republic of Iran: A case-control study. EMHJ—East. Mediterr. Health J. 2007, 13, 1265–1273. [Google Scholar] [CrossRef]
- Kim, Y.; Yoo, K.Y.; Goodman, M.T. Differences in Incidence, Mortality and Survival of Breast Cancer by Regions and Countries in Asia and Contributing Factors. Asian Pac. J. Cancer Prev. 2015, 16, 2857–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, C.; Mirabel, L.; Annane, K.; Mathelin, C. Breastfeeding and breast cancer. Gynecol. Obstet. Fertil. 2005, 33, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; An, Y.S.; Choi, J.Y.; Park, B.; Kang, D.; Lee, M.H.; Han, W.; Noh, D.Y.; Yoo, K.Y.; Park, S.K. Risk reduction of breast cancer by childbirth, breastfeeding, and their interaction in korean women: Heterogeneous effects across menopausal status, hormone receptor status, and pathological subtypes. J. Prev. Med. Public Health 2017, 50, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Xu, H.; Zeng, X. Induced abortion and breast cancer: An updated meta-analysis. Medicine 2018, 97, e9613. [Google Scholar] [CrossRef] [PubMed]
- Key, T.; Appleby, P.; Barnes, I.; Reeves, G. Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002, 94, 606–616. [Google Scholar]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J.; et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998, 90, 1371–1388. [Google Scholar] [CrossRef]
- Eliassen, A.H.; Missmer, S.A.; Tworoger, S.S.; Spiegelman, D.; Barbieri, R.L.; Dowsett, M.; Hankinson, S.E. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J. Natl. Cancer Inst. 2006, 98, 1406–1415. [Google Scholar] [CrossRef] [Green Version]
- Tworoger, S.S.; Eliassen, A.H.; Rosner, B.; Sluss, P.; Hankinson, S.E. Plasma prolactin concentrations and risk of postmenopausal breast cancer. Cancer Res. 2004, 64, 6814–6819. [Google Scholar] [CrossRef] [Green Version]
- Toniolo, P.; Bruning, P.F.; Akhmedkhanov, A.; Bonfrer, J.M.; Koenig, K.L.; Lukanova, A.; Shore, R.E.; Zeleniuch-Jacquotte, A. Serum insulin-like growth factor-I and breast cancer. Int. J. Cancer 2000, 88, 828–832. [Google Scholar] [CrossRef]
- Bhadoria, A.; Kapil, U.; Sareen, N.; Singh, P. Reproductive factors and breast cancer: A case-control study in tertiary care hospital of North India. Indian J. Cancer 2013, 50, 316–321. [Google Scholar]
- Fioretti, F.; Tavani, A.; Bosetti, C.; La Vecchia, C.; Negri, E.; Barbone, F.; Talamini, R.; Franceschi, S. Risk factors for breast cancer in nulliparous women. Br. J. Cancer 1999, 79, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Marchbanks, P.A.; McDonald, J.A.; Wilson, H.G.; Folger, S.G.; Mandel, M.G.; Daling, J.R.; Bernstein, L.; Malone, K.E.; Ursin, G.; Storm, B.L.; et al. Oral contraceptives and the risk of breast cancer. N. Engl. J. Med. 2002, 346, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Zolfaroli, I.; Tarín, J.J.; Cano, A. Hormonal contraceptives and breast cancer: Clinical data. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 230, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group of Hormonal Factors in Breast Cancer. Breast cancer and hormonal contraceptives: Collaborative reanalysis of individual data on 53,297 women with breast cancer and 100,239 women without breast cancer from 54 epidemiological studies. Lancet 1996, 347, 1713–1727. [Google Scholar] [CrossRef] [Green Version]
- Beral, V.; Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 2003, 362, 419–427. [Google Scholar] [CrossRef]
- Beral, V.; Bull, D.; Doll, R.; Key, T.; Peto, R.; Reeves, G. Breast cancer and hormone replacement therapy: Collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet 1997, 350, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.K.; Paganini-Hill, A.; Wan, P.C.; Pike, M.C. Effect of hormone replacement therapy on breast cancer risk: Estrogen versus estrogen plus progestin. J. Natl. Cancer Inst. 2000, 92, 328–332. [Google Scholar] [CrossRef]
- Magnusson, C.; Persson, I.; Adami, H.O. More about: Effect of hormone replacement therapy on breast cancer risk: Estrogen versus estrogen plus progestin. J. Natl. Cancer Inst. 2000, 92, 1183–1184. [Google Scholar] [CrossRef] [Green Version]
- Colditz, G.A. Estrogen, estrogen plus progestin therapy, and risk of breast cancer. Clin. Cancer Res. 2005, 11, 909s–917s. [Google Scholar]
- Taheripanah, R.; Balash, F.; Anbiaee, R.; Mahmoodi, M.; Akbari, S.A. Breast Cancer and Ovulation Induction Treatments. Clin. Breast Cancer 2018, 18, 395–399. [Google Scholar] [CrossRef]
