Neuroprotective Effects of Growth Hormone (GH) and Insulin-Like Growth Factor Type 1 (IGF-1) after Hypoxic-Ischemic Injury in Chicken Cerebellar Cell Cultures

 , and
, and 
Abstract
:1. Introduction
2. Results
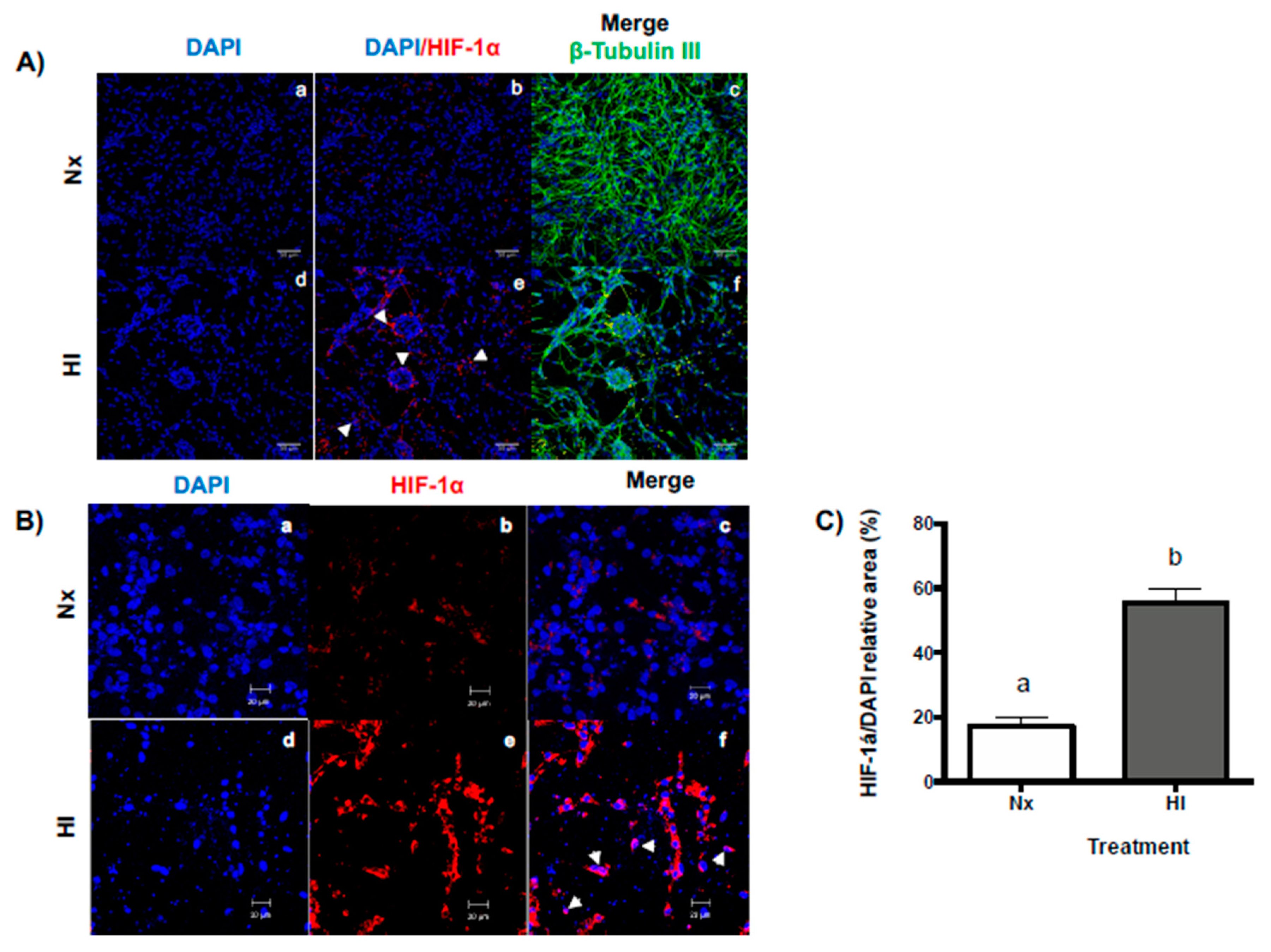
2.1. Hypoxic-Ischemic Conditions Increase HIF-1α Expression in Primary Cerebellar Cell Cultures
2.2. Effects of Hypoxic-Ischemic (Acute Injury) and Reoxygenation (Subacute Injury) Incubation Conditions on Cell Viability, Apoptosis, and Necrosis in Primary Cerebellar Cultures
