Inflammation-Induced Protein Unfolding in Airway Smooth Muscle Triggers a Homeostatic Response in Mitochondria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
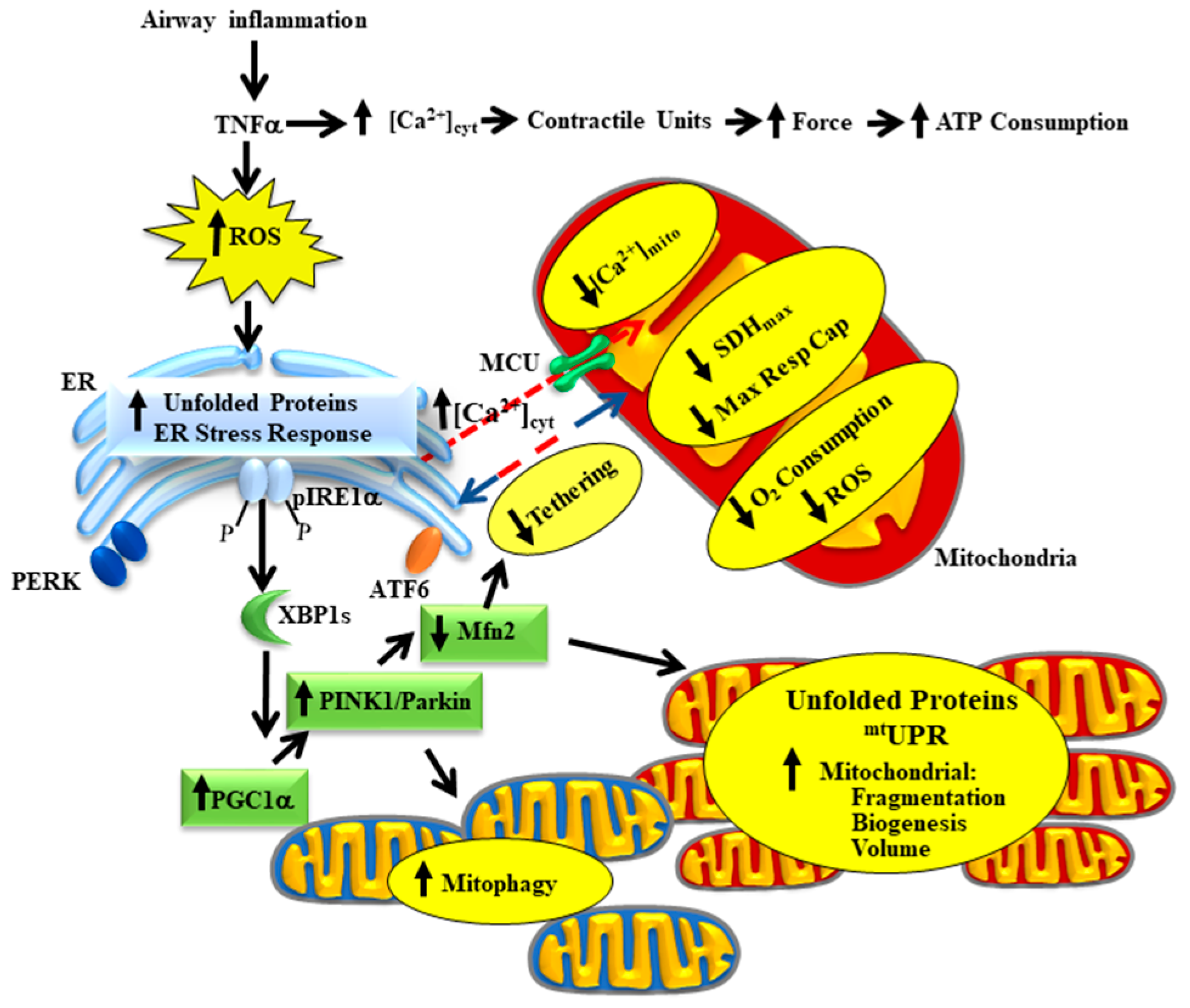
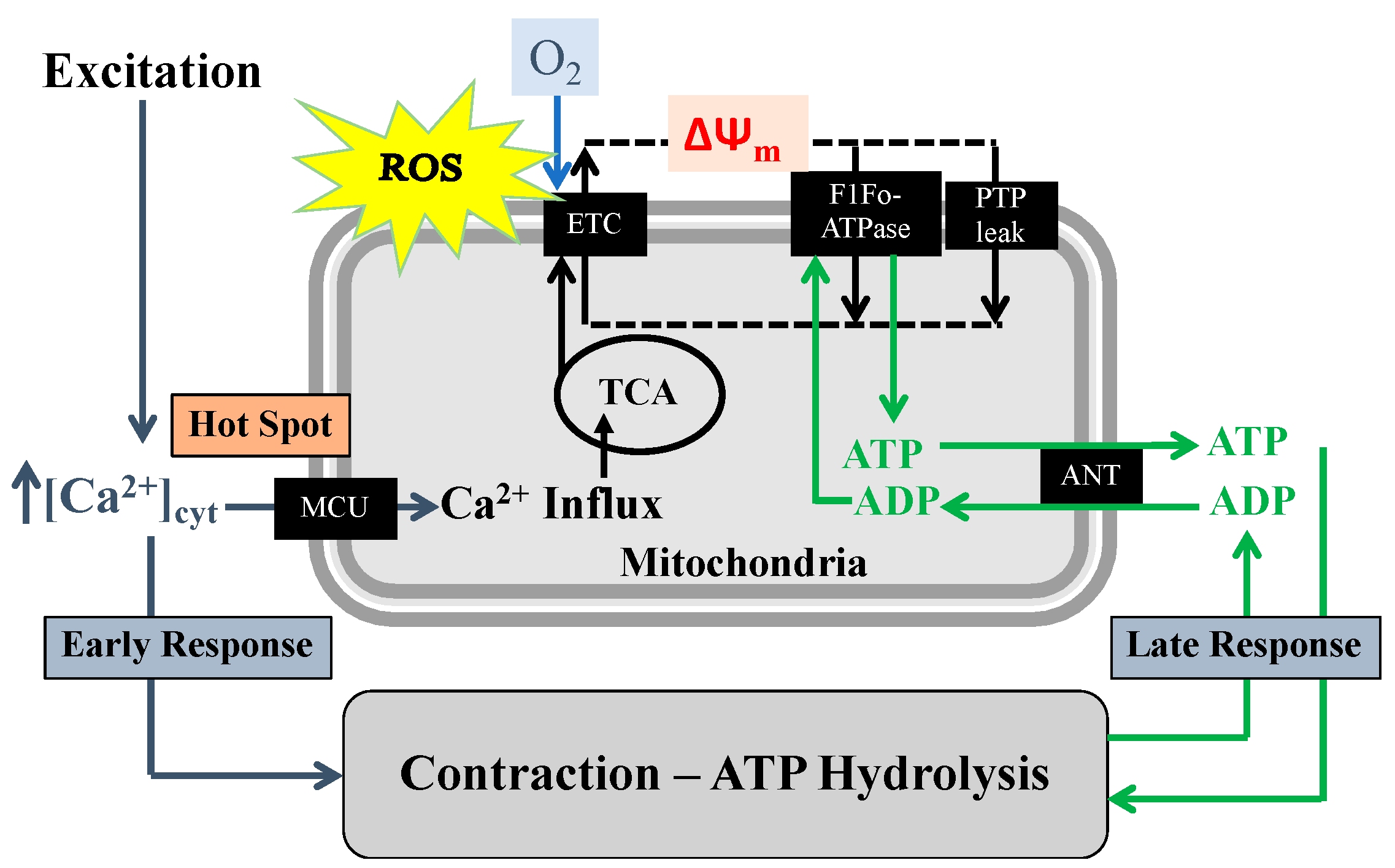
2. TNFα Increases ASM Force Generation, ATP Consumption and Tension Cost
3. TNFα Induced Oxidative Stress Triggers Protein Unfolding in the ER and Mitochondria
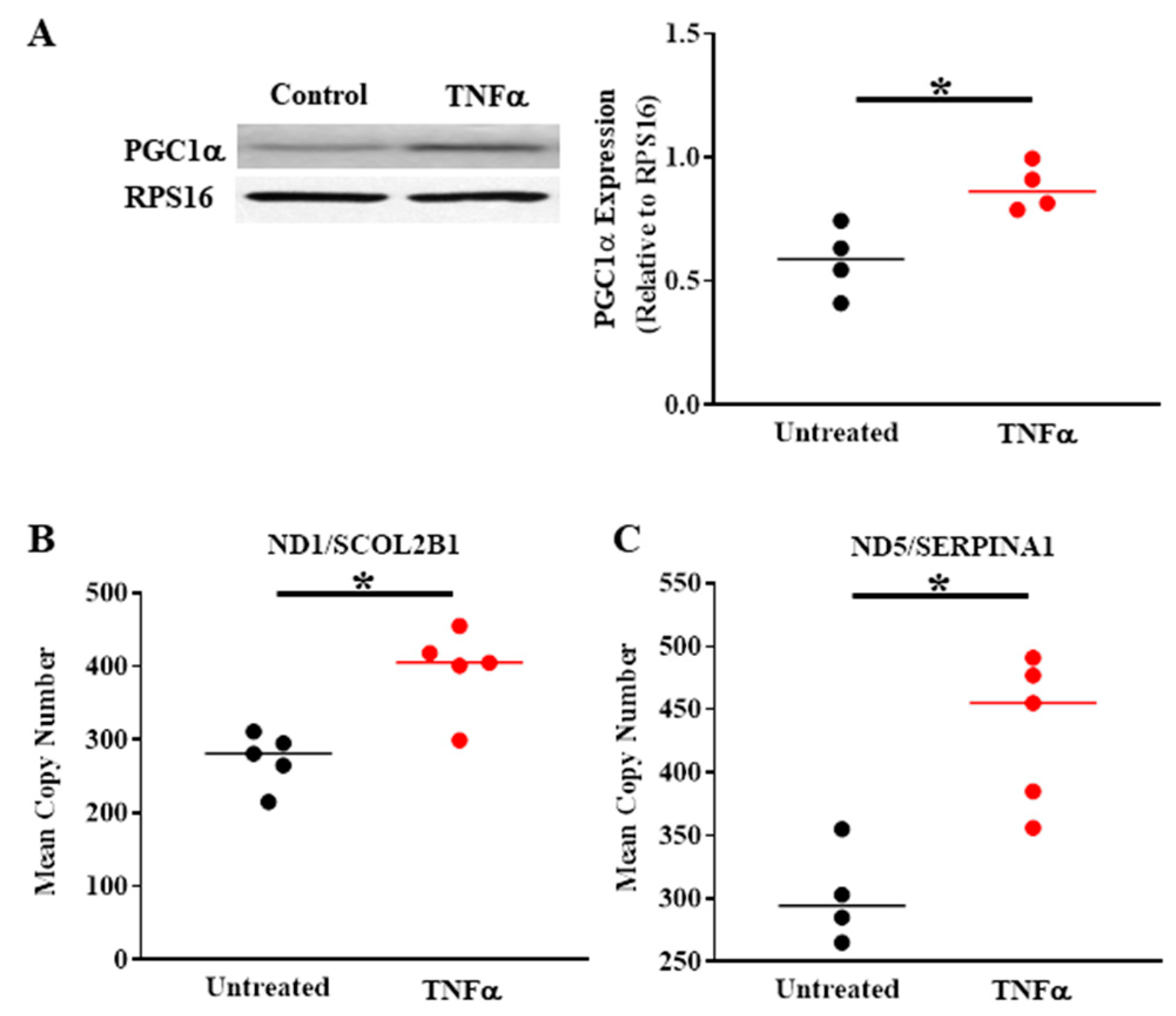
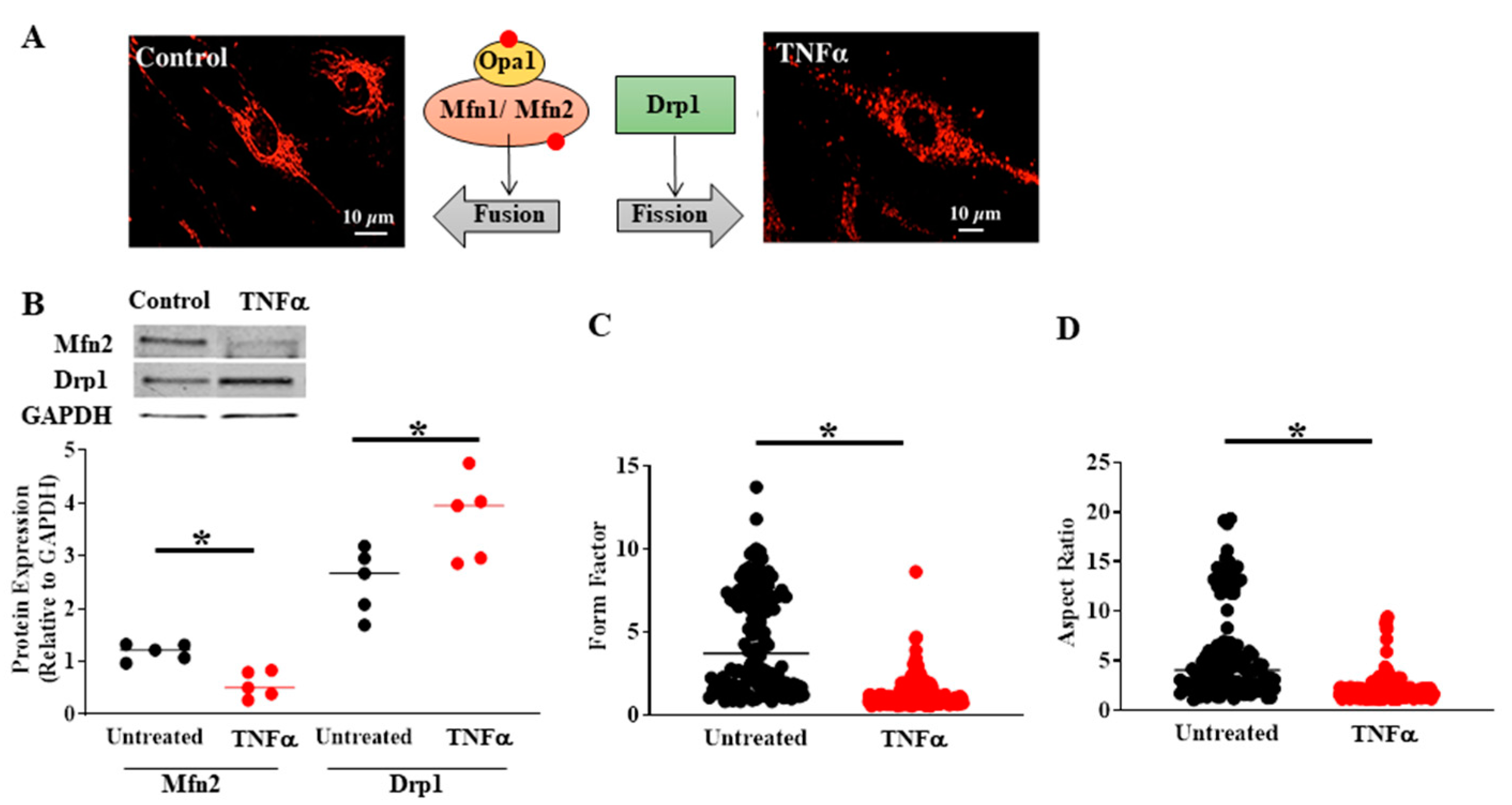
4. TNFα-Induced Activation of pIRE1α/XBP1s ER Stress Pathway Increases PGC1α Expression, Mediating Mitochondrial Fragmentation, and an Increase in Mitochondrial Biogenesis
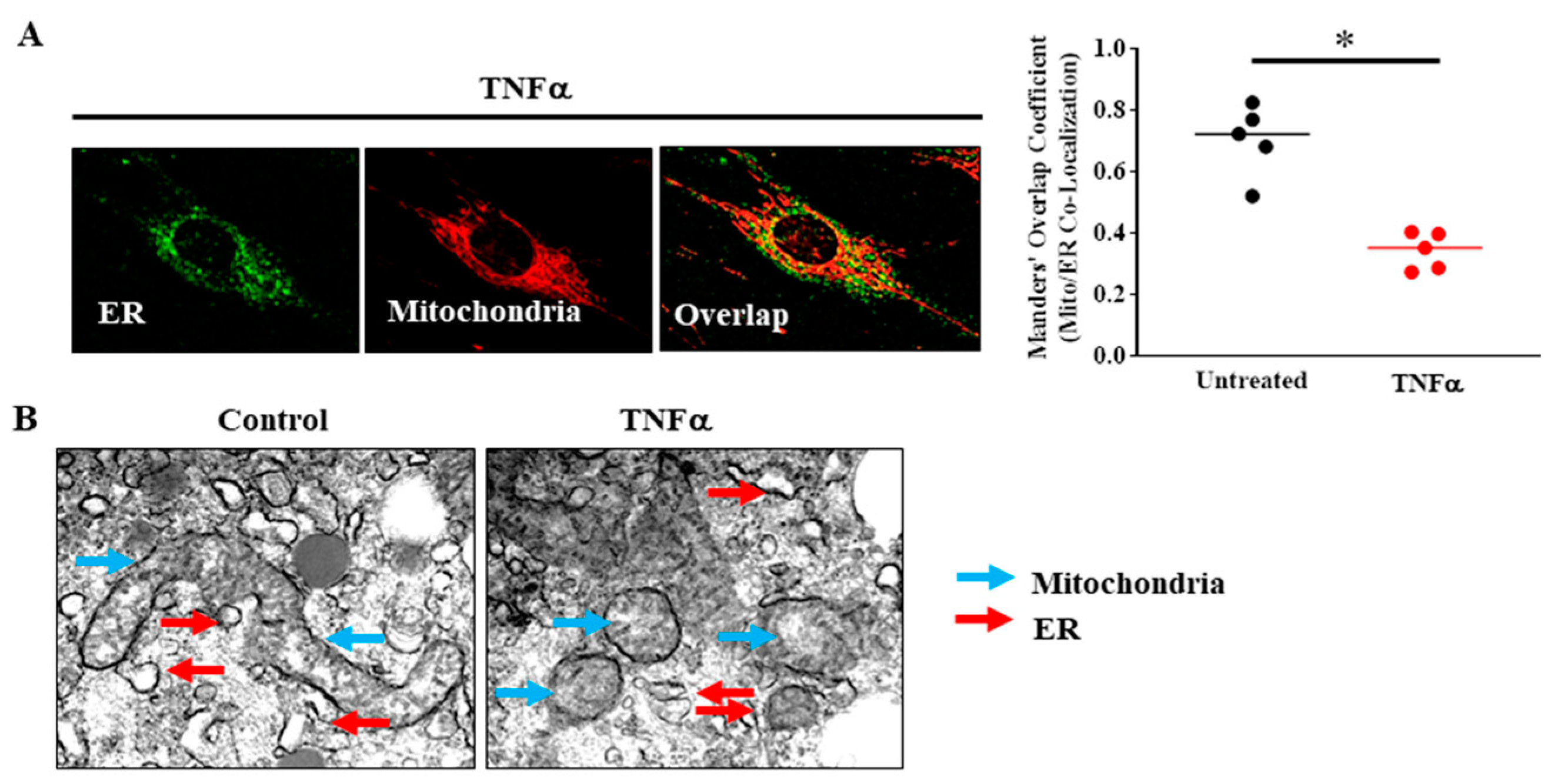
5. TNFα-Induced Reduction in Mfn2 Disrupts Mitochondrial Tethering to ER and Reduces Mitochondrial Ca2+ Influx
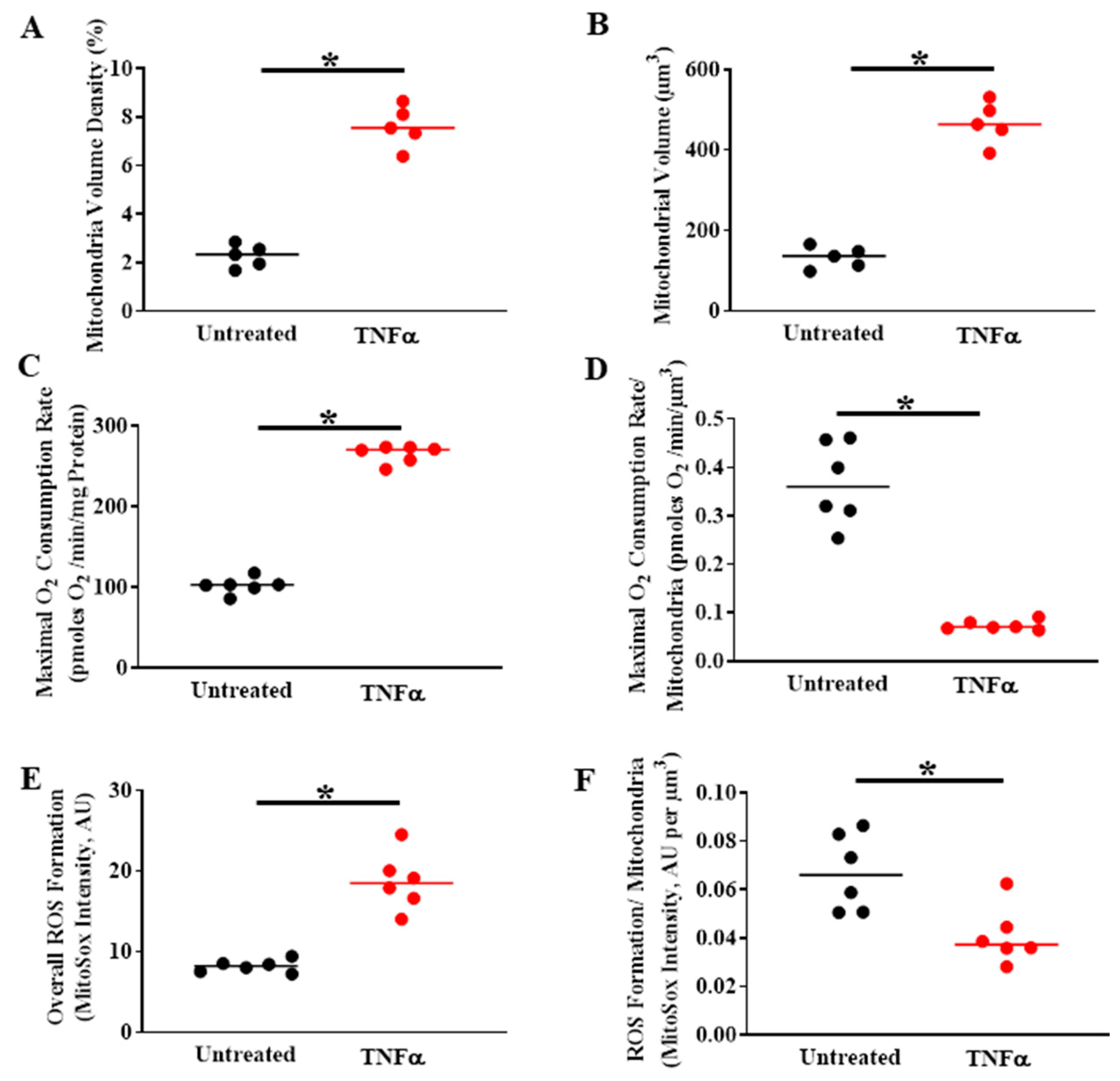
6. TNFα-Induced Mitochondrial Biogenesis and Increased Volume Density Increases O2 Consumption and ATP Production While Mitigating ROS Formation
7. The Homeostatic Response to TNFα in Asthmatic Human ASM
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| ASM | airway smooth muscle cells |
| ATF6 | activating transcription factor 6 |
| Drp1 | dynamin-related protein-1 |
| IRE1α | inositol-requiring enzyme 1 |
| MCU | mitochondrial Ca2+ uniporter |
| Mfn2 | mitofusin-2 |
| rMLC20 | regulatory myosin light chain |
| MLCK | myosin light chain kinase |
| mtUPR | mitochondrial unfolded protein response |
| MyHC | myosin heavy chain |
| PERK | protein kinase RNA-like ER kinase |
| PGC1α | peroxisome proliferator-activated receptor-gamma coactivator 1 alpha |
| PINK1 | PTEN-induced putative kinase 1 |
| ROS | reactive oxygen species |
| XBP1 | X-box protein 1 |
References
