Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
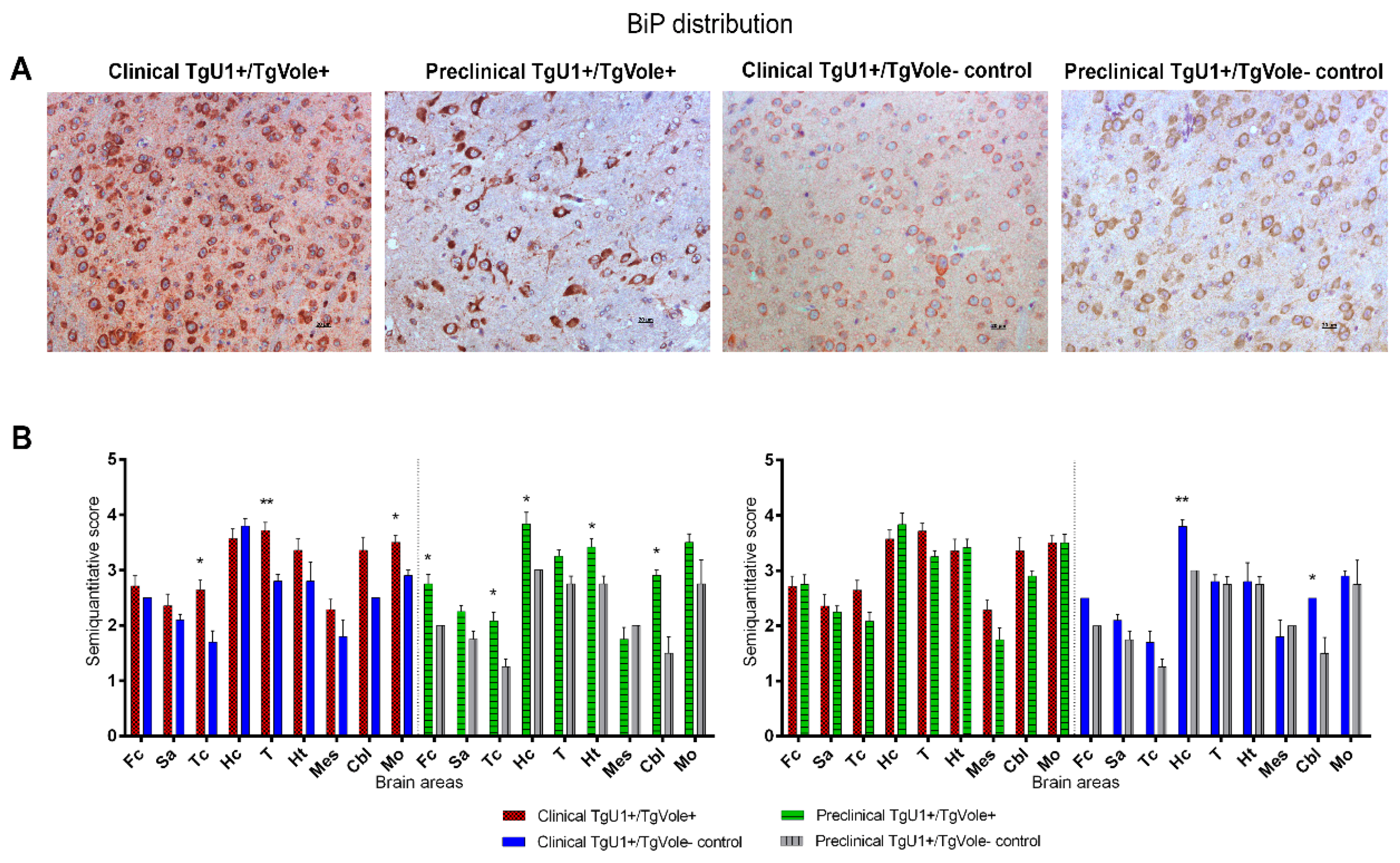
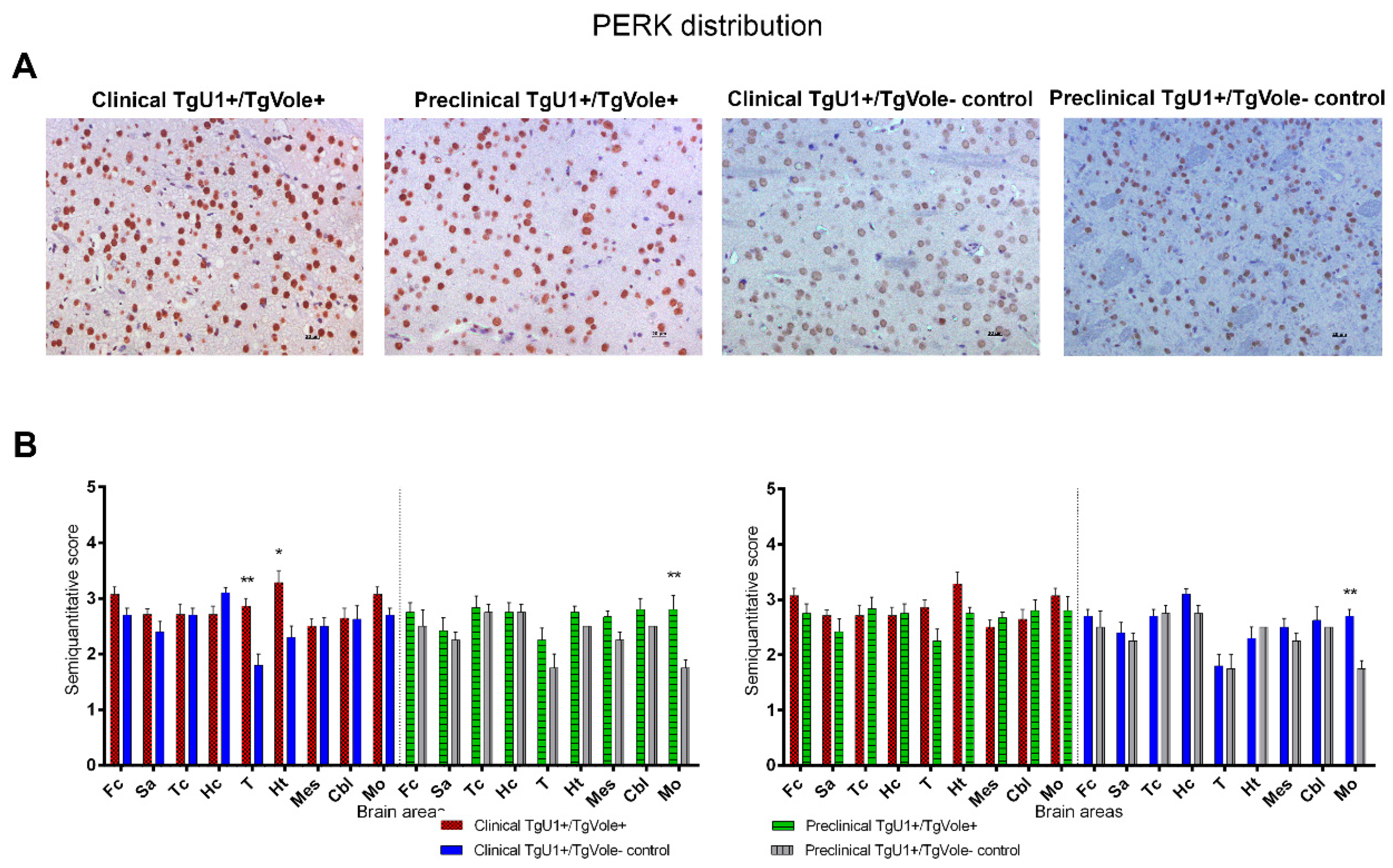
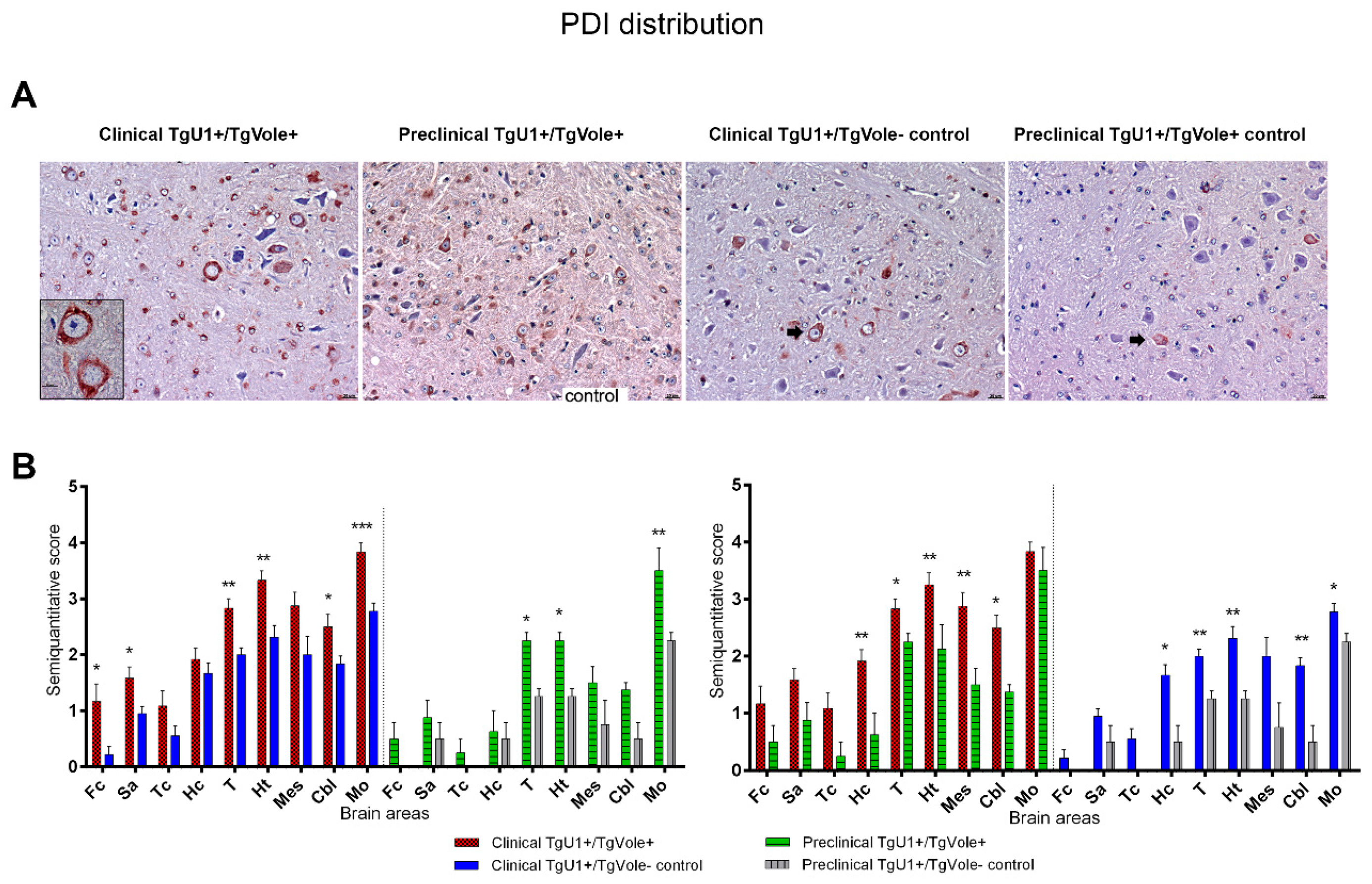
2.1. Mice Affected by the Spontaneous Prion Disease Show Increased Accumulation of ER Stress Markers in Certain Brain Areas
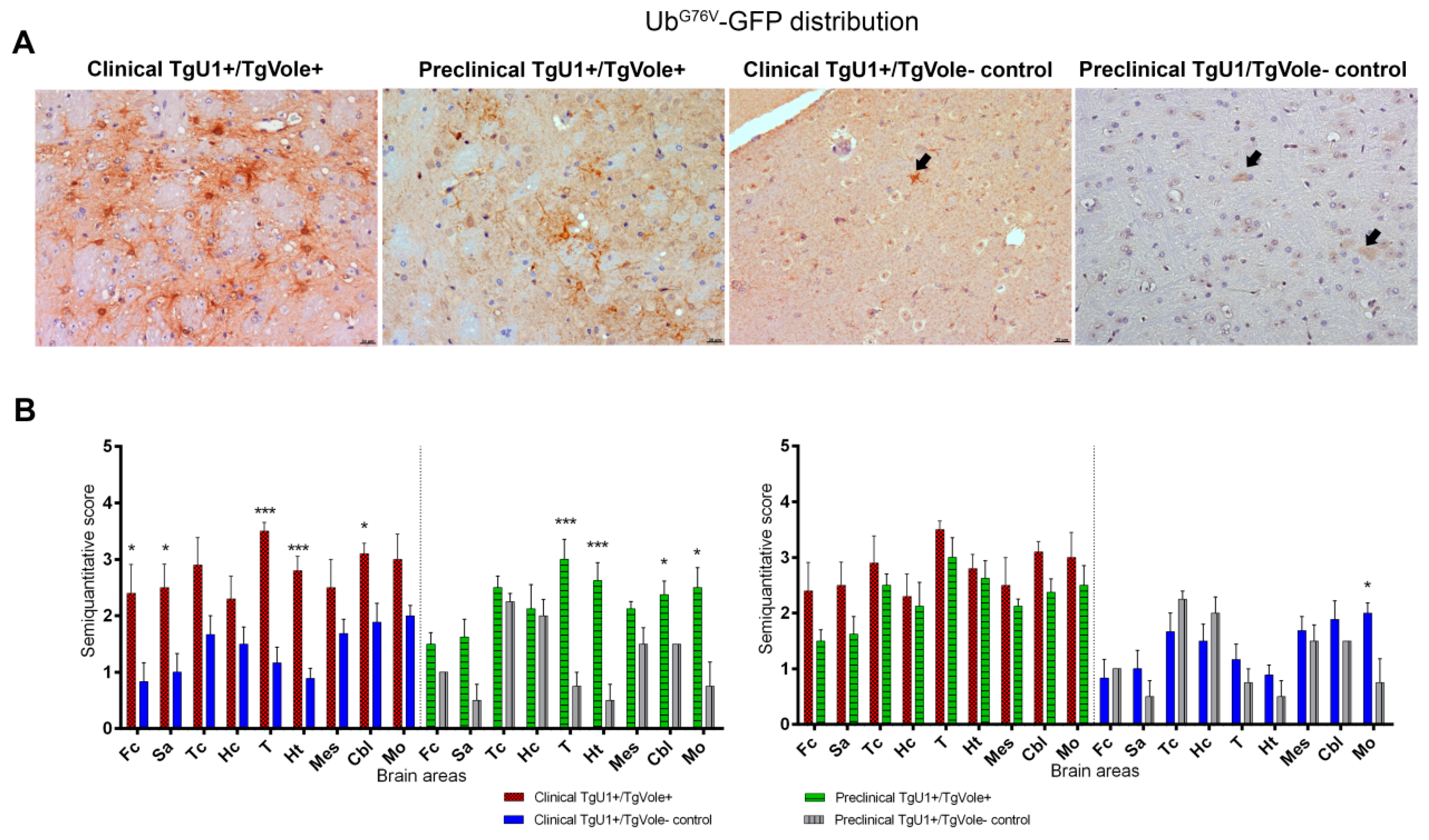
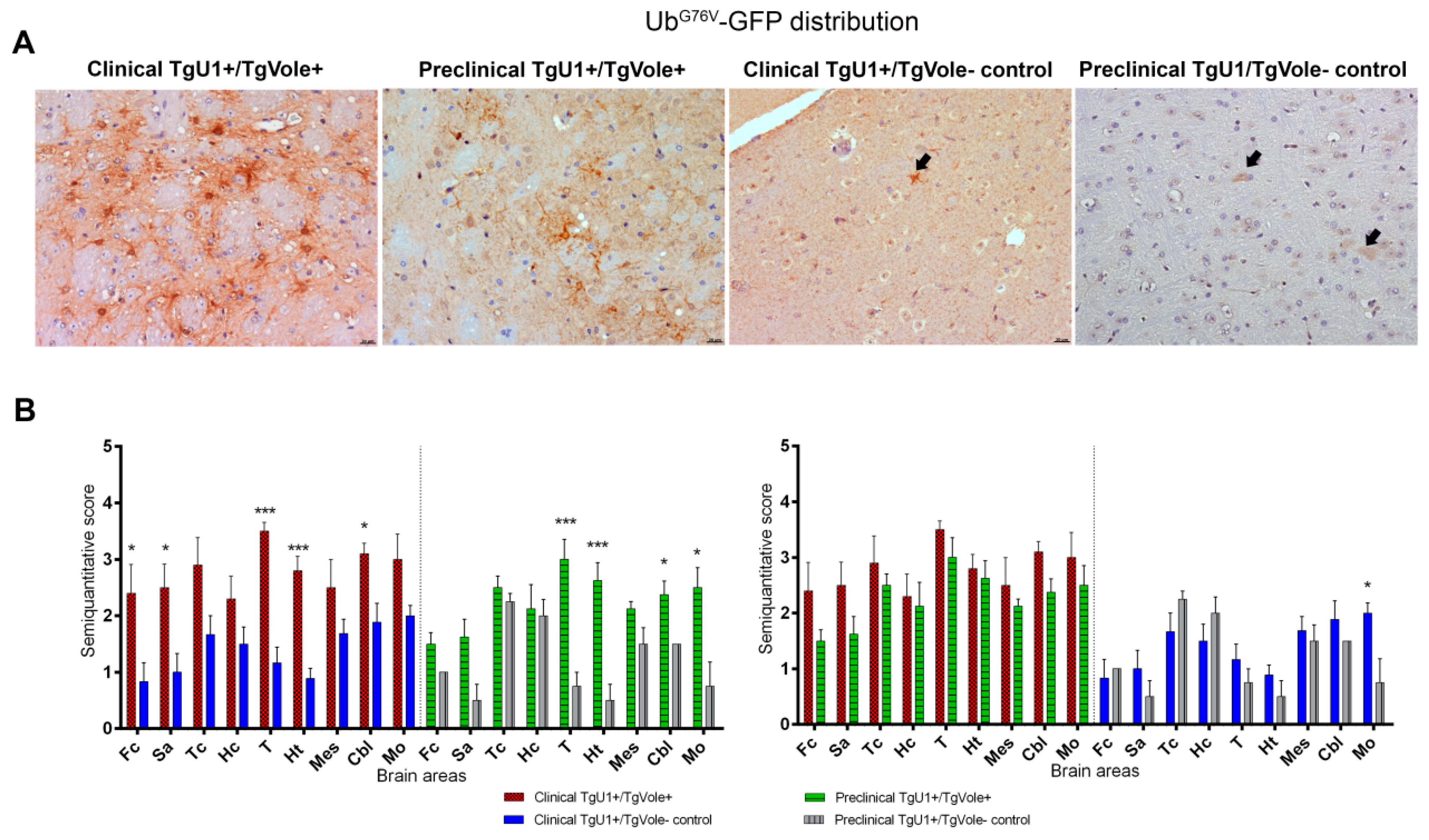
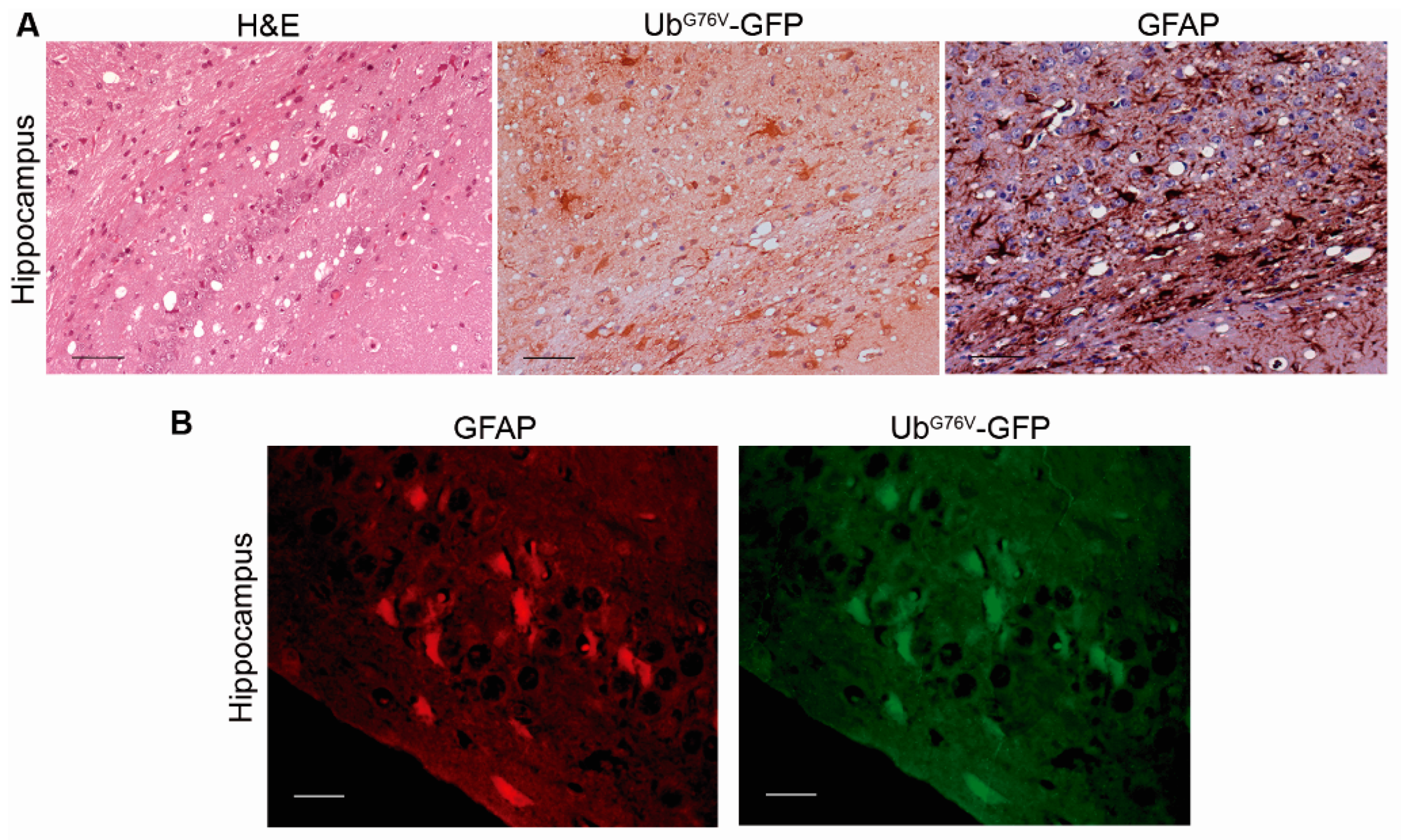
2.2. Mice Affected by the Spontaneous Prion Disease Show UbG76V-GFP accumulation in Brain Areas with Prion-Associated Neuropathology
2.3. Correlation between BiP, PERK, PDI and UbG76V-GFP Proteins and the Histopathological Features of the Disease
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Histological and Immunohistochemical Analyses
4.3. Immunofluorescence Staining
4.4. Data Analysis
4.5. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF6 | Activating transcription factor 6 |
| BiP/Grp78 | Binding immunoglobulin protein |
| CBL | Cerebellum |
| CNS | Central Nervous System |
| DAB | Diaminobenzidine |
| ER | Endoplasmic Reticulum |
| ERAD | Endoplasmic Reticulum Associated Degradation |
| FC | Frontal Cortex |