- Brinton, L.A.; Scoccia, B.; Moghissi, K.S.; Westhoff, C.L.; Althuis, M.D.; Mabie, J.E.; Lamb, E.J. Breast cancer risk associated with ovulation-stimulating drugs. Hum. Reprod. 2004, 19, 2005–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, K.; Stuckey, A. Breast Cancer Epidemiology and Risk Factors. Clin. Obstet. Gynecol. 2016, 59, 651–672. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.A.; Finch, A.; Poll, A.; Horsman, D.; Kim-Sing, C.; Scott, J.; Royer, R.; Sun, P.; Narod, S.A. Breast cancer risks in women with a family history of breast or ovarian cancer who have tested negative for a BRCA1 or BRCA2 mutation. Br. J. Cancer 2009, 100, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Cobain, E.F.; Milliron, K.J.; Merajver, S.D. Updates on breast cancer genetics: Clinical implications of detecting syndromes of inherited increased susceptibility to breast cancer. Semin. Oncol. 2016, 43, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Godet, I.; Gilkes, D.M. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr. Cancer Sci. Ther. 2017, 4, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, F.; Mahmood, N.; Shahid, S.; Hussain, Z.; Ahmed, I.; Jalal, A.; Ijaz, B.; Shahid, A.; Mujtaba, G.; Mustafa, T. Mutations in human interferon α2b gene and potential as risk factor associated with female breast cancer. Cancer Biother. Radiopharm. 2016, 31, 199–208. [Google Scholar] [CrossRef]
- Yari, K.; Rahimi, Z.; Moradi, M.T.; Rahimi, Z. The MMP-2-735 C Allele is a risk factor for susceptibility to breast cancer. Asian Pac. J. Cancer Prev. 2014, 15, 6199–6203. [Google Scholar] [CrossRef] [Green Version]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.; Xue, X.; Anderson, G.L.; et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009, 101, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, I.; Mahmood, H.; Faheem, M. Type 2 Diabetes Mellitus as a risk factor for female breast cancer in the population of northern Pakistan. Asian Pac. J. Cancer Prev. 2016, 17, 3255–3258. [Google Scholar]
- Larsson, S.C.; Mantzoros, C.S.; Wolk, A. Diabetes mellitus and risk of breast cancer: A meta-analysis. Int. J. Cancer 2007, 121, 856–862. [Google Scholar] [CrossRef]
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Steinberg, G.R.; Blandino, G.; Schünemann, H.J.; Muti, P. Association of metformin with breast cancer incidence and mortality in patients with type 2 diabetes: A GRADE assessed systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2018, 27, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.J.; Wu, W.Y.; Yen, A.M.; Fann, J.C.; Chen, S.L.; Chiu, S.Y.; Chen, H.H.; Chiou, S.T. Body mass index and breast cancer: Analysis of a nation-wide population-based prospective cohort study on 1,393,985 Taiwanese women. Int. J. Obes. 2016, 40, 524–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahmann, P.H.; Hoffmann, K.; Allen, N.; Van Gils, C.H.; Khaw, K.T.; Tehard, B.; Berrino, F.; Tjønneland, A.; Bigaard, J.; Olsen, A. Body size and breast cancer risk: Findings from the European prospective investigation into cancer and nutrition. Int. J. Cancer 2004, 111, 762–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Key, T.J.; Reeves, G.K. Adiposity and breast cancer risk in postmenopausal women: Results from the UK biobank prospective cohort. Int. J. Cancer 2018, 143, 1037–1046. [Google Scholar] [CrossRef]
- Pimentel, I.; Lohmann, A.E.; Goodwin, P.J. Normal weight adiposity and postmenopausal breast cancer risk. JAMA Oncol. 2019, 5, 150–151. [Google Scholar] [CrossRef]
- Taylor, E.F.; Burley, V.J.; Greenwood, D.C.; Cade, J.E. Meat consumption and risk of breast cancer in the UK women’s cohort study. Br. J. Cancer 2007, 96, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Sieri, S.; Krogh, V.; Ferrari, P.; Berrino, F.; Pala, V.; Thiébaut, A.C.; Tjønneland, A.; Olsen, A.; Overvad, K.; Jakobsen, M.U.; et al. Dietary fat and breast cancer risk in the European Prospective Investigation into Cancer and Nutrition. Am. J. Clin. Nutr. 2008, 88, 1304–1312. [Google Scholar]