- Dogan, M.; Han, Y.S.; Delmotte, P.; Sieck, G.C. TNFalpha enhances force generation in airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L994–L1002. [Google Scholar] [CrossRef]
- Sieck, G.C.; Dogan, M.; Young-Soo, H.; Osorio Valencia, S.; Delmotte, P. Mechanisms underlying TNFalpha-induced enhancement of force generation in airway smooth muscle. Physiol. Rep. 2019, 7, e14220. [Google Scholar] [CrossRef] [Green Version]
- Yap, J.; Chen, X.; Delmotte, P.; Sieck, G.C. TNFα selectively activates the IRE1α/XBP1 endoplasmic reticulum stress pathway in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L483–l493. [Google Scholar] [CrossRef]
- Delmotte, P.F.; Marin Mathieu, N.; Sieck, G.C. TNFα Induces Mitochondrial Fragmentation and Biogenesis in Human Airway Smooth Muscle. Am. J. Physiol. Lung. Cell Mol. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Münch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [Green Version]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Delmotte, P.; Sieck, C.G. Interaction between endoplasmic/sarcoplasmic reticulum stress (ER/SR stress), mitochondrial signaling and Ca(2+) regulation in airway smooth muscle (ASM). Can. J. Physiol. Pharmacol. 2015, 93, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Delmotte, P.; Sieck, C.G. Endoplasmic reticulum stress and mitochondrial function in airway smooth muscle. Front. Cell Dev. Biol. 2020, 7, 374. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell. 2010, 38, 280–290. [Google Scholar] [CrossRef]