| GFAP | Glial fibrillary acidic protein |
| Hc | Hippocampus |
| Ht | Hypothalamus |
| IRE1 | Inositol-requiring enzyme 1 |
| Mes | Mesencephalon |
| Mo | Medulla oblongata |
| PERK | PKR-like endoplasmic reticulum kinase |
| PDI | Protein disulfide isomerase |
| PrPSc | Pathological prion protein |
| PrPC | Cellular prion protein |
| Sa | Septal Area |
| PSP | Progressive Supranuclear Palsy |
| sCJD | Sporadic Creutzfeldt-Jakob Disease |
| T | Thalamus |
| TC | Cortex at the level of the thalamus |
| Tg | Transgenic |
| TSE | Transmissible Spongiform Encephalopathies |
| Ub | Ubiquitin |
| UPR | Unfolded Protein Response |
| UPS | Ubiquitin-Proteasome System |
| vCJD | Variant Creutzfeldt-Jakob Disease |
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collinge, J. Prion Diseases of Humans and Animals: Their Causes and Molecular Basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Glimcher, L.H. Fine-Tuning of the Unfolded Protein Response: Assembling the IRE1α Interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.; Castillo, K.; Armisén, R.; Stutzin, A.; Soto, C.; Hetz, C. Prion Protein Misfolding Affects Calcium Homeostasis and Sensitizes Cells to Endoplasmic Reticulum Stress. PLoS ONE 2010, 5, e15658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-B.; Shi, Q.; Xu, Y.; Xie, W.