- Berkey, C.S.; Rockett, H.R.; Willett, W.C.; Colditz, G.A. Milk, dairy fat, dietary calcium, and weight gain: A longitudinal study of adolescents. Arch. Pediatrics Adolesc. Med. 2005, 159, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Hatse, S.; Lambrechts, D.; Verstuyf, A.; Smeets, A.; Brouwers, B.; Vandorpe, T.; Brouckaert, O.; Peuteman, G.; Laenen, A.; Verlinden, L. Vitamin D status at breast cancer diagnosis: Correlation with tumor characteristics, disease outcome, and genetic determinants of vitamin D insufficiency. Carcinogenesis 2012, 33, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.M.; Sandler, D.P.; Taylor, J.A.; Weinberg, C.R. Serum vitamin D and risk of breast cancer within five years. Environ. Health Perspect. 2017, 125, 077004. [Google Scholar] [CrossRef]
- Hamajima, N.; Hirose, K.; Tajima, K.; Rohan, T.; Calle, E.E.; Heath, C.W., Jr.; Coates, R.J.; Liff, J.M.; Talamini, R.; Chantarakul, N.; et al. Alcohol, tobacco and breast cancer—Collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br. J. Cancer 2002, 87, 1234–1245. [Google Scholar] [PubMed]
- Romieu, I.; Scoccianti, C.; Chajès, V.; de Batlle, J.; Biessy, C.; Dossus, L.; Baglietto, L.; Clavel-Chapelon, F.; Overvad, K.; Olsen, A.; et al. Alcohol intake and breast cancer in the European prospective investigation into cancer and nutrition. Int. J. Cancer. 2015, 137, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Margolis, K.L.; Wactawski-Wende, J.; Horn, K.; Messina, C.; Stefanick, M.L.; Tindle, H.A.; Tong, E.; Rohan, T.E. Association of active and passive smoking with risk of breast cancer among postmenopausal women: A prospective cohort study. BMJ 2011, 342, d1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.H.; Li, Z.; Shi, J.; Li, H.M.; Wang, Y.; Fu, L.Y.; Liu, Y.P. Passive smoking exposure from partners as a risk factor for ER+/PR+ double positive breast cancer in never-smoking Chinese urban women: A hospital-based matched case control study. PLoS ONE 2014, 9, e97498. [Google Scholar] [CrossRef] [PubMed]
- The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. 2006. Available online: www.surgeongeneral.gov/library/secondhandsmoke/report/index.html (accessed on 11 October 2020).
- Mctiernan, A.; Kooperberg, C.; White, E.; Wilcox, S.; Coates, R.; Adams-Campbell, L.L.; Woods, N.; Ockene, J. Women’s health initiative cohort study recreational physical activity and the risk of breast cancer in postmenopausal women: The women’s health initiative cohort study. JAMA 2003, 290, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A. Meta-analysis of the association between physical activity and breast cancer mortality. Cancer Nurs. 2019, 42, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Benabu, J.C.; Stoll, F.; Gonzalez, M.; Mathelin, C. Night work, shift work: Breast cancer risk factor? Gynecol. Obstet. Fertil. 2015, 43, 791–799. [Google Scholar] [CrossRef]
- Stevens, R.G.; Davis, S. The melatonin hypothesis: Electric power and breast cancer. Environ. Health Perspect. 1996, 104 (Suppl. 1), 135–140. [Google Scholar]
- Megdal, S.P.; Kroenke, C.H.; Laden, F.; Pukkala, E.; Schernhammer, E.S. Night work and breast cancer risk: A systematic review and meta-analysis. Eur. J. Cancer 2005, 41, 2023–2032. [Google Scholar] [CrossRef]
- Orsini, M.; Trétarre, B.; Daurès, J.P.; Bessaoud, F. Individual socioeconomic status and breast cancer diagnostic stages: A French case-control study. Eur. J. Public Health 2016, 26, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Lundqvist, A.; Andersson, E.; Ahlberg, I.; Nilbert, M.; Gerdtham, U. Socioeconomic inequalities in breast cancer incidence and mortality in Europe-a systematic review and meta-analysis. Eur. J. Public Health 2016, 26, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulrahman, G.O.; Rahman, G.A. Epidemiology of Breast Cancer in Europe and Africa. J. Cancer Epidemiol. 2012, 2012, 915610. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.C.; Sellers, T.A.; Frost, M.H.; Lingle, W.L.; Degnim, A.C.; Ghosh, K.; Vierkant, R.A.; Maloney, S.D.; Pankratz, V.S.; Hillman, D.W.; et al. Benign breast disease and the risk of breast cancer. N. Engl. J. Med. 2005, 353, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinton, L.A.; Lubin, J.H.; Murray, M.C.; Colton, T.; Hoover, R.N. Mortality rates among augmentation mammoplasty patients: An update. Epidemiology 2006, 17, 162–169. [Google Scholar] [CrossRef]