- Pallafacchina, G.; Zanin, S.; Rizzuto, R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Zavaletta, V.A.; Thompson, M.A.; Prakash, Y.S.; Sieck, G.C. TNFalpha decreases mitochondrial movement in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L166–L176. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.A.; Gerthoffer, W.T.; Trevethick, M.A.; Singer, H.A. Ca2+-dependent regulatory mechanisms in smooth muscle. Bibl. Cardiol. 1984, 38, 99–107. [Google Scholar]
- Sieck, G.C.; Gransee, H.M. Respiratory muscles structure, function & regulation. In Colloquium Series on Integrated Systems Physiology, from Molecule to Function to Disease; Morgan & Claypool: San Rafael, CA, USA, 2012; p. 87. [Google Scholar]
- Sieck, G.C.; Han, Y.S.; Prakash, Y.S.; Jones, K.A. Cross-bridge cycling kinetics, actomyosin ATPase activity and myosin heavy chain isoforms in skeletal and smooth respiratory muscles. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 119, 435–450. [Google Scholar] [CrossRef]
- Squire, J. The actin-myosin interaction in muscle: Background and overview. Int. J. Mol. Sci. 2019, 20, 5715. [Google Scholar] [CrossRef] [Green Version]
- DuRose, J.B.; Tam, A.B.; Niwa, M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol. Biol. Cell. 2006, 17, 3095–3107. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.R.; Lee, Y. Endoplasmic reticulum stress and the related signaling networks in severe asthma. Allergy Asthma Immunol. Res. 2015, 7, 106–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.S.; Kim, S.R.; Cho, S.H.; Lee, Y.C. Endoplasmic reticulum stress and allergic diseases. Curr. Allergy Asthma Rep. 2017, 17, 82. [Google Scholar] [CrossRef] [Green Version]
- Pathinayake, P.S.; Hsu, A.C.; Waters, D.W.; Hansbro, P.M.; Wood, L.G.; Wark, P.A.B. Understanding the unfolded protein response in the pathogenesis of asthma. Front. Immunol. 2018, 9, 175. [Google Scholar] [CrossRef] [Green Version]