-L.; Zhang, J.; Tian, C.; Guo, Y.; Wang, K.; Zhang, B.-Y.; Chen, C.; et al. Protein Disulfide Isomerase Regulates Endoplasmic Reticulum Stress and the Apoptotic Process during Prion Infection and PrP Mutant-Induced Cytotoxicity. PLoS ONE 2012, 7, e38221. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M.U. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. eLife 2015, 4, e03522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nat. Cell Biol. 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Lewy, T.G.; Grabowski, J.M.; Bloom, M.E. BiP: Master Regulator of the Unfolded Protein Response and Crucial Factor in Flavivirus Biology. Yale J. Boil. Med. 2017, 90, 291–300. [Google Scholar]
- Hampton, R.Y. ER stress response: Getting the UPR hand on misfolded proteins. Curr. Biol. 2000, 10, R518–R521. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Kaufman, R.J. The unfolded protein response: A stress signaling pathway critical for health and disease. Neu-Rology 2005, 66 (Suppl. 1), S102–S109. [Google Scholar] [CrossRef]
- Hetz, C.; Russelakis-Carneiro, M.; Wälchli, S.; Carboni, S.; Vial-Knecht, E.; Maundrell, K.; Castilla, J.; Soto, C. The Disulfide Isomerase Grp58 Is a Protective Factor against Prion Neurotoxicity. J. Neurosci. 2005, 25, 2793–2802. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.C.; Krapfenbauer, K.; Cairns, N.; Belay, G.; Bajo, M.; Lubec, G. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt–Jakob disease. Neurosci. Lett. 2002, 334, 196–200. [Google Scholar] [CrossRef]
- Tang, Y.; Xiang, W.; Terry, L.; Kretzschmar, H.A.; Windl, O. Transcriptional Analysis Implicates Endoplasmic Reticulum Stress in Bovine Spongiform Encephalopathy. PLoS ONE 2010, 5, e14207. [Google Scholar] [CrossRef] [Green Version]
- Unterberger, U.; Höftberger, R.; Gelpi, E.; Flicker, H.; Budka, H.; Voigtländer, T. Endoplasmic Reticulum Stress Features Are Prominent in Alzheimer Disease but Not in Prion Diseases In Vivo. J. Neuropathol. Exp. Neurol. 2006, 65, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Lyles, M.M.; Gilbert, H.F. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: Depend-ence of the rate on the composition of the redox buffer. Biochemistry 1991, 30, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.B.; Hirst, T.R.; Tuite, M.F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci. 1994, 19, 331–336. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Wang, L. Protein disulfide–isomerase, a folding catalyst and a redox-regulated chaperone. Free. Radic. Biol. Med. 2015, 83, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Batten, M.R.; Lumb, R.A.; Bulleid, N.J. Quality control in the endoplasmic reticulum: PDI mediates the ER retention of unassembled procollagen C-propeptides. Curr. Biol. 2001, 11, 1114–1118. [Google Scholar] [CrossRef]
- Wang, C.C. Isomerase and chaperone activities of protein disulfide isomerase are both required for its function as a foldase. Biochem. Biokhimiia 1998, 63, 407–412. [Google Scholar]
- Wilson, R.; Lees, J.F.; Bulleid, N.J. Protein disulfide isomerase acts as a molecular chaperone during the assembly of pro-collagen. J. Biol. Chem. 1998, 273, 9637–9643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.; Medinas, D.B.; Matamala, J.M.; Woehlbier, U.; Cornejo, V.H.; Soldà, T.; Andreu, C.; Rozas, P.; Matus, S.; Muñoz, N.; et al. The Protein-disulfide Isomerase ERp57 