Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of mRNAs, lncRNAs, miRNAs and circRNAs in Response to Salt Stress in Sugar Beet (Beta vulgaris)


Abstract
:1. Introduction
2. Results
2.1. Effects of Salinity on Sugar Beet Physiological Indices
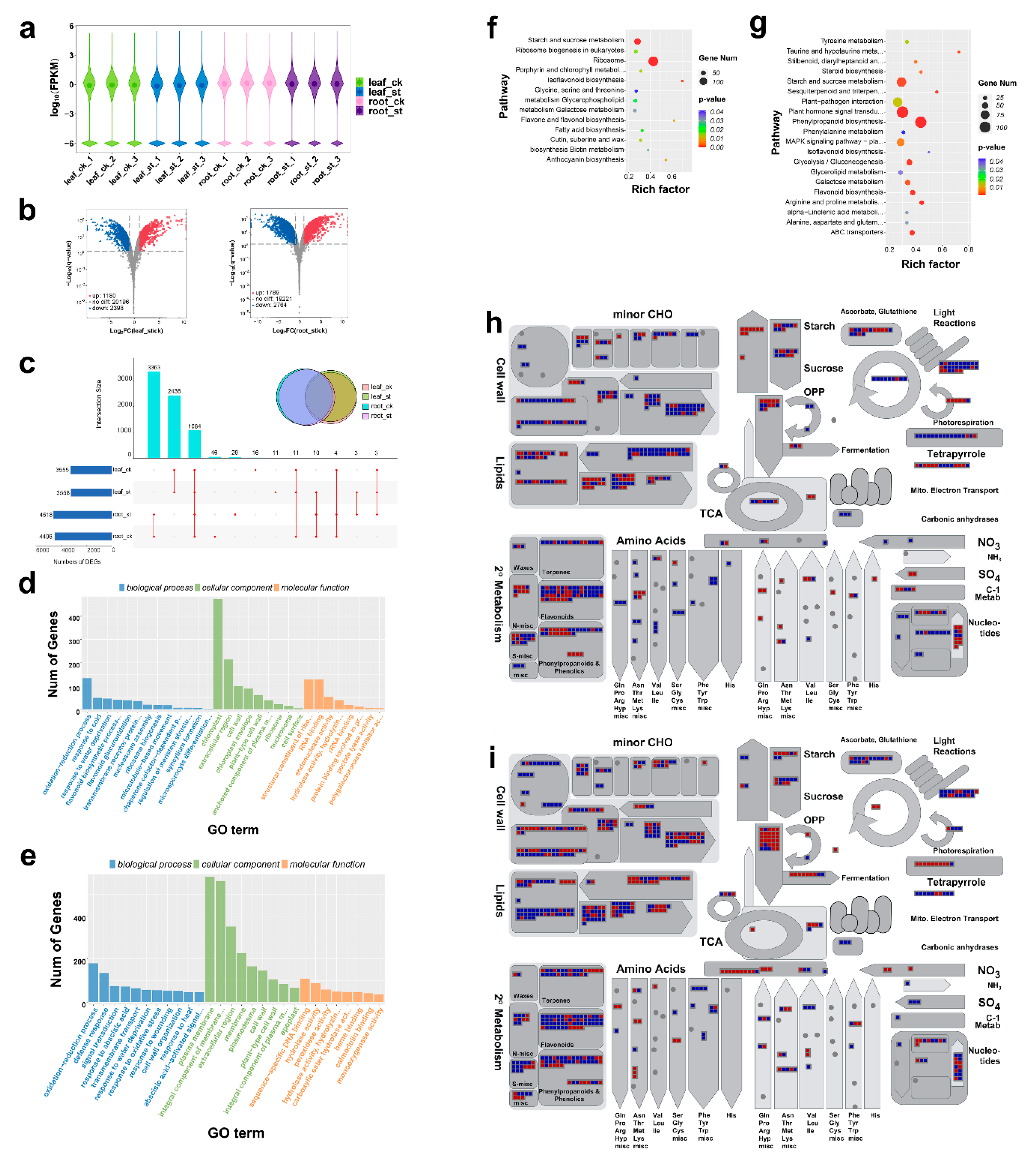
2.2. Global Response of mRNA to Salt Stress
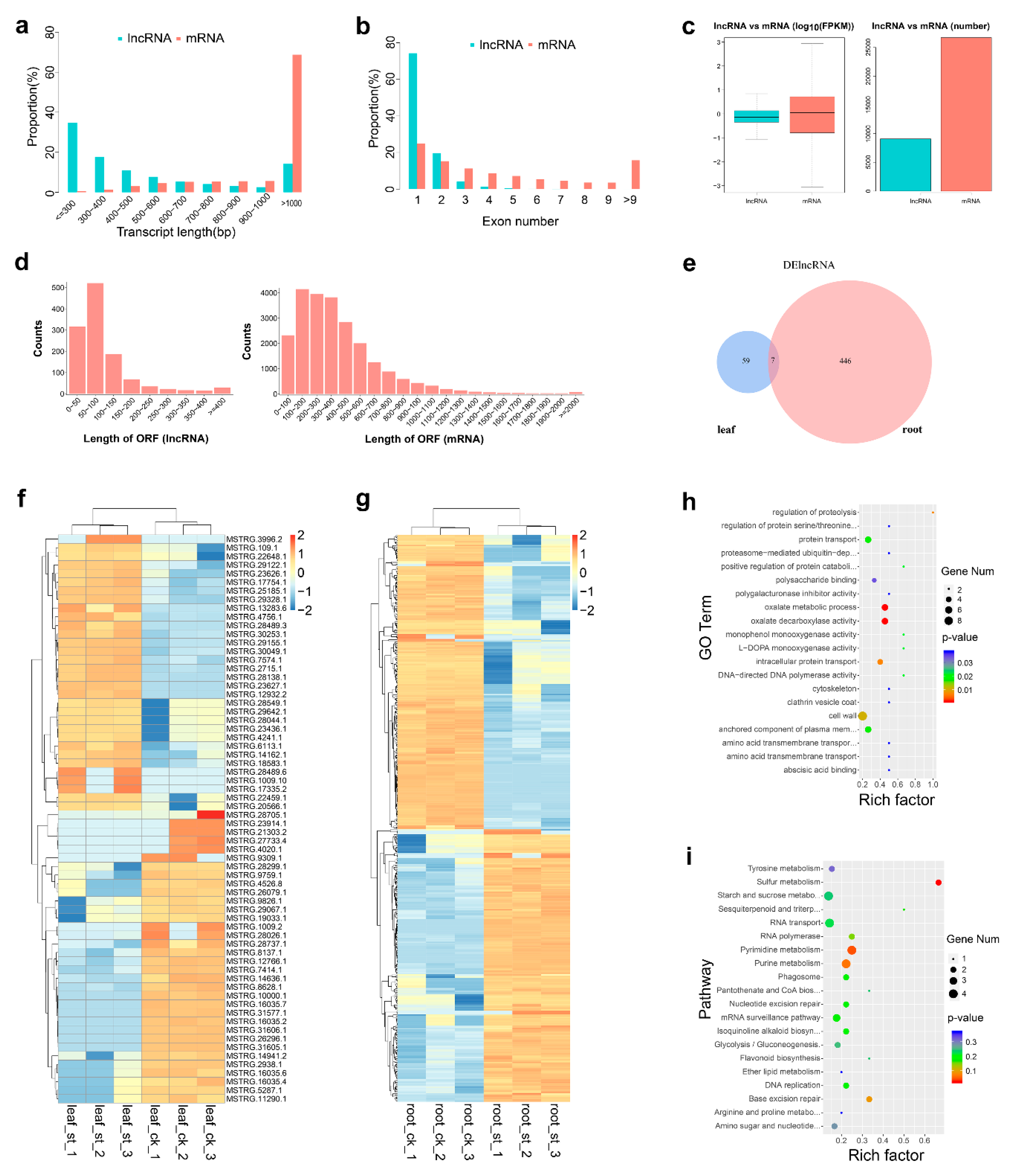
2.3. Global Response of lncRNA to Salt Stress
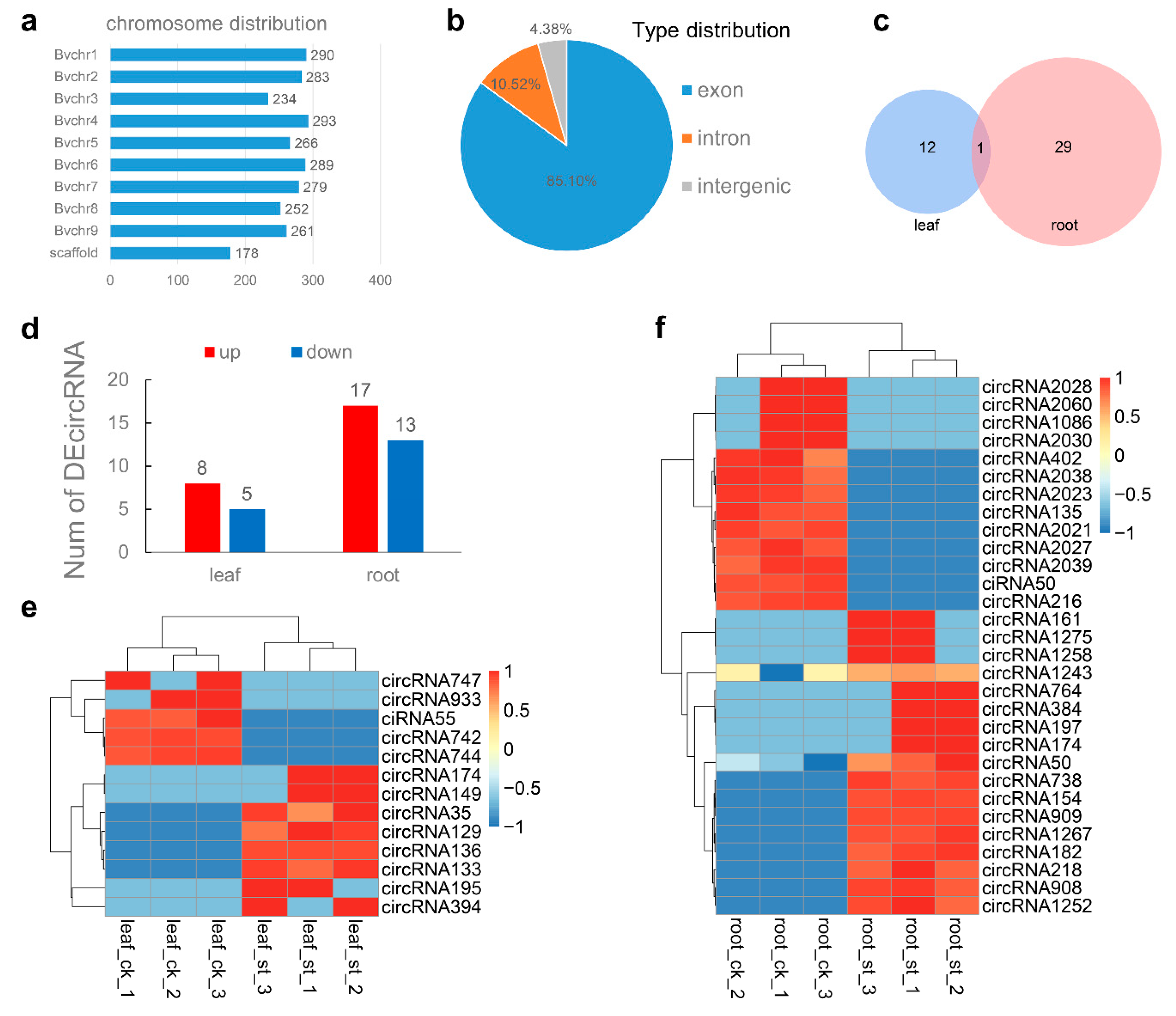
2.4. Global Responses of circRNA to Salt Stress
2.5. Global Responses of microRNA to Salt Stress
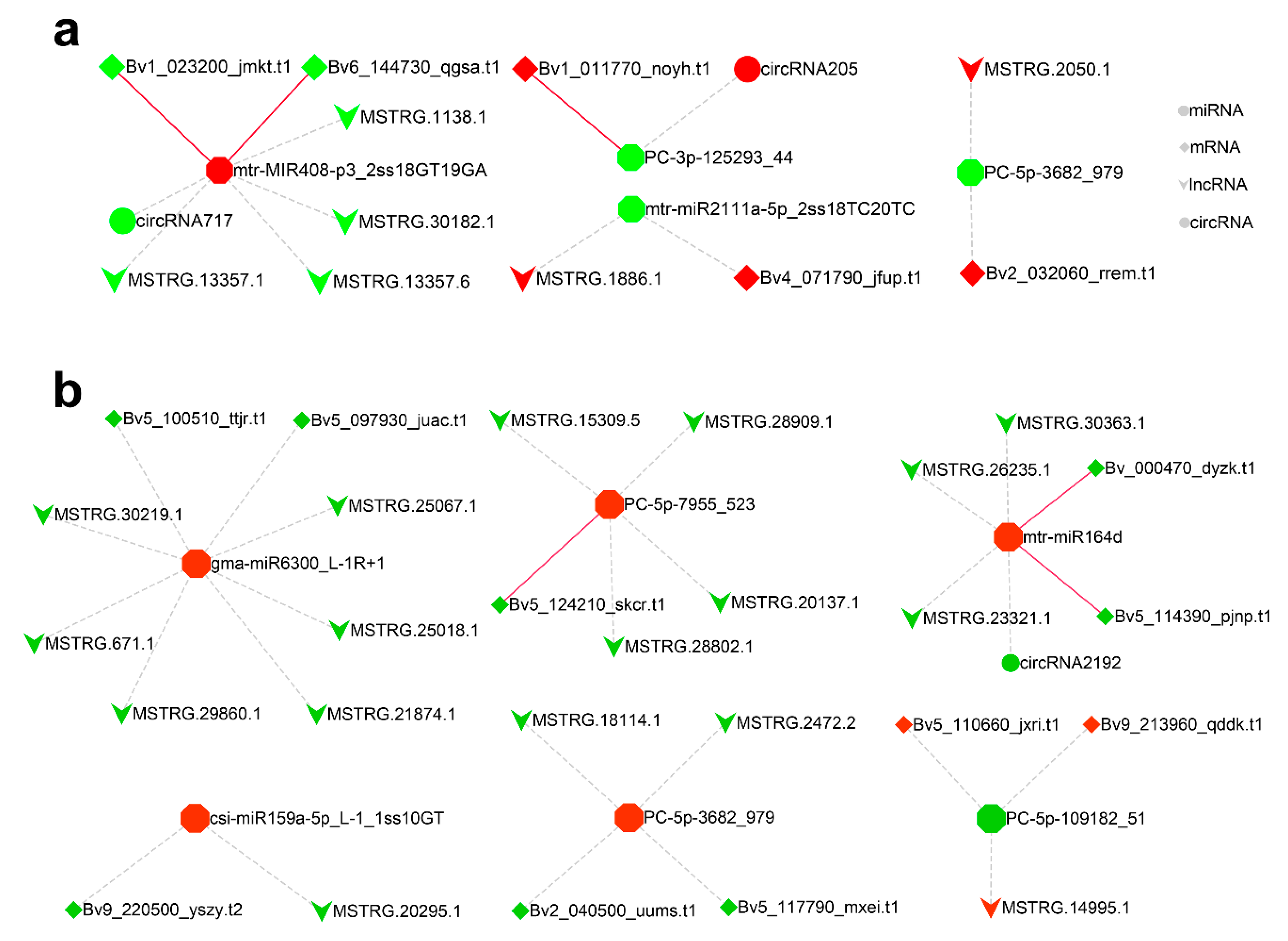
2.6. CeRNA Regulatory Network in Response to Salt Stress
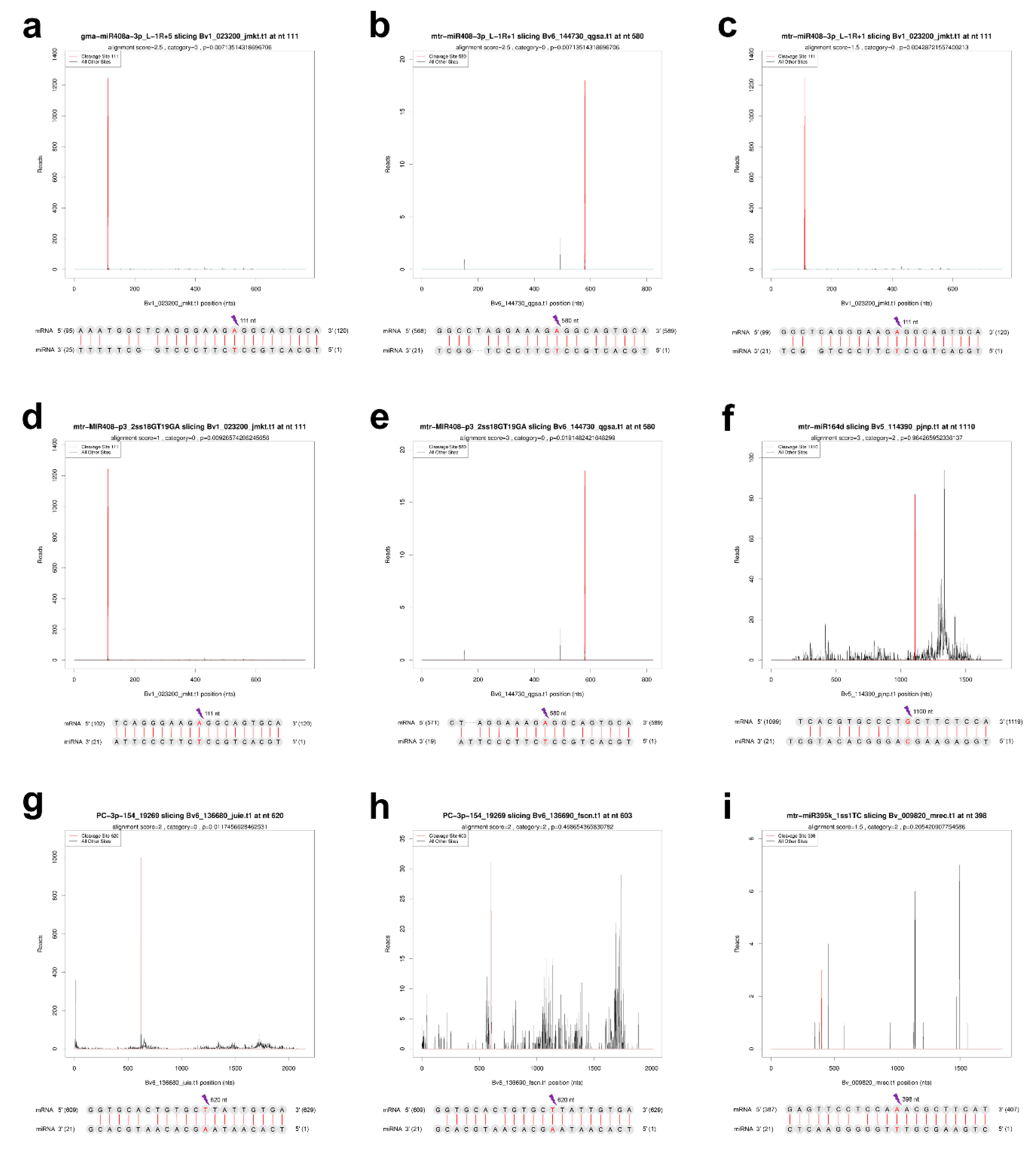
2.7. Verification of the Cleavage of miRNA to ceRNAs by Degradome Sequencing
2.8. qRT-PCR Validated Expression Correlation between miRNAs and ceRNAs under Salt Stress
3. Discussion
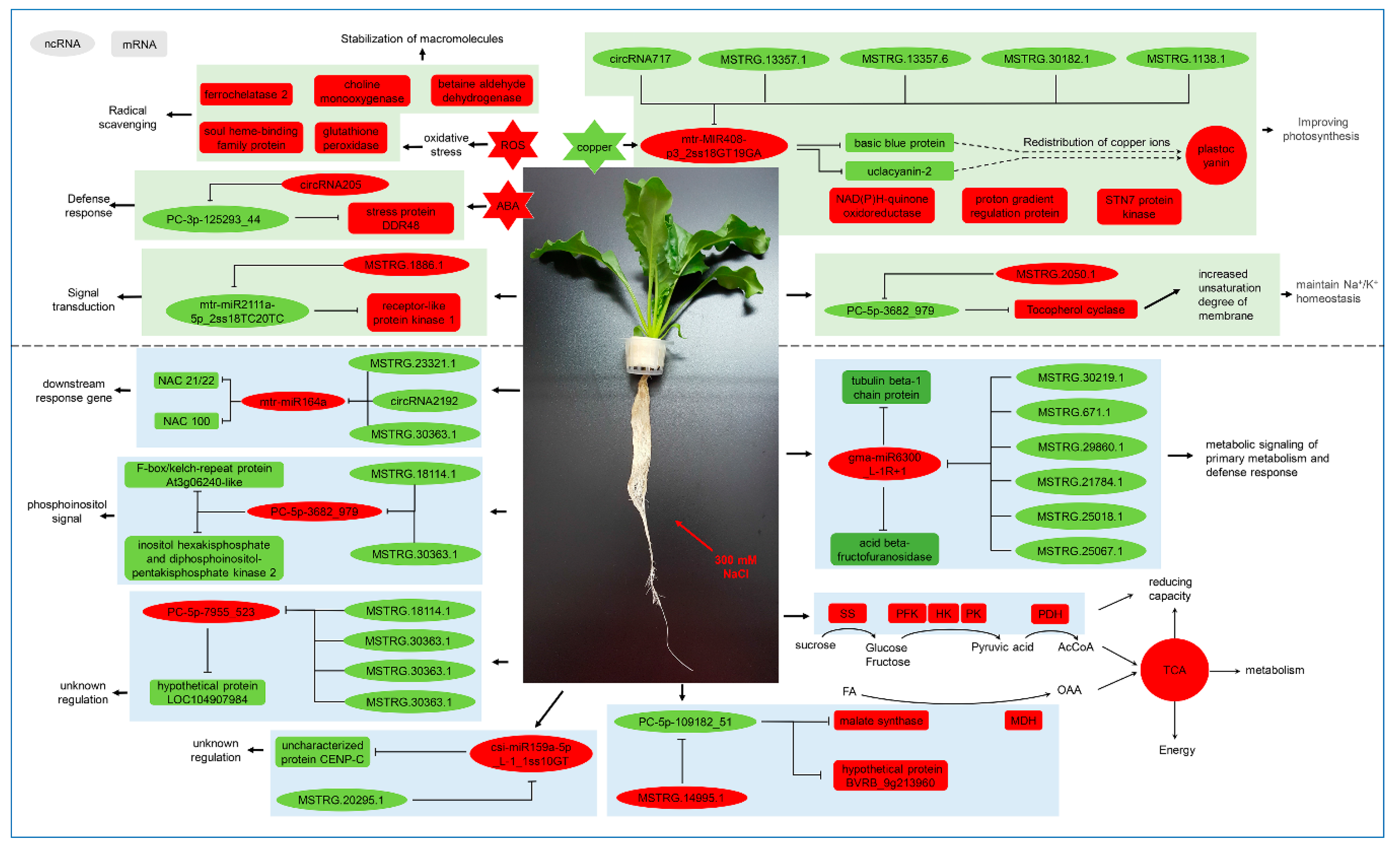
3.1. Analysis of Salt Stress Response in Sugar Beet Leaves
3.2. Analysis of Salt Stress Response in Sugar Beet Roots
4. Materials and Methods
4.1. Plant Cultivation and Treatments
4.2. Measurement of Physiologic Indicators and Harvest
4.3. RNA Extraction, Library Preparation, and RNA Sequencing
4.4. Read Mapping and Transcriptome Assembly
4.5. Differentially Expressed mRNA and Bioinformatics Analysis
4.6. Identification and Analysis of lncRNAs
4.7. Identification and Analysis of CircRNAs
4.8. Identification and Analysis of miRNAs
4.9. Degradome Sequencing Validation of the Cleavage of miRNA to Target Genes
4.10. Construction and Analysis of ceRNAs Regulatory Network
4.11. qRT-PCR Validation of DEmRNAs, DElncRNAs, and DEmiRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
Availability of Data and Materials
Abbreviations
| DEmRNA | Differentially expressed mRNA |
| DElncRNA | Differentially expressed lncRNA |
| DEcircRNA | Differentially expressed circRNA |
| DEmiRNA | Differentially expressed miRNA |
| DEG | Differentially expressed gene |
| MDA | malonaldehyde |
| POD | peroxidase |
| SOD | superoxide dismutase |
| CAT | catalase |
| ABA | abscisic acid |
| ROS | reactive oxygen species |
| FPKM | fragments per kilo-base per million reads |
| GO | gene ontology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| qRT-PCR | quantitative real-time polymerase chain reaction |
References
- Rengasamy, P. Soil processes affecting crop production in salt-affected soils. Funct. Plant Biol. 2010, 37, 613–620. [Google Scholar] [CrossRef]