2.3. Treatments with GH and IGF-1 Protect Primary Cerebellar Cell Cultures from Damage When Administered during or after HI and HI + Ox Incubation Conditions
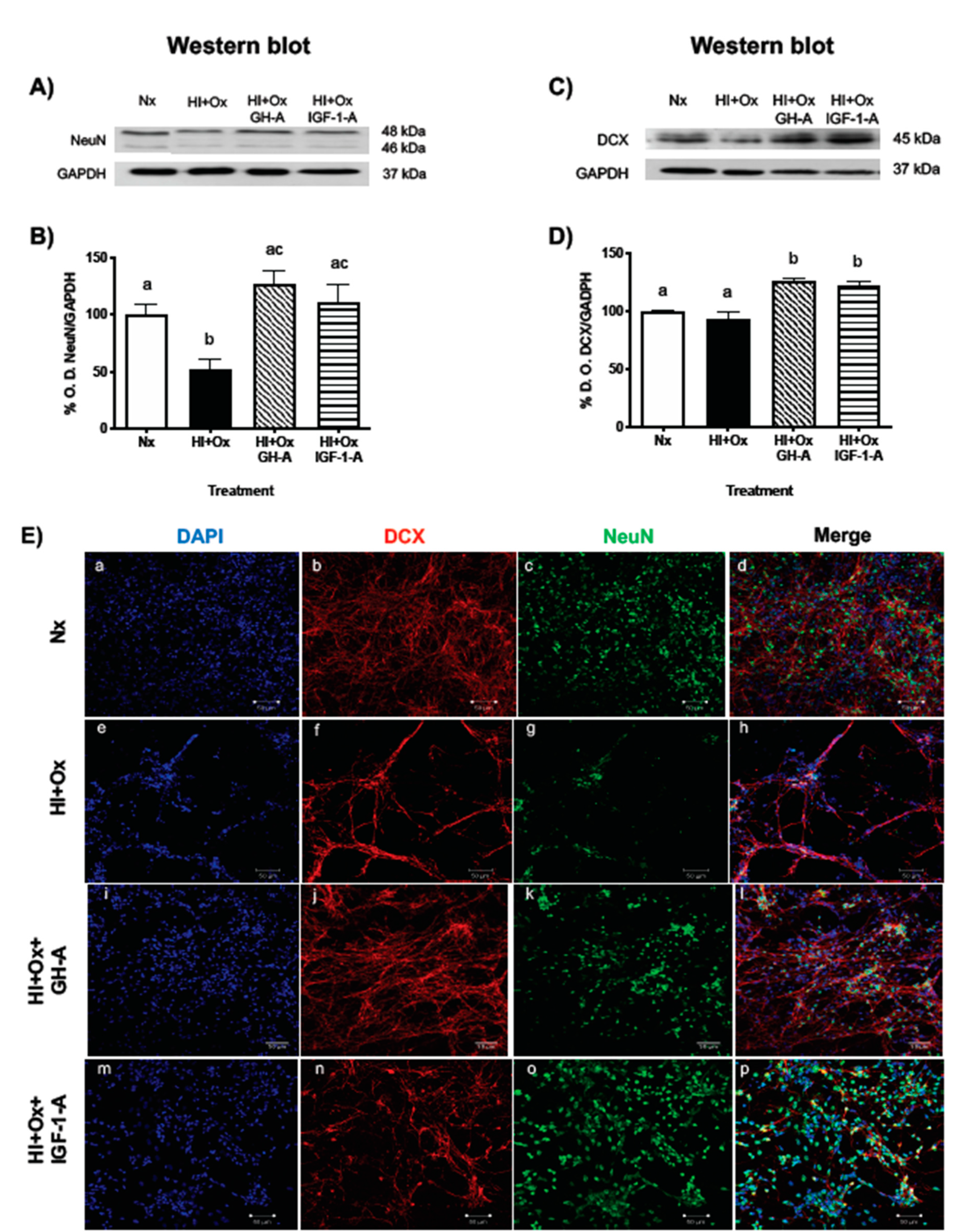
2.4. Treatments with GH and IGF-1 Protect Mature Neurons and Neuronal Precursors after an Acute Hypoxic-Ischemic Injury
2.5. Effects of GH and IGF-1 Administration after Hypoxia-Ischemia upon PI3K/Akt, MAPK/ERK1/2, and Bcl-2 Pathways
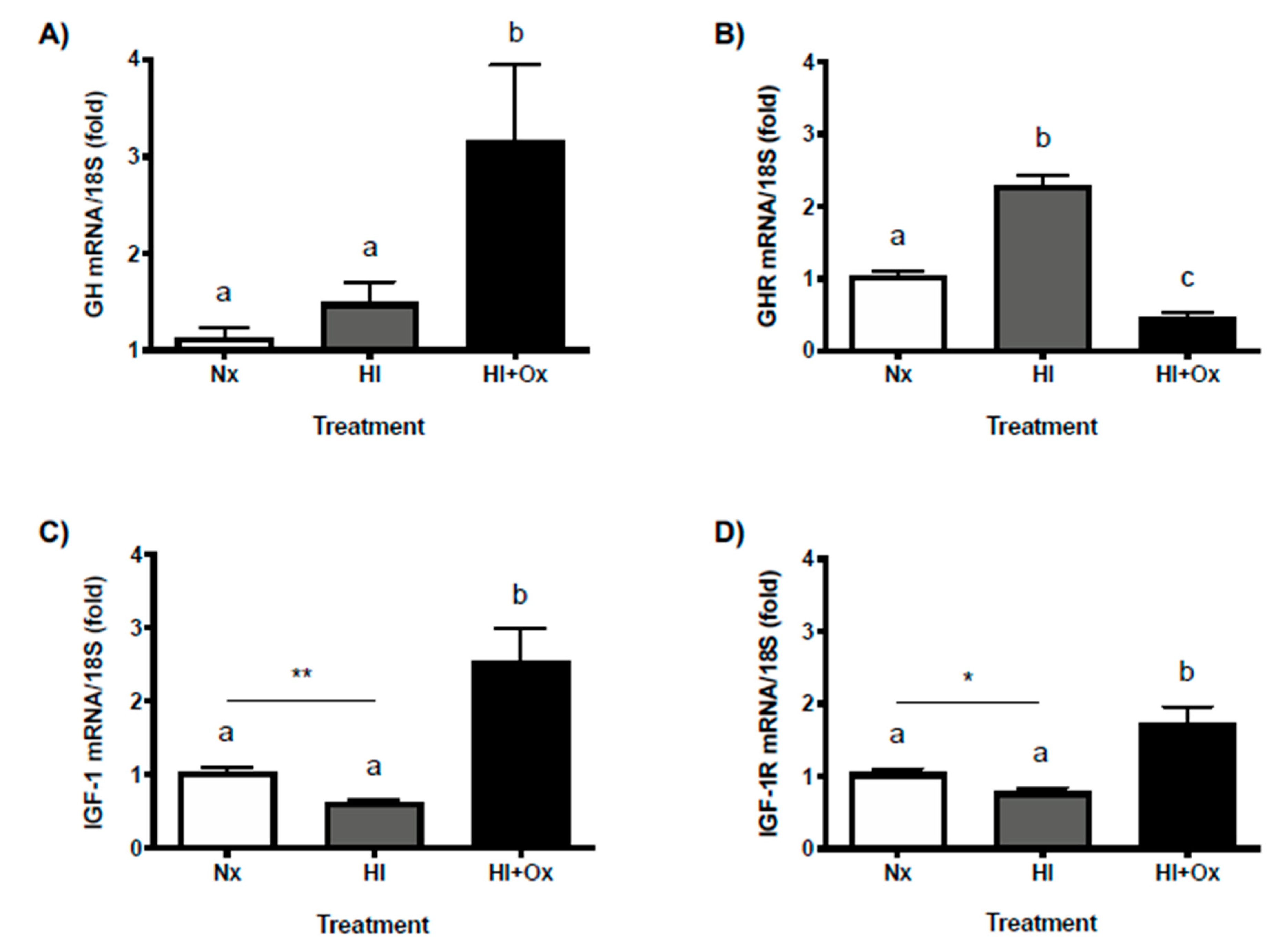
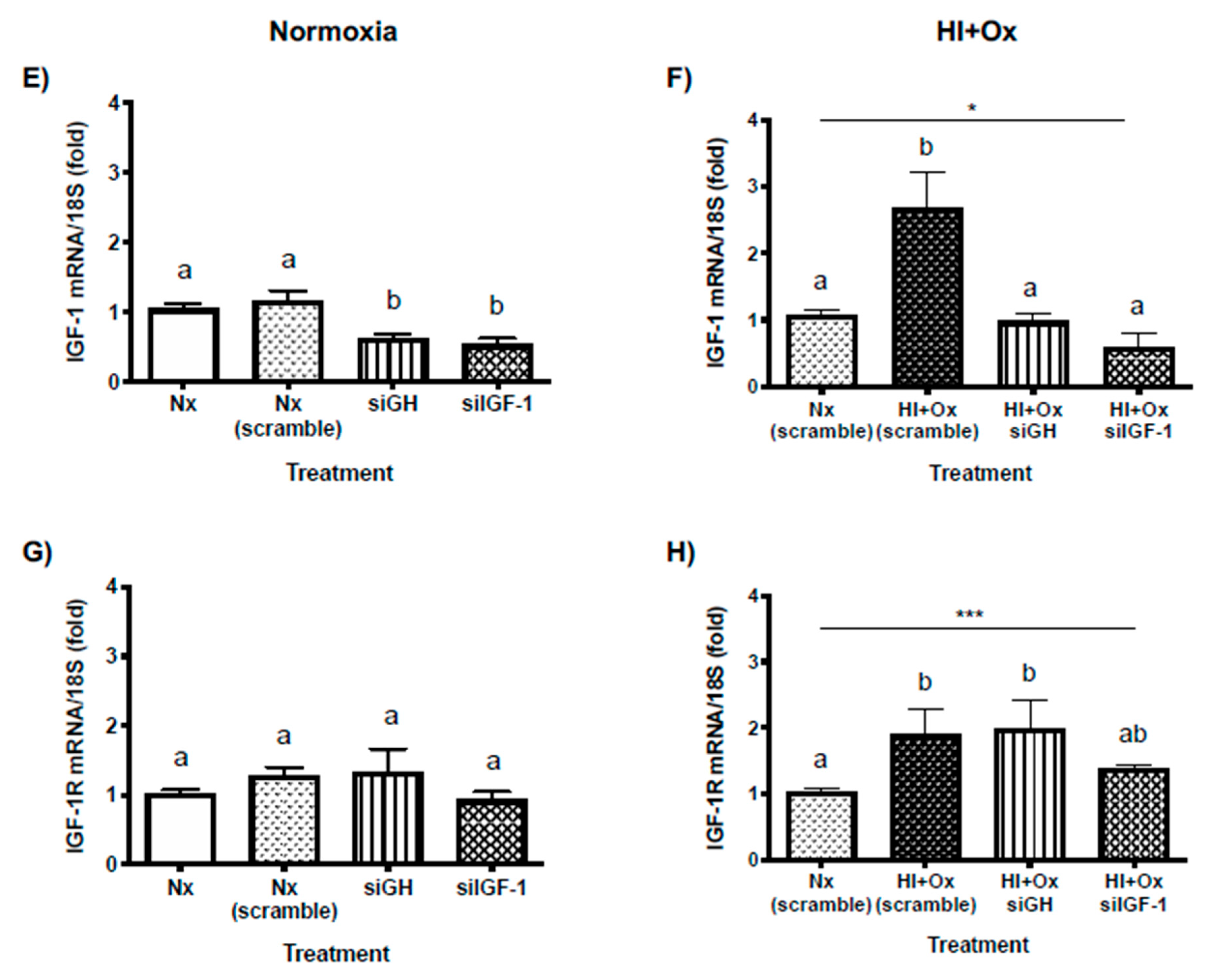