Regulates the Steady-state Levels of the Prion Protein. J. Biol. Chem. 2015, 290, 23631–23645. [Google Scholar] [CrossRef] [Green Version]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Ironside, J.W.; McCardle, L.; Hayward, P.A.; Bell, J.E. Ubiquitin immunocytochemistry in human spongiform encephalopa-thies. Neuropathol. Appl. Neurobiol. 1993, 19, 134–140. [Google Scholar] [CrossRef]
- Suenaga, T.; Hirano, A.; Llena, J.F.; Ksiezak-Reding, H.; Yen, S.-H.; Dickson, D.W. Ubiquitin immunoreactivity in kuru plaques in Creutzfeldt-Jakob disease. Ann. Neurol. 1990, 28, 174–177. [Google Scholar] [CrossRef]
- Kang, S.-C.; Brown, D.R.; Whiteman, M.; Li, R.; Pan, T.; Perry, G.; Wisniewski, T.; Sy, M.-S.; Wong, B.-S. Prion protein is ubiquitinated after developing protease resistance in the brains of scrapie-infected mice. J. Pathol. 2004, 203, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; Van Leeuwen, F.W.; Menéndez-Benito, V.; Dantuma, N.P.; et al. Disease-Associated Prion Protein Oligomers Inhibit the 26S Proteasome. Mol. Cell 2007, 26, 175–188. [Google Scholar] [CrossRef]