- Boyd, N.F.; Guo, H.; Martin, L.J.; Sun, L.; Stone, J.; Fishell, E.; Jong, R.A.; Hislop, G.; Chiarelli, A.; Minkin, S.; et al. Mammographic density and the risk and detection of breast cancer. N. Engl. J. Med. 2007, 356, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Nazari, S.S.; Mukherjee, P. An overview of mammographic density and its association with breast cancer. Breast Cancer 2018, 25, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Tamimi, R.M.; Byrne, C.; Colditz, G.A.; Hankinson, S.E. Endogenous hormone levels, mammographic density, and subsequent risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2007, 99, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Land, C.E.; Tokunaga, M.; Koyama, K.; Soda, M.; Preston, D.L.; Nishimori, I.; Tokuoka, S. Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950–1990. Radiat. Res. 2003, 160, 707–717. [Google Scholar] [CrossRef]
- Henderson, T.O.; Moskowitz, C.S.; Chou, J.F.; Bradbury, A.R.; Neglia, J.P.; Dang, C.T.; Onel, K.; Friedman, D.N.; Bhatia, S.; Strong, L.C.; et al. Breast cancer risk in childhood cancer survivors without a history of chest radiotherapy: A report from the Childhood Cancer Survivor Study. J. Clin. Oncol. 2016, 34, 910–918. [Google Scholar] [CrossRef]
- Horwich, A.; Swerdlow, A.J. Second primary breast cancer after Hodgkin’s disease. Br. J. Cancer 2004, 90, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumors. Nature 2000, 17, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 2014, 106, dju055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, S.; Purohit, P.; Misra, R.; Lingeswaran, M.; Vishnoi, J.R.; Pareek, P.; Sharma, P.; Misra, S. Application of single-cell omics in breast cancer in single-cell omics. Appl. Biomed. Agric. 2019, 2, 69–103. [Google Scholar]
- Aftimos, P.; Azim, H.A., Jr.; Sotiriou, C. Molecular biology of breast cancer. In Molecular Pathology, 2nd ed.; Coleman, W.B., Tsongalis, G.J., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 569–588. [Google Scholar]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Colozza, M.; Azambuja, E.; Cardoso, F.; Sotiriou, C.; Larsimont, D.; Piccart, M.J. Proliferative markers as prognostic and predictive tools in early breast cancer: Where are we now? Ann. Oncol. 2005, 16, 1723–1739. [Google Scholar] [CrossRef]
- Lim, W.; Mayer, B.; Pawson, T. Cell Signaling: Principles and Mechanisms; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef]
- Paduch, M.; Jelen, F.; Otlewski, J. Structure of small G proteins and their regulators. Acta Biochim. Pol. 2001, 48, 829–850. [Google Scholar] [CrossRef] [Green Version]
- Yudushkin, I. Getting the Akt together: Guiding intracellular Akt activity by PI3K. Biomolecules 2019, 9, 67. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [Green Version]
- Lehninger, A.; Nelson, D.L.; Cox, M.C.; Freeman, W.H. Lehninger Principles of Biochemistry; W.H. Freeman: New York, NY, USA, 2012. [Google Scholar]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falasca, M.; Hughes, W.E.; Dominguez, V.; Sala, G.; Fostira, F.; Fang, M.Q.; Cazzolli, R.; Shepherd, P.R.; James, D.E.; Maffucci, T. The role of phosphoinositide 3-kinase C2α in insulin signaling. J. Biol. Chem. 2007, 282, 28226–28236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backer, J. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Dummler, B.; Hemmings, B.A. Physiological roles of PKB/Akt isoforms in development and disease. Biochem. Soc. Trans. 2007, 35, 231–235. [Google Scholar] [CrossRef]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New insights into protein kinase B/Akt signaling: Role of localized Akt activation and compartment-specific target proteins for the cellular radiation response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One kinase, many modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef]
- Luo, C.T.; Li, M. Foxo transcription factors in T cell biology and tumor immunity. Semin. Cancer Biol. 2018, 50, 13–20. [Google Scholar] [CrossRef]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Woodgett, J.R. Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr. Top. Dev. Biol. 2017, 123, 277–302. [Google Scholar] [PubMed]
- Dokken, B.B.; Sloniger, J.A.; Henriksen, E.J. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1188–E1194. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.A.; Coghlan, M.; Rice, S.Q.; Sutherland, C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes 2001, 50, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Luo, L.; Chen, J. Roles of mTOR signaling in tissue regeneration. Cells 2019, 8, 1075. [Google Scholar] [CrossRef] [Green Version]
- Kakumoto, K.; Ikeda, J.; Okada, M.; Morii, E.; Oneyama, C. mLST8 promotes mTOR-mediated tumor progression. PLoS ONE 2015, 10, e0119015. [Google Scholar] [CrossRef]