- Munns, R.; Gilliham, M. Salinity tolerance of crops-what is the cost? New Phytol. 2015, 208, 668–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tester, M.; Davenport, R. Na+ Tolerance and Na+ Transport in Higher Plants. Ann. Bot. 2003, 91, 503–527. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of Salinity Tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flowers, T.J.; Colmer, T.D. Plant salt tolerance: Adaptations in halophytes. Ann. Bot. 2015, 115, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garthwaite, A.J.; Bothmer, R.V.; Colmer, T.D. Salt tolerance in wild Hordeum species is associated with restricted entry of Na+ and Cl- into the shoots. J. Exp. Bot. 2005, 56, 2365–2378. [Google Scholar] [CrossRef]
- Deinlein, U.; Stephan, A.B.; Horie, T.; Luo, W.; Xu, G.; Schroeder, J.I. Plant salt-tolerance mechanisms. Trends Plant Sci. 2014, 19, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Feng, K.; Yu, J.; Cheng, Y.; Ruan, M.; Wang, R.; Ye, Q.; Zhou, G.; Li, Z.; Yao, Z.; Yang, Y.; et al. The SOD Gene Family in Tomato: Identification, Phylogenetic Relationships, and Expression Patterns. Front. Plant Sci. 2016, 7, 1279. [Google Scholar] [CrossRef] [Green Version]
- Kapilan, R.; Vaziri, M.; Zwiazek, J.J. Regulation of aquaporins in plants under stress. Biol. Res. 2018, 51, 4. [Google Scholar] [CrossRef]
- Liu, X.S.; Feng, S.J.; Zhang, B.Q.; Wang, M.Q.; Cao, H.W.; Rono, J.K.; Chen, X.; Yang, Z.M. OsZIP1 functions as a metal efflux transporter limiting excess zinc, copper and cadmium accumulation in rice. BMC Plant Biol. 2019, 19, 283. [Google Scholar] [CrossRef]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Zhang, Y.; Chen, X.; Chen, Y. Plant Noncoding RNAs: Hidden Players in Development and Stress Responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs, Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Jin, J.; Qian, Q.; Huang, K.; Ding, Y. Small RNA and degradome profiling reveals miRNA regulation in the seed germination of ancient eudicot Nelumbo. Nucifera. BMC Genom. 2016, 17, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X. A MicroRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Sci. N. Y. 2004, 303, 2022–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like Genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Zhang, H.; Zheng, Y.; Ding, Y. Comparative expression profiling of miRNAs between the cytoplasmic male sterile line MeixiangA and its maintainer line MeixiangB during rice anther development. Planta 2015, 241, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Chinnusamy, V.; Zhu, J.; Zhu, J.-K. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007, 12, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, Y.-L.; Zhao, J.-H.; Wang1, S.; Jin1, Y.; Chen, Z.-Q.; Fang, Y.-Y.; Hua, C.-L.; Ding, S.-W.; Guo, H.-S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, H.; Chua, N.-H. Long noncoding RNA transcriptome of plants. Plant Biotechnol. J. 2015, 13, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Shuai, P.; Liang, D.; Tang, S.; Zhang, Z.; Ye, C.-Y.; Su, Y.; Xia, X.; Yin, W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus Trichocarpa. J. Exp. Bot. 2014, 65, 4975–4983. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ding, Z.; Tan, D.; Han, B.; Sun, X.; Zhang, J. Genome-wide discovery and functional prediction of salt-responsive lncRNAs in duckweed. BMC Genom. 2020, 21, 212. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, J.; Deng, F.; Wang, W.; Cheng, Y.; Song, L.; Hu, M.; Shen, J.; Xu, Q.; Shen, F. The long non-coding RNA lncRNA973 is involved in cotton response to salt stress. BMC Plant Biol. 2019, 19, 459. [Google Scholar] [CrossRef]
- Sun, Y.; Hao, P.; Lv, X.; Tian, J.; Wang, Y.; Zhang, X.; Xu, X.; Han, Z.; Wu, T. A long non-coding apple RNA, MSTRG.85814.11, acts as a transcriptional enhancer of SAUR32 and contributes to the Fe-deficiency response. Plant J. 2020, 103, 53–67. [Google Scholar] [CrossRef]
- Chen, G.; Cui, J.; Wang, L.; Zhu, Y.; Lu, Z.; Jin, B. Genome-Wide Identification of Circular RNAs in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 1678. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Chen, L.; Liu, C.; Zhu, Q.; Fan, L. Widespread noncoding circular RNAs in plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, M.; Wei, S.; Qin, F.; Zhao, H.; Suo, B. Identification of Circular RNAs and Their Targets in Leaves of Triticum aestivum L. under Dehydration Stress. Front. Plant Sci. 2016, 7, 2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, J.; Wang, Q.; Zhu, B.; Luo, Y.; Gao, L. Deciphering the roles of circRNAs on chilling injury in tomato. Biochem. Biophys Res. Commun. 2016, 479, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qin, S.; Bao, L.; Guo, Z.; Zhao, L. Identification and functional prediction of circRNAs in Populus Euphratica Oliv. heteromorphic leaves. Genomics 2020, 112, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Li, J.; Luo, M.; Li, H.; Chen, Q.; Wang, L.; Song, S.; Zhao, L.; Xu, W.; Zhang, C.; et al. Characterization and Cloning of Grape Circular RNAs Identified the Cold Resistance-Related Vv-circATS1. Plant Physiol. 2019, 180, 966–985. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Deng, S.; Li, Z.; Wu, J.; Liu, Q.; Liu, W.; Yu, W.; Zhang, Y.; Shi, W.; Zhou, J.; et al. Competing Endogenous RNA Networks Underlying Anatomical and Physiological Characteristics of Poplar Wood in Acclimation to Low Nitrogen Availability. Plant Cell Physiol. 2019, 60, 2478–2495. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, X.; Qiu, J.; Zhou, X.; Yuan, M.; He, Y.; Chun, C.; Cao, L.; Ling, L.; Peng, L. Whole-transcriptome RNA sequencing reveals the global molecular responses and ceRNA regulatory network of mRNAs, lncRNAs, miRNAs and circRNAs in response to copper toxicity in Ziyang Xiangcheng (Citrus junos Sieb. Ex Tanaka). BMC Plant Biol. 2019, 19, 509. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Guo, S.; Wang, Y.; Wang, L.; Shu, S.; Sun, J. Systematic identification and analysis of heat-stress-responsive lncRNAs, circRNAs and miRNAs with associated co-expression and ceRNA networks in cucumber (Cucumis sativus L.). Physiol. Plant 2020, 168, 736–754. [Google Scholar] [CrossRef]
- Skorupa, M.; Gołębiewski, M.; Kurnik, K.; Niedojadło, J.; Kęsy, J.; Klamkowski, K.; Wójcik, K.; Treder, W.; Tretyn, A.; Tyburski, J. Salt stress vs. salt shock-the case of sugar beet and its halophytic ancestor. BMC Plant Biol. 2019, 19, 57. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Cui, J.; Lu, Z.; Cheng, D.; Luo, C. Screening of tolerance to NaCl in sugar beet germplasms. China Beet Sugar 2008, 4, 7–9. [Google Scholar]
- Dohm, J.C.; Minoche, A.E.; Holtgra¨we, D.; Capella-Gutie’rrez, S.; Zakrzewski, F.; Tafer, H.; Rupp, O.; RosleffSo¨rensen, T.; Stracke, R.; Reinhardt, R.; et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 2014, 505, 546–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and Functional Diversification of Small RNA Pathways in Plants. PLoS Biol. 2004, 2, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, T.; Kato, T.; Nishio, M.; Goto, K.; Atagi, S.; Hosomi, Y.; Yamamoto, N.; Hida, T.; Maemondo, M.; Nakagawa, K.; et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): An open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014, 15, 1236–1244. [Google Scholar] [CrossRef]
- Ala, U.; Karreth, F.A.; Bosia, C.; Pagnani, A.; Taulli, R.; Léopold, V.; Tay, Y.; Provero, P.; Zecchina, R.; Pandolfi, P.P. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc. Natl. Acad. Sci. USA 2013, 110, 7154–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, S.; Murphy, D.; Bhave, M. Glycine betaine biosynthesis in saltbushes (Atriplex spp.) under salinity stress. Biologia 2013, 68, 879–895. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, M.; Yu, D.; Teng, Y.; Wei, T.; Chen, C.; Song, W. Identification of Glutathione Peroxidase (GPX) Gene Family in Rhodiola crenulata and Gene Expression Analysis under Stress Conditions. Int. J. Mol. Sci. 2018, 19, 3329. [Google Scholar] [CrossRef] [Green Version]