- McKinnon, C.; Goold, R.G.; Andre, R.; Devoy, A.; Ortega, Z.; Moonga, J.; Linehan, J.M.; Brandner, S.; Lucas, J.J.; Collinge, J.; et al. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin–proteasome system. Acta Neuropathol. 2016, 131, 411–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsi, A.; Fioriti, L.; Chiesa, R.; Sitia, R. Conditions of Endoplasmic Reticulum Stress Favor the Accumulation of Cytosolic Prion Protein. J. Biol. Chem. 2006, 281, 30431–30438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.-W.; Rane, N.S.; Kim, S.J.; Garrison, J.L.; Taunton, J.; Hegde, R.S. Substrate-Specific Translocational Attenuation during ER Stress Defines a Pre-Emptive Quality Control Pathway. Cell 2006, 127, 999–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez-Benito, V.; Verhoef, L.G.; Masucci, M.G.; Dantuma, N.P. Endoplasmic reticulum stress compromises the ubiquitin–proteasome system. Hum. Mol. Genet. 2005, 14, 2787–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunziante, M.; Ackermann, K.; Dietrich, K.; Wolf, H.; Gädtke, L.; Gilch, S.; Vorberg, I.; Groschup, M.; Schatzl, H.M. Proteasomal Dysfunction and Endoplasmic Reticulum Stress Enhance Trafficking of Prion Protein Aggregates through the Secretory Pathway and