- Mahoney, R.E.; Azpurua, J.; Eaton, B.A. Insulin signaling controls neurotransmission via the 4eBP-dependent modification of the exocytotic machinery. eLife 2016, 5, e16807. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, D.; Walther, T.C. TORC2 Plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol. Biol. Cell 2009, 20, 1565–1575. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; Bose, M.E.; Mielke, M.R.; Müller, U.; Fox, C.A. Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for FKH1 and FKH2 in Saccharomyces cerevisiae. Genetics 2000, 154, 1533–1548. [Google Scholar]
- Cabrera-Ortega, A.; Feinberg, D.; Liang, Y.; Rossa, J.C.; Graves, D.T. The role of Forkhead Box 1 (FOXO1) in the immune system: Dendritic cells, T cells, B cells, and hematopoietic stem cells. Crit. Rev. Immunol. 2017, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xin, Z.; Hu, W.; Jiang, S.; Yang, Z.; Yan, X.; Li, X.; Yang, Y.; Chen, F. Forkhead box O proteins: Crucial regulators of cancer EMT. Semin. Cancer Biol. 2018, 50, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Forkhead transcription factors: Formulating a FOXO target for cognitive loss. Curr. Neurovascular Res. 2017, 14, 415–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cretella, D.; Digiacomo, G.; Giovannetti, E.; Cavazzoni, A. PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers 2019, 11, 1318. [Google Scholar] [CrossRef] [Green Version]
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN tumor-suppressor: The dam of stemness in cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef] [Green Version]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Alihemmati, A.; Rahmati, M.; Nozad-Charoudeh, H. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef]
- Maehama, T.; Taylor, G.S.; Dixon, J.E. PTEN and myotubularin: Novel phosphoinositide phosphatases. Annu. Rev. Biochem. 2001, 70, 247–279. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Tajmir, P.; Lin, C.H.; Liadis, N.; Zhu, X.D.; Eweida, M.; Tolasa-Karaman, G.; Cai, F.; Wang, R.; Kitamura, T.; et al. Essential role of PTEN in body size determination and pancreatic beta-cell homeostasis in vivo. Mol. Cell. Biol. 2006, 26, 4511–4518. [Google Scholar] [CrossRef] [Green Version]
- Abraham, J. PI3K/AKT/mTOR pathway inhibitors: The ideal combination partners for breast cancer therapies? Expert Rev. Anticancer Ther. 2015, 15, 51–68. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, C.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Razavi, P.; Johnson, J.L.; Shao, H.; Shah, H.; Antoine, A.; Ladewig, E.; Gorelick, A.N.; Lin, T.-Y.; Toska, E.; et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science 2019, 366, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.A.; Yisheng, L.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Ptak, N.J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; Willson, J.K.V.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Rand, A.; Yardena, S. PIK3CA in cancer: The past 30 years. Semin. Cancer. Biol. 2019, 59, 36–49. [Google Scholar]
- Saal, L.H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmström, P.O.; Mansukhani, M.; Enoksson, J.; et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005, 1, 2554–2559. [Google Scholar] [CrossRef] [Green Version]
- Bachman, K.E.; Argani, P.; Samuels, Y.; Silliman, N.; Ptak, J.; Szabo, S.; Konishi, H.; Karakas, B.; Blair, B.G.; Lin, C.; et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 2004, 3, 772–775. [Google Scholar] [CrossRef] [Green Version]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Yinghui, G.; Guan, Y.; Sahin, A.; et al. An Integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.Y.; Rong, M.; Grieu, F.; Iacopetta, B. PIK3CA mutations in breast cancer are associated with poor outcome. Breast Cancer Res. Treat. 2005, 96, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.-H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and predictive implications of PTEN in breast cancer: Unfulfilled promises but intriguing perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobral-Leite, M.; Salomon, I.; Opdam, M.; Kruger, D.T.; Beelen, K.J.; Van Der Noort, V.; Van Vlierberghe, R.L.P.; Blok, E.J.; Giardiello, D.; Sanders, J.; et al. Cancer-immune interactions in ER-positive breast cancers: PI3K pathway alterations and tumor-infiltrating lymphocytes. Breast Cancer Res. 2019, 21, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zardavas, D.; Te Marvelde, L.; Milne, R.L.; Fumagalli, D.F.; Fountzilas, G.; Kotoula, V.; Razis, E.; Papaxoinis, G.; Joensuu, H.; Moynahan, M.E.; et al. Tumor PIK3CA genotype and prognosis in early-stage breast cancer: A pooled analysis of individual patient data. J. Clin. Oncol. 2018, 1, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, D.; Xuehua, Z.; Yun, S.; Jiemin, W.; Xiaorong, Z.; Jiayuan, L.; Min, H.; Hong, Z. Prevalence and prognostic role of PIK3CA/AKT1 mutations in chinese breast cancer patients. Cancer. Res. Treat. 2019, 51, 128–140. [Google Scholar]