- Bashir, K.; Rasheed, S.; Kobayashi, T.; Seki, M.; Nishizawa, N.K. Regulating Subcellular Metal Homeostasis: The Key to Crop Improvement. Front. Plant Sci. 2016, 7, 1192. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cui, J.; Cheng, D.; Dai, C.; Liu, T.; Wang, C.; Luo, C. iTRAQ protein profile analysis of sugar beet under salt stress: Different coping mechanisms in leaves and roots. BMC Plant Biol. 2020, 20, 347. [Google Scholar] [CrossRef]
- Ma, C.; Burd, S.; Lers, A. miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 2015, 84, 169–187. [Google Scholar] [CrossRef]
- Hajyzadeh, M.; Turktas, M.; Khawar, K.M.; Unver, T. miR408 overexpression causes increased drought tolerance in chickpea. Gene 2015, 555, 186–193. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Miao, X.; Hu, X.; Nan, S.; Wang, J.; Fu, H. Effect of salt stress on fatty acid and alpha-tocopherol metabolism in two desert shrub species. Planta 2018, 247, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yang, D.; Ha, Y.; Shin, H.-Y.; Nam, K.H. Receptor-like protein kinases RPK1 and BAK1 sequentially form complexes with the cytoplasmic kinase OST1 to regulate ABA-induced stomatal closure. J. Exp. Bot. 2020, 71, 1491–1502. [Google Scholar] [PubMed]
- Wei, M.; Zhuang, Y.; Li, H.; Li, P.; Huo, H.; Shu, D.; Huang, W.; Wang, S. The cloning and characterization of hypersensitive to salt stress mutant, affected in quinolinate synthase, highlights the involvement of NAD in stress-induced accumulation of ABA and proline. Plant J. 2020, 102, 85–98. [Google Scholar] [CrossRef]
- Ruan, Y.-L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Mclaughlin, J.E.; Boyer, J.S. Sugar-responsive gene expression, invertase activity, and senescence in aborting maize ovaries at low water potentials. Ann. Bot. 2004, 94, 675–689. [Google Scholar] [CrossRef]
- Roitsch, T.; Balibrea, M.E.; Hofmann, M.; Proels, R.; Sinha, A.K. Extracellular invertase: Key metabolic enzyme and PR protein. J. Exp. Bot. 2003, 54, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Liu, M. The microtubule cytoskeleton acts as a sensor for stress response signaling in plants. Mol. Biol. Rep. 2019, 46, 5603–5608. [Google Scholar] [CrossRef]