- Siddesha, J.M.; Nakada, E.M.; Mihavics, B.R.; Hoffman, S.M.; Rattu, G.K.; Chamberlain, N.; Cahoon, J.M.; Lahue, K.G.; Daphtary, N.; Aliyeva, M.; et al. Effect of a chemical chaperone, tauroursodeoxycholic acid, on HDM-induced allergic airway disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1243–L1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.; Rosenthal, P.; Beppu, A.; Mueller, J.L.; Hoffman, H.M.; Tam, A.B.; Doherty, T.A.; McGeough, M.D.; Pena, C.A.; Suzukawa, M.; et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J. Immunol. 2014, 192, 3475–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.R.; Kim, D.I.; Kang, M.R.; Lee, K.S.; Park, S.Y.; Jeong, J.S.; Lee, Y.C. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating nuclear factor kappaB activation. J. Allergy Clin. Immunol. 2013, 132, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Makhija, L.; Krishnan, V.; Rehman, R.; Chakraborty, S.; Maity, S.; Mabalirajan, U.; Chakraborty, K.; Ghosh, B.; Agrawal, A. Chemical chaperones mitigate experimental asthma by attenuating endoplasmic reticulum stress. Am. J. Respir. Cell Mol. Biol. 2014, 50, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell. Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Al Baghdadi, R.J.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell. 2016, 27, 1536–1551. [Google Scholar] [CrossRef]
- Yung, H.W.; Colleoni, F.; Dommett, E.; Cindrova-Davies, T.; Kingdom, J.; Murray, A.J.; Burton, G.J. Noncanonical mitochondrial unfolded protein response impairs placental oxidative phosphorylation in early-onset preeclampsia. Proc. Natl. Acad. Sci. USA 2019, 116, 18109–18118. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef] [Green Version]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007, 12, 815–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.M.; Murphy, E. Role of mitochondrial calcium and the permeability transition pore in regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Corona, J.C.; Duchen, M.R. PPARγ and PGC-1α as therapeutic targets in Parkinson’s. Neurochem. Res. 2015, 40, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol. Ther. 2020, 107705. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Arensdorf, A.M.; Diedrichs, D.; Rutkowski, D.T. Regulation of the transcriptome by ER stress: Non-canonical mechanisms and physiological consequences. Front. Genet. 2013, 4, 256. [Google Scholar] [CrossRef] [Green Version]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leites, E.P.; Morais, V.A. Mitochondrial quality control pathways: PINK1 acts as a gatekeeper. Biochem. Biophys. Res. Commun. 2018, 500, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2018, 93, 933–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, G.W.; Kitsis, R.N. The mitochondrial dynamism-mitophagy-cell death interactome: Multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 2015, 116, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, P.; Song, M.; Dorn, G.W. Dissociation of mitochondrial from sarcoplasmic reticular stress in Drosophila cardiomyopathy induced by molecularly distinct mitochondrial fusion defects. J. Mol. Cell Cardiol. 2015, 80, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [Green Version]
- Ngoh, G.A.; Papanicolaou, K.N.; Walsh, K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 20321–20332. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium. 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C. ER-mitochondria communication. How privileged? Physiology 2007, 22, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maack, C.; O’Rourke, B. Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 2007, 102, 369–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca(2+) Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef] [PubMed]
- Vallese, F.; Barazzuol, L.; Maso, L.; Brini, M.; Calì, T. ER-Mitochondria calcium transfer, organelle contacts and neurodegenerative diseases. Adv. Exp. Med. Biol. 2020, 1131, 719–746. [Google Scholar]
- Fan, Y.; Simmen, T. Mechanistic connections between endoplasmic reticulum (ER) redox control and mitochondrial metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.; Walker, J.E.; Saheki, T.; Satrústegui, J.; et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Punzi, G.; Obre, E.; Lacombe, D.; De Grassi, A.; Pierri, C.L.; Rossignol, R. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim. Biophys. Acta 2016, 1863, 2394–2412. [Google Scholar] [CrossRef]
- Rueda, C.B.; Llorente-Folch, I.; Traba, J.; Amigo, I.; Gonzalez-Sanchez, P.; Contreras, L.; Juaristi, I.; Martinez-Valero, P.; Pardo, B.; Del Arco, A.; et al. Glutamate excitotoxicity and Ca2+-regulation of respiration: Role of the Ca2+ activated mitochondrial transporters (CaMCs). Biochim. Biophys. Acta 2016, 1857, 1158–1166. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R. Structural changes in the transport cycle of the mitochondrial ADP/ATP carrier. Curr. Opin. Struct. Biol. 2019, 57, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Schaible, N.; Delmotte, P.; Sieck, G.C. Mitochondrial Excitation-Energy Coupling in Airway Smooth Muscle. In Mitochondrial Function in Lung Health and Disease; Natarajan, V., Parinandi, N.L., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2014; pp. 93–116. [Google Scholar]