2.6. Effects of Acute (HI) and Subacute (HI + Ox) Injury upon Local GH, GHR, IGF-1 and IGF-1R mRNAs Expression in Primary Cerebellar Cultures
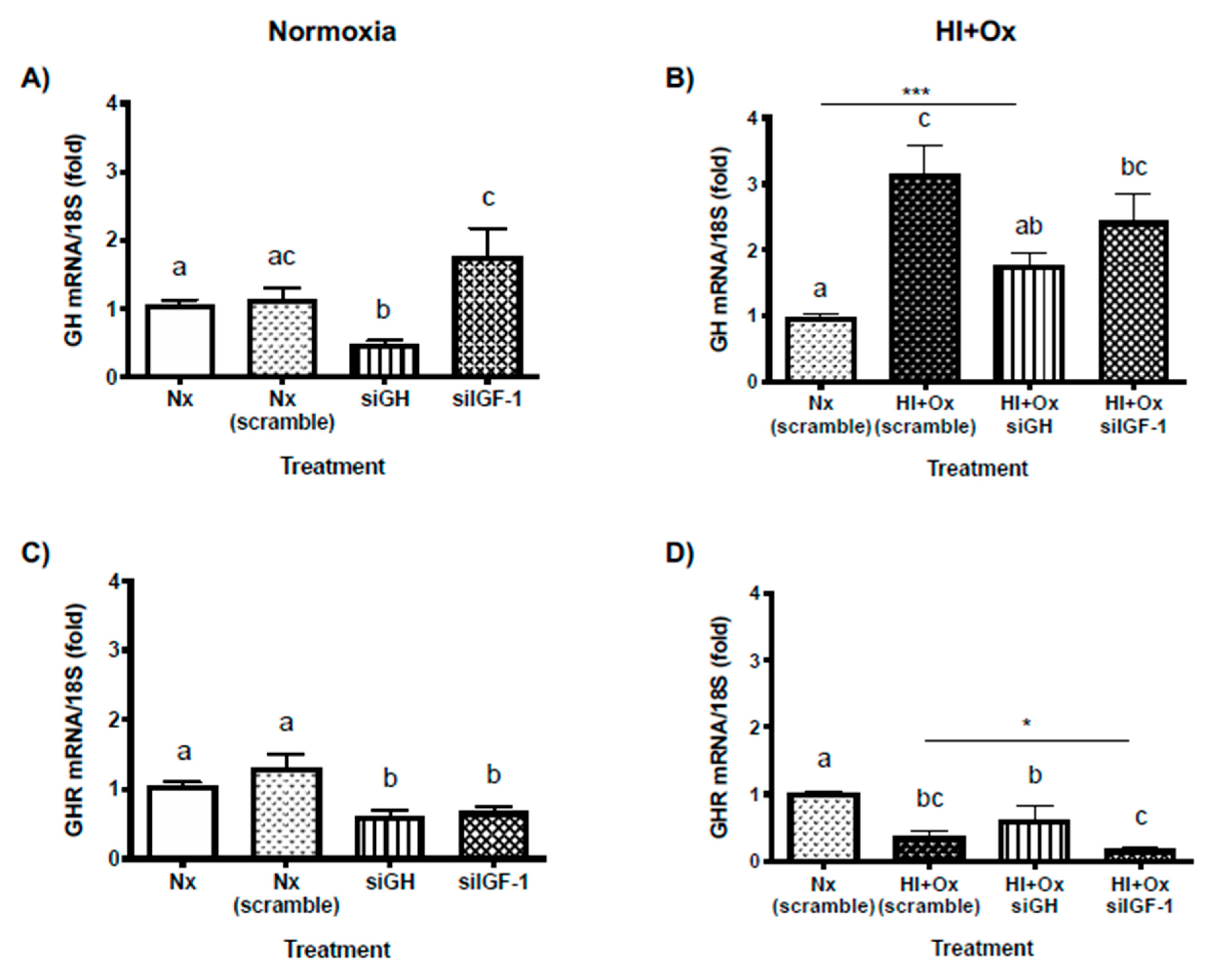
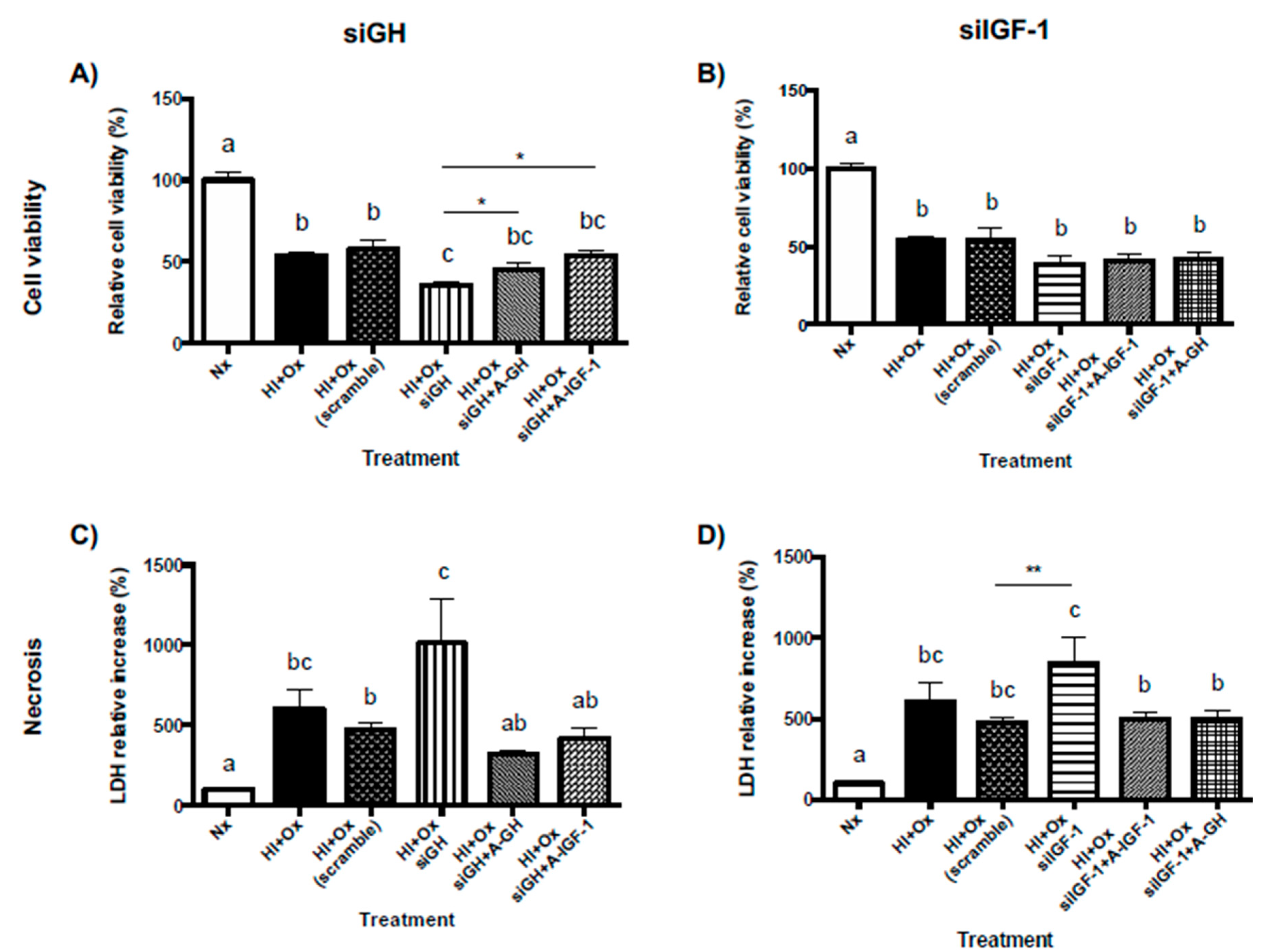
2.7. Effects from GH and IGF-1 Gene Silencing by Specific siRNAs in Cerebellar Cell Cultures under Normal and Hypoxic-Ischemic Conditions, upon GH, GHR, IGF-1 and IGF-1R mRNAs Expression
2.8. Role of Locally-Expressed and Exogenously-Added GH and IGF-1 in the Neuroprotective Response to Hypoxic-Ischemic Injury in Primary Cerebellar Cultures
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Primary Cerebellar Cell Culture
4.3. Treatments
4.4. Determination of Cell Viability
- (a)
- Trypan blue exclusion assay: Cells were harvested and resuspended in 1 mL medium, then a 10 µL aliquot was mixed with a 10 µL 0.05% trypan blue solution (Gibco 15250061, Grand Island, NY, USA), placed into a Neubauer chamber, and several fields were observed under a microscope (Olympus CX41). At least 100 cells (in duplicate) were analyzed for viability, and the mean percentage of living cells was calculated [70].