- Pua, E.-C.; Chandramouli, S.; Han, P.; Liu, P. Malate synthase gene expression during fruit ripening of Cavendish banana (Musa acuminata cv. Williams). J. Exp. Bot. 2003, 54, 309–316. [Google Scholar] [CrossRef]
- Mao, C.; Lu, S.; Lv, B.; Zhang, B.; Shen, J.; He, J.; Luo, L.; Xi, D.; Chen, X.; Ming, F. A Rice NAC Transcription Factor Promotes Leaf Senescence via ABA Biosynthesis. Plant Physiol. 2017, 174, 1747–1763. [Google Scholar] [CrossRef] [Green Version]
- Thirumalaikumar, V.P.; Devkar, V.; Mehterov, N.; Ali, S.; Ozgur, R.; Turkan, I.; Mueller-Roeber, B.; Balazadeh, S. NAC transcription factor JUNGBRUNNEN1 enhances drought tolerance in tomato. Plant Biotechnol. J. 2018, 16, 354–366. [Google Scholar] [CrossRef]
- Liu, J.; Fan, H.; Wang, Y.; Han, C.; Wang, X.; Yu, J.; Li, D.; Zhang, Y. Genome-Wide microRNA Profiling Using Oligonucleotide Microarray Reveals Regulatory Networks of microRNAs in Nicotiana benthamiana During Beet Necrotic Yellow Vein Virus Infection. Viruses 2020, 12, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hichri, I.; Muhovski, Y.; Clippe, A.; Žižková, E.; Dobrev, P.I.; Motyka, V.; Lutts, S. SlDREB2, a tomato dehydration-responsive element-binding 2 transcription factor, mediates salt stress tolerance in tomato and Arabidopsis. Plant Cell Env. 2016, 39, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, D.; Pirrello, J.; Amor, H.B.; Hammami, A.; Charfeddine, M.; Dhieb, A.; Bouzayen, M.; Gargouri-Bouzid, R. Ectopic expression of dehydration responsive element binding proteins (StDREB2) confers higher tolerance to salt stress in potato. Plant Physiol. Biochem. 2012, 60, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J. Plant Physiology Experiment Guide; Higher Education Press: Beijing, China, 2006. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic. Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Salzberg, S.L. TopHat-Fusion: An algorithm for discovery of novel fusion transcripts. Genome. Biol. 2011, 12, R72. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, H.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Dong, R.; Zhang, Y.; Zhang, J.; Luo, Z.; Zhang, J.; Chen, L.-L.; Yang, L. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome. Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome. Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome. Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic. Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef]