- Anderson, A.J.; Mollon, L.E.; Dean, J.L.; Warholak, T.L.; Aizer, A.; Platt, E.A.; Tang, D.H.; Lisa, E.; Davis, L.E. A systematic review of the prevalence and diagnostic workup of PIK3CA mutations in HR+/HER2− metastatic breast cancer. Int. J. Breast. Cancer 2020, 2020, 3759179. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gómez, H.L.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 8, 406. [Google Scholar] [CrossRef] [Green Version]
- Dieci, M.V.; Miglietta, F.; Griguolo, G.; Guarneri, V. Biomarkers for HER2-positive metastatic breast cancer: Beyond hormone receptors. Cancer Treat. Rev. 2020, 88, 102064. [Google Scholar] [CrossRef]
- Toss, A.; Cristofanilli, M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 receptor in breast cancer: Pathophysiology, clinical use, and new advances in therapy. Chemother. Res. Pract. 2012, 2012, 743193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliato, D.D.M.; Jardim, D.L.F.; Marchesi, M.S.P.; Hortobagyi, G.N. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 2016, 7, 64431–64446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schettini, F.; Buono, G.; Cardalesi, C.; Desideri, I.; De Placido, S.; Del Mastro, L. Hormone receptor/human epidermal growth factor receptor 2-positive breast cancer: Where we are now and where we are going. Cancer Treat. Rev. 2016, 46, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babak, N.; Hamid, M.; Zhixiang, W. Mechanisms underlying the action and synergism of trastuzumab and pertuzumab in targeting HER2-positive breast cancer. Cancers 2018, 10, 342. [Google Scholar]
- Kechagioglou, P.; Papi, R.; Provatopoulou, X.; Kalogera, E.; Papadimitriou, E.; Grigoropoulos, P.; Nonni, A.; Zografos, G.; Kyriakidis, D.A.; Gounaris, A. Tumor suppressor PTEN in breast cancer: Heterozygosity, mutations and protein expression. Anticancer Res. 2014, 34, 1387–1400. [Google Scholar]
- Ruiz-Saenz, A.; Dreyer, C.; Campbell, M.R.; Steri, V.; Gulizia, N.; Moasser, M. HER2 amplification in tumors activates PI3K/AKT signaling independent of HER3. Cancer Res. 2018, 78, 3655–3658. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Li, N.; Li, X.; Lei, L.; Wang, X. The prognostic impact of hormonal receptor and HER-2 expression discordance in metastatic breast cancer patients. OncoTargets Ther. 2020, 13, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer-An overview and update. Mol. Cell. Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef] [Green Version]
- García-Becerra, R.; Santos-Martínez, N.; Díaz, L.; Camacho, J. Mechanisms of resistance to endocrine therapy in breast cancer: Focus on signaling pathways, miRNAs and genetically based resistance. Int. J. Mol. Sci. 2013, 14, 108–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K.; Schiff, R. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghayad, S.E.; Vendrell, J.A.; Larbi, S.B.; Dumontet, C.; Bieche, I.; Cohen, P.A. Endocrine resistance associated with activated ErbB system in breast cancer cells is reversed by inhibiting MAPK or PI3K/Akt signaling pathways. Int. J. Cancer 2010, 126, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R. Enhancing endocrine therapy for hormone receptor-positive advanced breast cancer: Cotargeting signaling pathways. J. Natl. Cancer Inst. 2015, 107, djv212. [Google Scholar] [CrossRef]
- Lange, C.A.; Yee, D. Killing the second messenger: Targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr.-Relat. Cancer 2011, 18, C19–C24. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, M.; Schiff, R.; Osborne, C.K.; Trivedi, M.V. Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast 2011, 20, S42–S49. [Google Scholar] [CrossRef]
- Cook, K.L.; Shajahan, A.N.; Clarke, R. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer Ther. 2011, 11, 1283–1294. [Google Scholar] [CrossRef]