- (b)
- MTT assay was performed according to manufacturer’s instructions: In brief, culture media in the plates were substituted with DMEM media without phenol red, and then 500 µL of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Thermo-Fisher Scientific M6494, Waltham, MA, USA) labeling reagent, at a final concentration of 0.5 mg/mL, was added to each well and incubated for 4 h at 39 °C. The resulting formazan crystals were dissolved using an equal volume of the solubilization solution (1 g/mL SDS in 0.01 N HCl), and the plates were incubated for another 4 h in a humidified atmosphere at 39 °C. Aliquots (200 µL) of soluble formazan product were placed in a 96-well plate and optical density was analyzed at 570 nm in a microplate reader (Bio-Rad, Mod. 550, Hercules, CA, USA).
4.5. Determination of Apoptosis by Caspase-3 Activity
4.6. Determination of Necrosis by Lactate Dehydrogenase (LDH) Release
4.7. Immunocytochemistry
4.8. Western Blot Analysis
4.9. RT-PCR
4.10. Quantitative PCR (qPCR)
4.11. Knockdown of GH and IGF-1 RNA Expression by Small-Interfering RNA (siRNA)
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ajo, R.; Cacicedo, L.; Navarro, C.; Sanchez-Franco, F. Growth hormone action on proliferation and differentiation of cerebral cortical cells from fetal rat. Endocrinology 2003, 144, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberg, D. Role of the growth hormone/insulin-like growth factor 1 axis in neurogenesis. Endocr. Dev. 2010, 17, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Frago, L.M.; Pañeda, C.; Dickinson, L.; Hewson, A.K.; Argente, J.; Chowen, J.A. Growth hormone (GH) and GH-releasing peptide-6 increase brain insulin-like growth factor-I expression and activate intracellular signaling pathways involved in neuroprotection. Endocrinology 2002, 143, 4113–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyberg, F.; Hallberg, M. Growth hormone and cognitive function. Nat. Rev. Endocrinol. 2013, 9, 357–365. [Google Scholar] [CrossRef]
- Devesa, J.; Almengló, C.; Devesa, P. Multiple effects of growth hormone in the body: Is it really the hormone for growth. Clin. Med. Insights Endocrinol. Diabetes 2016, 9, 47–71. [Google Scholar] [CrossRef] [Green Version]
- Wasinski, F.; Frazão, R.; Donato, J., Jr. Effects of growth hormone in the central nervous system. Arch. Endocrinol. Metab. 2019, 63, 549–556. [Google Scholar] [CrossRef]
- Alba-Betancourt, C.; Arámburo, C.; Ávila-Mendoza, J.; Ahumada-Solórzano, S.M.; Carranza, M.; Rodríguez-Méndez, A.J.; Harvey, S.; Luna, M. Expression, cellular distribution, and heterogeneity of growth hormone in the chicken cerebellum during development. Gen. Comp. Endocrinol. 2011, 170, 528–540. [Google Scholar] [CrossRef]
- Arámburo, C.; Alba-Betancourt, C.; Luna, M.; Harvey, S. Expression and function of growth hormone in the nervous system: A brief review. Gen. Comp. Endocrinol. 2014, 203, 35–42. [Google Scholar] [CrossRef]
- Devesa, J.; Núñez, I.; Agra, C.; Bejarano, A.; Devesa, P. Treatment with growth hormone (GH) increased the metabolic activity of the brain in an elder patient, not GH-deficient, who suffered mild cognitive alterations and had an ApoE 4/3 genotype. Int. J. Mol. Sci. 2018, 19, 2294. [Google Scholar] [CrossRef] [Green Version]
- Isgaard, J.; Aberg, D.; Nilsson, M. Protective and regenerative effects of the GH/IGF-1 axis on the brain. Minerva Endocrinol. 2007, 32, 103–113. [Google Scholar] [PubMed]
- Alba-Betancourt, C.; Luna-Acosta, J.L.; Ramírez-Martínez, C.E.; Ávila-González, D.; Granados-Ávalos, E.; Carranza, M.; Martínez-Coria, H.; Arámburo, C.; Luna, M. Neuro-protective effects of growth hormone (GH) after hypoxia-ischemia injury in embryonic chicken cerebellum. Gen. Comp. Endocrinol. 2013, 183, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Devesa, P.; Reimunde, P.; Gallego, R.; Devesa, J.; Arce, V.M. Growth hormone (GH) treatment may cooperate with locally-produced GH in increasing the proliferative response of hippocampal progenitors to kainate-induced injury. Brain Inj. 2011, 25, 503–510. [Google Scholar] [CrossRef]
- Devesa, P.; Gelabert, M.; González-Mosquera, T.; Gallego, R.; Relova, J.L.; Devesa, J.; Arce, V.M. Growth hormone treatment enhances the functional recovery of sciatic nerves after transection and repair. Muscle Nerve 2012, 45, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Devesa, P.; Agasse, F.; Xapelli, S.; Almengló, C.; Devesa, J.; Malva, O.; Arce, V.M. Growth hormone pathways signaling for cell proliferation and survival in hippocampal neural precursors from postnatal mice. BMC Neurosci. 2014, 15, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Magalhaes Filho, C.D.; Kappeler, L.; Dupont, J.; Solinc, J.; Villapol, S.; Denis, C.; Nosten-Bertrand, M.; Billard, J.-M.; Blaise, A.; Tronche, F.; et al. Deleting IGF-1 receptor from forebrain neurons confers neuroprotection during stroke and upregulates endocrine somatotropin. J. Cereb. Blood Flow Metab. 2017, 37, 396–412. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Moreno, C.G.; Calderón-Vallejo, D.; Harvey, S.; Arámburo, C.; Quintanar, J.L. Growth hormone (GH) and gonadotropin-releasing hormone (GnRH) in the central nervous system: A potential neurological combinatory therapy? Int. J. Mol. Sci. 2018, 19, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Moreno, C.G.; Fleming, T.; Carranza, M.; Ávila-Mendoza, J.; Luna, M.; Harvey, S.; Arámburo, C. Growth hormone protects against kainate excitotoxicity and induces BDNF and NT3 expression in chicken neuroretinal cells. Exp. Eye Res. 2018, 166, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nylander, E.; Zelleroth, S.; Nyberg, F.; Grönbladh, A.; Hallberg, M. The protective and restorative effects of growth hormone and insulin-like growth factor-1 on methadone-induced toxicity in vitro. Int. J. Mol. Sci. 2018, 19, 3627. [Google Scholar] [CrossRef] [Green Version]