- Ma, Z.; Coruh, C.; Axtell, M.J. Arabidopsis lyrata small RNAs: Transient MIRNA and small interfering RNA loci within the Arabidopsis genus. Plant Cell 2010, 22, 1090–1103. [Google Scholar] [CrossRef] [Green Version]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yin, L.; Ying, Q.; Song, H.; Xue, D.; Lai, T.; Xu, M.; Shen, B.; Wang, H.; Shi, X. High-Throughput Sequencing and Degradome Analysis Identify miRNAs and Their Targets Involved in Fruit Senescence of Fragaria ananassa. PLoS ONE 2013, 8, e70959. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Shao, C.; Wang, H.; Jin, Y. Target mimics_ an embedded layer of microRNA-involved gene regulatory networks in plants. Bmc Genom. 2012, 13, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideke, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.F. Stem-Loop RT-qPCR for miRNAs. Curr. Protoc. Mol. Biol. 2011, 95, 15.10.1–15.10.15. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; Preter, K.D.; Pattyn, F.; Poppe, B.; Roy, N.V.; Paepe, A.D.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome. Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Indices | ck (Leaf) | st (Leaf) | ck (Root) | st (Root) |
|---|---|---|---|---|
| Relative water content (%) | 90.17 ± 0.06 * | 87.73 ± 0.15 * | - | - |
| Chlorophyll (mg·g−1 FW) | 2.104 ± 0.069 * | 1.759 ± 0.096 * | - | - |
| Tocopherol (μg·g−1 FW) | 223.22 ± 14.70 * | 164.50 ± 10.87 * | - | - |
| Soluble sugar (mg·g−1 FW) | 0.3664 ± 0.0186 * | 0.5835 ± 0.0121 * | 0.2792 ± 0.0067 * | 0.3309 ± 0.0151 * |
| MDA (nmol·g−1 FW) | 8.3427 ± 0.1464 | 9.4161 ± 0.8450 | 6.6839 ± 0.0845 | 8.1476 ± 0.5915 |
| Proline (μg·g−1 FW) | 1.4131 ± 0.1915 * | 4.0466 ± 0.0335 * | 0.5334 ± 0.0213 | 0.5897 ± 0.0563 |
| POD activity (U·g−1 FW) | 48.14 ± 1.26 | 44.43 ± 1.29 | 121.85 ± 13.85 * | 206.85 ± 8.66 * |
| SOD activity (U·g−1 FW) | 24.57 ± 0.54 * | 41.07 ± 6.28 * | 24.62 ± 1.33 * | 64.09 ± 2.49 * |
| CAT activity (U·g−1 FW) | 568.78 ± 2.71 | 569.36 ± 2.89 | 36.45 ± 1.28 * | 21.07 ± 1.15 * |
| miR_name | up/down | Transcript | up/down | Tissue | Degradome Detection |
|---|---|---|---|---|---|
| mtr-miR408-3p_L-1R+1 | up | Bv6_144730_qgsa.t1 | down | leaf | Y |
| mtr-miR408-3p_L-1R+1 | up | Bv1_023200_jmkt.t1 | down | leaf | Y |
| gma-miR408a-3p_L-1R+5 | up | Bv1_023200_jmkt.t1 | down | leaf | Y |
| PC-3p-154_19269 | up | Bv6_136680_juie.t1 | down | leaf | Y |
| PC-3p-154_19269 | up | Bv6_136690_fscn.t1 | down | leaf | Y |
| mtr-miR395a_L-1 | down | Bv_009820_mrec.t1 | up | leaf | Y |
| mtr-miR395k_1ss1TC | down | Bv_009820_mrec.t1 | up | leaf | Y |
| gma-miR403a_1ss20TC | down | Bv4_094840_csri.t1 | up | leaf | Y |
| mtr-miR395a_L-1 | down | Bv_010680_tsww.t1 | up | leaf | N |