- Delmotte, P.; Yang, B.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S.; Sieck, G.C. Inflammation alters regional mitochondrial calcium in human airway smooth muscle cells. Am. J. Physiol. Cell Physiol. 2012, 303, C244–C256. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Min, K.T. The interface between ER and mitochondria: Molecular compositions and functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar] [PubMed]
- Facts about chickenpox. Paediatr. Child. Health 2005, 10, 413–414. [CrossRef] [PubMed] [Green Version]
- Girodet, P.O.; Ozier, A.; Bara, I.; Tunon de Lara, J.M.; Marthan, R.; Berger, P. Airway remodeling in asthma: New mechanisms and potential for pharmacological intervention. Pharmacol. Ther. 2011, 130, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Trian, T.; Benard, G.; Begueret, H.; Rossignol, R.; Girodet, P.O.; Ghosh, D.; Ousova, O.; Vernejoux, J.M.; Marthan, R.; Tunon-de-Lara, J.M.; et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J. Exp. Med. 2007, 204, 3173–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athari, S.S. Targeting cell signaling in allergic asthma. Signal. Transduct. Target. Ther. 2019, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Alashkar Alhamwe, B.; Miethe, S.; Pogge von Strandmann, E.; Potaczek, D.P.; Garn, H. Epigenetic regulation of airway epithelium immune functions in asthma. Front. Immunol. 2020, 11, 1747. [Google Scholar] [CrossRef]