- Heredia, M.; Rodríguez, N.; Sánchez Robledo, V.; Criado, J.M.; de la Fuente, A.; Devesa, J.; Devesa, P.; Sánchez Riolobos, A. Factors involved in the functional motor recovery of rats with cortical ablation after GH and rehabilitation treatment: Cortical cell proliferation and nestin and actin expression in the striatum and thalamus. Int. J. Mol. Sci. 2019, 20, 5770. [Google Scholar] [CrossRef] [Green Version]
- Aberg, N.D.; Brywe, K.G.; Isgaard, J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. Sci. World J. 2006, 6, 53–80. [Google Scholar] [CrossRef] [Green Version]
- Harvey, S.; Baudet, M.-L.; Sanders, E.J. Growth hormone-induced neuroprotection in the neural retina during chick embryogenesis. Ann. N. Y. Acad. Sci. 2009, 1163, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Sanders, E.J.; Parker, E.; Harvey, S. Endogenous growth hormone in human retinal ganglion cells correlates with cell survival. Mol. Vis. 2009, 15, 920–926. [Google Scholar] [PubMed]
- Sanders, E.J.; Lin, W.Y.; Parker, E.; Harvey, S. Growth hormone promotes the survival or retinal cells in vivo. Gen. Comp. Endocrinol. 2011, 172, 140–150. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.R.; Galli, C.; Ciotti, T.; Calissano, P. Induction of apoptosis in cerebellar granule neurons by low potassium: Inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA 1993, 90, 10989–10993. [Google Scholar] [CrossRef] [Green Version]
- Parrizas, M.; Saltiel, A.R.; Le Roith, D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J. Biol. Chem. 1997, 272, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Brywe, K.G.; Mallard, C.; Gustavsson, M.; Hedtjärn, M.; Leverin, A.L.; Wang, X.; Blomgren, K.; Isgaard, J.; Hagberg, H. IGF-I neuroprotection in the immature brain after hypoxia-ischemia, involvement of Akt and GSK3beta? Eur. J. Neurosci. 2005, 21, 1489–1502. [Google Scholar] [CrossRef]
- Sanders, E.J.; Lin, W.Y.; Parker, E.; Harvey, S. Growth hormone expression and neuroprotective activity in a quail neural retina cell line. Gen. Comp. Endocrinol. 2010, 165, 111–119. [Google Scholar] [CrossRef]
- Guan, J.; Bennet, L.; Gluckman, P.D.; Gunn, A. Insulin-like growth factor-1 and post-ischemic brain injury. Prog. Neurobiol. 2003, 70, 443–462. [Google Scholar] [CrossRef]
- Lee, W.H.; Wang, G.M.; Seaman, L.B.; Vannucci, S.J. Coordinate IGF-I and IGFBP5 gene expression in perinatal rat brain after hypoxia-ischemia. J. Cereb. Blood Flow Metab. 1996, 16, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Gluckman, P.; Klempt, N.D.; Guan, J.; Mallard, C.; Sirimanne, E.; Dragunow, M.; Klempt, M.; Singh, K.; Williams, C.; Nikolics, K. A role for IGF-1 in the rescue of CNS neurons following hypoxic-ischemic injury. Biochem. Biophys. Res. Commun. 1992, 182, 593–599. [Google Scholar] [CrossRef]
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The somatomedin hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef] [PubMed]
- Stroka, D.M.; Burkhardt, T.; Desbaillets, I.; Wenger, R.H.; Neil, D.A.; Bauer, C.; Gassmann, M.; Candinas, D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001, 15, 2445–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giusti, S.; Fiszer de Plazas, S. Neuroprotection by hypoxic preconditioning involves upregulation of hypoxia-inducible factor-1 in a prenatal model of acute hypoxia. J. Neurosci. Res. 2012, 90, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, P.; Shi, H. Ischemia induces different levels of hypoxia inducible factor-1α protein expression in interneurons and pyramidal neurons. Acta Neuropathol. Commun. 2014, 2, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Boie, G.; Doerr, H.G.; Trollmann, R. Oxygen-sensitive regulation and neuroprotective effects of growth hormone-dependent growth factors during early postnatal development. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R539–R548. [Google Scholar] [CrossRef]
- Sharp, F.R.; Bernaudin, M. HIF1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 2004, 5, 437–448. [Google Scholar] [CrossRef]
- Liang, X.; Liu, X.; Lu, F.; Zhang, Y.; Jiang, X.; Ferriero, D.M. HIF1α signaling in the endogenous protective responses after neonatal brain hypoxia-ischemia. Dev. Neurosci. 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Heijnen, C.J.; van der Kooij, M.A.; Groenendaal, F.; van Bel, F. The role and regulation of hypoxia-inducible factor-1alpha expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Res. Rev. 2009, 62, 99–108. [Google Scholar] [CrossRef]
- Nisimov, H.; Orenbuch, A.; Pleasure, S.J.; Golan, H.M. Impaired organization of GABAergic neurons following prenatal hypoxia. Neuroscience 2018, 384, 300–313. [Google Scholar] [CrossRef]
- Han, T.R.; Chun, M.H.; Jang, D.H.; Kim, K.S.; Lim, K.H.; Cho, H.J. Neuroprotective effects of growth hormone against hypoxic-ischemic brain injury in neonatal rats: 1H magnetic resonance spectroscopic study. J. Korean Med. Sci. 2007, 22, 122–126. [Google Scholar] [CrossRef]