| lus-MIR396e-p5 | down | Bv1_015660_nghm.t1 | up | leaf | N |
| PC-3p-726_3835 | down | MSTRG.2715.1 | up | leaf | N |
| PC-5p-109182_51 | down | Bv4_078360_ftyu.t1 | up | leaf | N |
| PC-5p-36464_148 | down | Bv1_011830_oexd.t1 | up | leaf | N |
| PC-5p-3682_979 | down | Bv5_114160_hsdp.t1 | up | leaf | N |
| PC-5p-3682_979 | down | Bv8_193090_kary.t1 | up | leaf | N |
| PC-5p-49652_112 | down | MSTRG.29155.1 | up | leaf | N |
| csi-miR156a-5p_R+1_1ss9GT | up | Bv6_136190_cygi.t1 | down | root | Y |
| mtr-miR164d | up | Bv5_114390_pjnp.t1 | down | root | Y |
| PC-5p-7955_523 | up | Bv5_124210_skcr.t1 | down | root | Y |
| gma-miR6300_L-1R+1 | up | Bv5_097930_juac.t1 | down | root | N |
| gma-miR6300_L-1R+1 | up | Bv5_100510_ttjr.t1 | down | root | N |
| gma-miR6300_L-1R+1 | up | Bv9_208900_gijo.t1 | down | root | N |
| PC-5p-3682_979 | up | Bv1_012390_utfq.t1 | down | root | N |
| PC-5p-3682_979 | up | Bv1_017030_kxfa.t1 | down | root | N |
| PC-5p-3682_979 | up | Bv5_117790_mxei.t1 | down | root | N |
| PC-5p-3682_979 | up | Bv6_132150_dsqx.t1 | down | root | N |
| PC-5p-3682_979 | up | Bv6_135930_aphq.t1 | down | root | N |
| PC-5p-3682_979 | up | Bv8_193090_kary.t1 | down | root | N |
| ptc-miR399e | down | Bv5_121080_kswa.t1 | up | root | N |
| PC-5p-109182_51 | down | Bv5_110660_jxri.t1 | up | root | N |
| PC-5p-109182_51 | down | Bv9_213960_qddk.t1 | up | root | N |
| PC-5p-130970_42 | down | MSTRG.29564.3 | up | root | N |
| PC-5p-36464_148 | down | MSTRG.25036.1 | up | root | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Cui, J.; Dai, C.; Liu, T.; Cheng, D.; Luo, C. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of mRNAs, lncRNAs, miRNAs and circRNAs in Response to Salt Stress in Sugar Beet (Beta vulgaris). Int. J. Mol. Sci. 2021, 22, 289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010289
Li J, Cui J, Dai C, Liu T, Cheng D, Luo C. Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of mRNAs, lncRNAs, miRNAs and circRNAs in Response to Salt Stress in Sugar Beet (Beta vulgaris). International Journal of Molecular Sciences. 2021; 22(1):289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010289
Chicago/Turabian StyleLi, Junliang, Jie Cui, Cuihong Dai, Tianjiao Liu, Dayou Cheng, and Chengfei Luo. 2021. "Whole-Transcriptome RNA Sequencing Reveals the Global Molecular Responses and CeRNA Regulatory Network of mRNAs, lncRNAs, miRNAs and circRNAs in Response to Salt Stress in Sugar Beet (Beta vulgaris)" International Journal of Molecular Sciences 22, no. 1: 289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010289