- Rosanna, D.P.; Salvatore, C. Reactive oxygen species, inflammation, and lung diseases. Curr. Pharm. Des. 2012, 18, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Hough, K.P.; Curtiss, M.L.; Blain, T.J.; Liu, R.M.; Trevor, J.; Deshane, J.S.; Thannickal, V.J. Airway remodeling in asthma. Front. Med. 2020, 7, 191. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, L. Status and prospects: Personalized treatment and biomarker for airway remodeling in asthma. J. Thorac. Dis. 2020, 12, 6090–6101. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, K.S.; Lee, S.J.; Kim, S.R.; Park, S.Y.; Jeon, M.S.; Lee, H.B.; Lee, Y.C. L-2-Oxothiazolidine-4-carboxylic acid or α-lipoic acid attenuates airway remodeling: Involvement of nuclear factor-κB (NF-κB), nuclear factor erythroid 2p45-related factor-2 (Nrf2), and hypoxia-inducible factor (HIF). Int. J. Mol. Sci. 2012, 13, 7915–7937. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Otenbaker, N.P.; Rose, B.A.; Salisbury, K.S. Molecular mechanisms of reactive oxygen species-related pulmonary inflammation and asthma. Mol. Immunol. 2013, 56, 57–63. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dasgupta, D.; Delmotte, P.; Sieck, G.C. Inflammation-Induced Protein Unfolding in Airway Smooth Muscle Triggers a Homeostatic Response in Mitochondria. Int. J. Mol. Sci. 2021, 22, 363. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010363
Dasgupta D, Delmotte P, Sieck GC. Inflammation-Induced Protein Unfolding in Airway Smooth Muscle Triggers a Homeostatic Response in Mitochondria. International Journal of Molecular Sciences. 2021; 22(1):363. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010363
Chicago/Turabian StyleDasgupta, Debanjali, Philippe Delmotte, and Gary C. Sieck. 2021. "Inflammation-Induced Protein Unfolding in Airway Smooth Muscle Triggers a Homeostatic Response in Mitochondria" International Journal of Molecular Sciences 22, no. 1: 363. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010363