- Shin, D.H.; Lee, E.; Kim, J.W.; Kwon, B.S.; Jung, M.K.; Jee, Y.H.; Kim, J.; Bae, S.R.; Chang, Y.P. Protective effect of growth hormone on neuronal apoptosis after hypoxia-ischemia in the neonatal rat brain. Neurosci. Lett. 2004, 354, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Nylander, E.; Grönbladh, A.; Zelleroth, S.; Diwakarla, S.; Nyberg, F.; Hallberg, M. Growth hormone is protective against acute methadone-induced toxicity by modulating the NMDA receptor complex. Neuroscience 2016, 339, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Yang, S.N. Perinatal hypoxic-ischemic encephalopathy. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Marle, G.; Antony, J.M.; Silva, C.; Sullivan, A.; Power, C. Aberrant cortical neurogenesis in a pediatric neuroAIDS model: Neurotrophic effects of growth hormone. AIDS 2005, 19, 1781–1791. [Google Scholar] [CrossRef]
- Möderscheim, T.A.; Christophidis, L.J.; Williams, C.E.; Scheepens, A. Distinct neuronal growth hormone receptor ligand specificity in the rat brain. Brain Res. 2007, 1137, 29–34. [Google Scholar] [CrossRef]
- Christophidis, L.J.; Gorba, T.; Gustavsson, M.; Williams, C.E.; Werther, G.A.; Russo, V.C.; Scheepens, A. Growth hormone receptor immunoreactivity is increased in the subventricular zone of juvenile rat brain after focal ischemia: A potential role for growth hormone in injury-induced neurogenesis. Growth Horm. IGF Res. 2009, 19, 497–506. [Google Scholar] [CrossRef]
- McLenachan, S.; Lum, M.G.; Waters, M.J.; Turnley, A.M. Growth hormone promotes proliferation of adult neurosphere cultures. Growth Horm. IGF Res. 2009, 19, 212–218. [Google Scholar] [CrossRef]
- Sanchez-Bezanilla, S.; Åberg, N.D.; Crock, P.; Walker, F.R.; Nilsson, M.; Isgaard, J.; Ong, L.K. Growth hormone promotes motor function after experimental stroke and enhances recovery-promoting mechanisms within the peri-infarct area. Int. J. Mol. Sci. 2020, 21, 606. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Fan, L.W.; Rhodes, P.G.; Cai, Z. Intranasal administration of IGF-1 attenuates hypoxic-ischemic brain injury in neonatal rats. Exp. Neurol. 2009, 217, 361–370. [Google Scholar] [CrossRef] [Green Version]
- D’Ercole, A.J.; Ye, P.; Calikoglu, A.S.; Gutiérrez-Ospina, G. The role of the insulin-like growth factors in the central nervous system. Mol. Neurobiol. 1996, 13, 227–255. [Google Scholar] [CrossRef]
- D’Ercole, A.J.; Ye, P. Expanding the mind: Insulin-like growth factor I and brain development. Endocrinology 2008, 149, 5958–5962. [Google Scholar] [CrossRef] [Green Version]
- Costoya, J.A.; Finidori, J.; Moutoussamy, S.; Seãris, R.; Devesa, J.; Arce, V.M. Activation of growth hormone receptor delivers an antiapoptotic signal: Evidence for a role of Akt in this pathway. Endocrinology 1999, 140, 5937–5943. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.W.; Windebank, A.J.; Schenone, A.; Feldman, E.L. Insulin-like growth factor-I prevents apoptosis in neurons after nerve growth factor withdrawal. J. Neurobiol. 1998, 36, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Peruzzi, F.; Prisco, M.; Dews, M.; Salomoni, P.; Grassilli, E.; Romano, G.; Calabretta, B.; Baserga, R. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol. Cell. Biol. 1999, 19, 7203–7215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.H.; Kar, S.; Doré, S.; Quirion, R. Insulin-like growth factor-1 (IGF-1): A neuroprotective trophic factor acting via the Akt kinase pathway. J. Neural Transm. Suppl. 2000, 60, 261–272. [Google Scholar] [CrossRef]
- Yang, X.; Wei, A.; Liu, Y.; He, G.; Zhou, Z.; Yu, Z. IGF-1 protects retinal ganglion cells from hypoxia-induced apoptosis by activating the Erk-1/2 and Akt pathways. Mol. Vis. 2013, 19, 1901–1912. [Google Scholar] [PubMed]
- Zhao, B.; Zheng, Z. Insulin growth factor 1 protects neural stem cells against apoptosis induced by hypoxia through Akt/Mitogen-activated protein kinase/Extracellular signal-regulated kinase (Akt/MAPK/ERK) pathway in hypoxia-ischemic encephalopathy. Med. Sci. Monit. 2017, 23, 1872–1879. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, C.; Qiu, L.; Hagberg, H.; Sandberg, M.; Blomgren, K. Activation of ERK1/2 after neonatal rat cerebral hypoxia-ischaemia. J. Neurochem. 2003, 86, 351–362. [Google Scholar] [CrossRef]
- Thei, L.; Rocha-Ferreira, E.; Peebles, D.; Raivich, G.; Hristova, M. Extracellular signal-regulated kinase 2 has duality in function between neuronal and astrocyte expression following neonatal hypoxic-ischaemic cerebral injury. J. Physiol. 2018, 596, 6043–6062. [Google Scholar] [CrossRef]
- Fleming, T.; Balderas-Márquez, J.E.; Epardo, D.; Ávila-Mendoza, J.; Carranza, M.; Luna, M.; Harvey, S.; Arámburo, C.; Martínez-Moreno, C.G. Growth hormone neuroprotection against kainate excitotoxicity in the retina is mediated by Notch/PTEN/Akt signaling. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4532–4547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, K.; Hagberg, H.; Bengtsson, B.A.; Brantsing, C.; Isgaard, J. Possible protective role of growth hormone in hypoxia-ischemia in neonatal rats. Pediatr. Res. 1999, 45, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arellanes-Licea, E.C.; Ávila-Mendoza, J.; Ramírez-Martínez, E.C.; Ramos, E.; Uribe-González, N.; Arámburo, C.; Morales, T.; Luna, M. Upregulation of GH, but not IGF1, in the hippocampus of the lactating dam after kainic acid injury. Endocr. Connect. 2018, 7, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devesa, J.; Reimunde, P.; Devesa, P.; Barberá, M.; Arce, V. Growth hormone (GH) and brain trauma. Horm. Behav. 2013, 63, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.K.; Chow, W.Z.; TeBay, C.; Kluge, M.; Pietrogrande, G.; Zalewska, K.; Crock, P.; Åberg, N.D.; Bivard, A.; Johnson, S.J.; et al. Growth hormone improves cognitive function after experimental stroke. Stroke 2018, 49, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Novosyadlyy, R.; Wu, Y.; Yakar, S.; LeRoith, D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm. IGF Res. 2010, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gosney, E.S.; Jara, A.; Basu, A.; Kopchick, J.J. GH in the central nervous system: Lessons from the growth hormone receptor knockout mouse. Open Endocrinol. J. 2012, 6, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Donahue, C.P.; Kosik, K.S.; Shors, T.J. Growth hormone is produced within the hippocampus where it responds to age, sex, and stress. Proc. Natl. Acad. Sci. USA 2006, 103, 6031–6036. [Google Scholar] [CrossRef] [Green Version]