- Mackey, J.; Kaufman, B.; Clemens, M.; Bapsy, P.P. Trastuzumab prolongs progression free survival in hormone-dependent and HER2-positive metastatic breast cancer. In Breast Cancer Research and Treatment, Proceedings of the 29th Annual San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 14–17 December 2006; Springer: Berlin/Heidelberg, Germany, 2006; Volume 100. [Google Scholar]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Rizel, S. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, R.M.; Paplomata, E. New and emerging treatments for estrogen receptor-positive breast cancer: Focus on everolimus. Ther. Clin. Risk Manag. 2013, 9, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, R.; Achinger-Kawecka, J.; Winter, S.; Fritz, P.; Lo, W.-Y.; Schroth, W.; Brauch, H. Increased expression of miR-126 and miR-10a predict prolonged relapse-free time of primary oestrogen receptor-positive breast cancer following tamoxifen treatment. Eur. J. Cancer 2013, 49, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, O.; Filkowski, J.; Meservy, J.; Ilnytskyy, Y.; Tryndyak, V.P.; Chekhun, V.F.; Pogribny, I.P. Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol. Cancer Ther. 2008, 7, 2152–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luqmani, Y.A.; Al Saleh, S.; Sharaf, L.H. Signalling pathways involved in endocrine resistance in breast cancer and associations with epithelial to mesenchymal transition. Int. J. Oncol. 2011, 38, 1197–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creighton, C.J.; Fu, X.; Hennessy, B.T.; Casa, A.J.; Zhang, Y.; Gonzalez-Angulo, A.M.; Lluch, A.; Gray, J.W.; Brown, P.H.; Hilsenbeck, S.G.; et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010, 12, R40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavazzoni, A.; Bonelli, M.; Fumarola, C.; La Monica, S.; Airoud, K.; Bertoni, R.; Alfieri, R.; Galetti, M.; Tramonti, S.; Galvani, E.; et al. Overcoming acquired resistance to letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell clones. Cancer Lett. 2012, 323, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R. New strategies in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2010, 16, 1979–1987. [Google Scholar] [CrossRef] [Green Version]
- Houghton, P.J. Everolimus. Clin. Cancer Res. 2010, 16, 1368–1372. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.X.; Loh, K.; Yap, Y.-S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., III; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnan, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Ruiz, I. Fulvestrant plus vistusertib vs. fulvestrant plus everolimus vs. fulvestrant alone for women with hormone receptor–positive metastatic breast cancer: The MANTA phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 1556–1563. [Google Scholar] [CrossRef]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.-M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.-C.; Debled, M.; Spaëth, D.; et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2–negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO Study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef] [PubMed]
- André, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Dalenc, F.; Campone, M.; O’Regan, R.M.; Tjan-Heijnen, V.C.; Gligorov, J.; Llombart, A.; Jhangiani, H.; Mirshahidi, H.R.; Tan-Chiu, E.; et al. A phase 2 study of everolimus combined with trastuzumab and paclitaxel in patients with HER2-overexpressing advanced breast cancer that progressed during prior trastuzumab and taxane therapy. Breast Cancer Res. Treat. 2013, 141, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulay, A.; Rudloff, J.; Ye, J.; Zumstein-Mecker, S.; O’Reilly, T.; Evans, D.B.; Chen, S.; Lane, H.A. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin. Cancer Res. 2005, 11, 5319–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Z.; Ma, F.; Liu, B.; Guan, X.; Li, L.; Li, C.; Qian, H.; Xu, B. Everolimus in hormone receptor-positive metastatic breast cancer: PIK3CA mutation H1047R was a potential efficacy biomarker in a retrospective study. BMC Cancer 2019, 19, 442. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Toral-Barza, L.; Discafani, C.; Zhang, W.G.; Skotnicki, J.; Frost, P.; Gibbons, J.J. mTOR, a novel target in breast cancer: The effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr.-Relat. Cancer 2001, 8, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.C.; Lazar, A.A.; Bondarenko, I.; Garin, A.M.; Brincat, S.; Chow, L.; Sun, Y.; Neskovic-Konstantinovic, Z.; Guimaraes, R.C.; Fumoleau, P.; et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2013, 31, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Fleming, G.F.; Ma, C.X.; Huo, D.; Sattar, H.; Tretiakova, M.; Lin, L.; Hahn, O.M.; Olopade, F.O.; Nanda, R.; Hoffman, P.C.; et al. Phase II trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res. Treat. 2012, 136, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Sadler, T.M.; Gavriil, M.; Annable, T.; Frost, P.; Greenberger, L.M.; Zhang, Y. Combination therapy for treating breast cancer using antiestrogen, ERA-923, and the mammalian target of rapamycin inhibitor, temsirolimus. Endocr.-Relat. Cancer 2006, 13, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharvva, G.S.; Biswas, J.; Singh, J.K.; Singh, M.; Govindbabu, K.; Ranade, A.; Malhotra, H.; Parikh, P.; Shahid, T.; Basu, S. Reversal of tamoxifen resistance (hormone resistance) by addition of Sirolimus (mTOR Inhibitor) in metastatic breast cancer. Eur. J. Cancer 2011, 47, 9. [Google Scholar] [CrossRef]
- Seiler, M.; Ray-Coquard, I.; Melichar, B.; Yardley, D.A.; Wang, R.X.; Dodion, P.F.; Lee, M.A. Oral Ridaforolimus plus Trastuzumab for patients with HER2+ trastuzumab-refractory metastatic breast cancer. Clin. Breast Cancer 2015, 15, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.J.; Chen, S.M.; Guo, C.L.; Li, Y.X.; Ding, J.; Meng, L.H. The mTOR inhibitor AZD8055 overcomes tamoxifen resistance in breast cancer cells by down-regulating HSPB8. Acta Pharmacol. Sin. 2018, 39, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.J.; Dutkowski, C.M.; Barrow, D.; Mottram, H.J.; Hutcheson, I.R.; Nicholson, R.I.; Guichard, S.M.; Gee, J.M.W. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res. 2014, 23, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrossian, K.; Nguyen, D.; Lo, C.; Kanaya, N.; Somlo, G.; Cui, Y.X.; Huang, C.-S.; Chen, S. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res. Treat. 2018, 170, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Bostner, J.; Alayev, A.; Berman, A.Y.; Fornander, T.; Nordenskjöld, B.; Holz, M.K.; Stål, O. Raptor localization predicts prognosis and tamoxifen response in estrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2018, 168, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Li, X.; Shi, L.; Wu, J.; Qian, J.; Xia, T.; Zhou, W.-B.; Sun, X.; Xu-Jie, Z.; Wei, J.-F.; et al. Rapamycin enhances the sensitivity of ER‑positive breast cancer cells to tamoxifen by upregulating p73 expression. Oncol. Rep. 2019, 41, 455–464. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| mTOR Inhibitors | Type of Breast Cancer | Type of Study | References |
|---|---|---|---|
| Everolimus + exemestane | hormone-receptor-positive advanced breast cancer | Phase 3, randomized trial | [180] |
| Everolimus + fulvestrant | estrogen receptor-positive breast cancer | Phase 2 Manta trial | [181] |
| Everolimus + tamoxifen | metastatic breast cancer | Phase II Randomized trial | [182] |
| Everolimus + plustrastuzumab + vinorelbine | HER2-positive breast cancer | Phase 3 trial (Bolero-3) | [183] |
| Everolimus + trastuzumab + paclitaxel | HER2-positive advanced breast cancer | Phase 2 multicenter study | [184] |
| Everolimus | metastatic breast cancer | Retrospective study | [186] |
| Temsirolimus + letrozole | hormone receptor-positive metastatic breast cancer | Phase III randomized trial | [188] |
| Temsirolimus | metastatic breast cancer | Phase II trial | [189] |
| Sirolimus + Tamoxifen | hormone receptor-positive and HER2-negative breast cancer | Phase I/II trial | [191] |
| Ridaforolimus + trastuzumab | Human epidermal growth factor receptor 2–positive (HER2+) trastuzumab-refractory metastatic breast cancer | Phase IIb trail | [192] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2021, 22, 173. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010173
Miricescu D, Totan A, Stanescu-Spinu I-I, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. International Journal of Molecular Sciences. 2021; 22(1):173. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010173
Chicago/Turabian StyleMiricescu, Daniela, Alexandra Totan, Iulia-Ioana Stanescu-Spinu, Silviu Constantin Badoiu, Constantin Stefani, and Maria Greabu. 2021. "PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects" International Journal of Molecular Sciences 22, no. 1: 173. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010173