- Hallberg, M.; Nyberg, F. Growth hormone receptors in the brain and their potential as therapeutic targets in central nervous system disorders. Open Endocrinol. J. 2012, 6, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Heredia, M.; Fuente, A.; Criado, J.; Yajeya, J.; Devesa, J.; Riolobos, A.S. Early growth hormone (GH) treatment promotes relevant motor functional improvement after severe frontal cortex lesion in adult rats. Behav. Brain Res. 2013, 247, 48–58. [Google Scholar] [CrossRef]
- Tennant, J.R. Evaluation of the trypan blue technique for determination of cell viability. Transplantation 1964, 2, 685–694. [Google Scholar] [CrossRef]
- Luna-Acosta, J.L.; Alba-Betancourt, C.; Martínez-Moreno, C.G.; Ramírez, C.; Carranza, M.; Luna, M.; Arámburo, C. Direct antiapoptotic effects of growth hormone are mediated by PI3K/Akt pathway in the chicken bursa of Fabricius. Gen. Comp. Endocrinol. 2015, 224, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Vassault, A. (Ed.) Methods of Enzymatic Analysis; Academic Press: New York, NY, USA, 1983; pp. 118–126. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Baudet, M.L.; Rattray, D.; Martin, B.T.; Harvey, S. Growth hormone promotes axon growth in the developing nervous system. Endocrinology 2009, 150, 2758–2766. [Google Scholar] [CrossRef] [Green Version]
- Ahumada-Solórzano, S.M.; Martínez-Moreno, C.G.; Carranza, M.; Ávila-Mendoza, J.; Luna-Acosta, J.L.; Harvey, S.; Luna, M.; Arámburo, C. Autocrine/paracrine proliferative effect of ovarian GH and IGF-I in chicken granulosa cell cultures. Gen. Comp. Endocrinol. 2016, 234, 47–56. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Host/Type | Dilution | Source | Cat. No. |
|---|---|---|---|---|
| DCX | guinea pig/polyclonal | 1:1000 | Sigma-Aldrich | AB2253 |
| NeuN | mouse/monoclonal | 1:1000 | Sigma-Aldrich | MAB377 |
| HIF-1α | rabbit/polyclonal | 1:1000 | Cell Signaling | 3716S |
| β-Tubulin III | mouse/monoclonal | 1:2000 | Sigma-Aldrich | T8578 |
| Phospho-Akt(S437) | rabbit/monoclonal | 1:1000 | Thermo Fisher Scientific | 44-621G |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | rabbit/monoclonal | 1:1000 | Cell Signaling | 4370S |
| Bcl-2 | rabbit/polyclonal | 1:1000 | Invitrogen | 138800 |
| GAPDH | rabbit/monoclonal | 1:2000 | Cell Signaling Technology | 14C10 |
| Goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, HRP | goat/polyclonal | 1:5000 | Thermo Fisher Scientific | G-21040 |
| Goat anti-rabbit Ig (H + L) secondary antibody, HRP | goat/polyclonal | 1:5000 | Invitrogen | 656120 |
| Goat anti-guinea pig IgG antibody, HRP conjugate | goat/polyclonal | 1:5000 | Millipore | AP108P |
| Rabbit anti-mouse IgG (H + L) secondary antibody, FITC | rabbit/polyclonal | 1:2000 | Invitrogen | A16161 |
| Goat anti-rabbit IgG antibody, Cy3 conjugate | goat/polyclonal | 1:5000 | Millipore | AP132C |
| Target | Primer | Sequence (5′–3′) | Size | Accession Number |
|---|---|---|---|---|
| cGH | Fwd Rev | CGCACCTATATTCCGGAGGAC GGCAGCTCCATGTCTGACT | 128 bp | NM_204359 |
| cGHR | Fwd Rev | ACTTCACCATGGACAATGCCTA GGGGTTTCTGCCATTGAAGCTC | 180 bp | NM_001001293.1 |
| cIGF-1 | Fwd Rev | TACCTTGGCCTGTGTTTGCT CCCTTGTGGTGTAAGCGTCT | 170 bp | NM_001004384 |
| cIGF-1R | Fwd Rev | TCCAACACAACACTGAAGAATC ACCATATTCCAGCTATTGGAGC | 166 bp | NM_205032.1 |
| c18S | Fwd Rev | CTCTTTCTCGATTCCGTGGGT TTAGCATGCCAGAGTCTCGT | 100 bp | M59389 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baltazar-Lara, R.; Ávila-Mendoza, J.; Martínez-Moreno, C.G.; Carranza, M.; Pech-Pool, S.; Vázquez-Martínez, O.; Díaz-Muñoz, M.; Luna, M.; Arámburo, C. Neuroprotective Effects of Growth Hormone (GH) and Insulin-Like Growth Factor Type 1 (IGF-1) after Hypoxic-Ischemic Injury in Chicken Cerebellar Cell Cultures. Int. J. Mol. Sci. 2021, 22, 256. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010256
Baltazar-Lara R, Ávila-Mendoza J, Martínez-Moreno CG, Carranza M, Pech-Pool S, Vázquez-Martínez O, Díaz-Muñoz M, Luna M, Arámburo C. Neuroprotective Effects of Growth Hormone (GH) and Insulin-Like Growth Factor Type 1 (IGF-1) after Hypoxic-Ischemic Injury in Chicken Cerebellar Cell Cultures. International Journal of Molecular Sciences. 2021; 22(1):256. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010256
Chicago/Turabian StyleBaltazar-Lara, Rosario, José Ávila-Mendoza, Carlos G. Martínez-Moreno, Martha Carranza, Santiago Pech-Pool, Olivia Vázquez-Martínez, Mauricio Díaz-Muñoz, Maricela Luna, and Carlos Arámburo. 2021. "Neuroprotective Effects of Growth Hormone (GH) and Insulin-Like Growth Factor Type 1 (IGF-1) after Hypoxic-Ischemic Injury in Chicken Cerebellar Cell Cultures" International Journal of Molecular Sciences 22, no. 1: 256. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010256