Increase Accumulation of Pathologic Prion Protein. J. Biol. Chem. 2011, 286, 33942–33953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglio, E.; Restelli, E.; Garofoli, A.; Dossena, S.; De Luigi, A.; Tagliavacca, L.; Imperiale, D.; Migheli, A.; Salmona, M.; Sitia, R.; et al. Expression of Mutant or Cytosolic PrP in Transgenic Mice and Cells Is Not Associated with Endoplasmic Reticulum Stress or Proteasome Dysfunction. PLoS ONE 2011, 6, e19339. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.C.; Giles, K.; Stöhr, J.; Oehler, A.; Bhardwaj, S.; Grillo, S.K.; Patel, S.; DeArmond, S.J.; Prusiner, S.B. Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc. Natl. Acad. Sci. USA 2012, 109, 3498–3503. [Google Scholar] [CrossRef] [Green Version]
- Otero, A.; Hedman, C.; Fernández-Borges, N.; Eraña, H.; Marín, B.; Monzón, M.; Sánchez-Martín, M.A.; Nonno, R.; Badiola, J.J.; Bolea, R.; et al. A Single Amino Acid Substitution, Found in Mammals with Low Susceptibility to Prion Diseases, Delays Propagation of Two Prion Strains in Highly Susceptible Transgenic Mouse Models. Mol. Neurobiol. 2019, 56, 6501–6511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsten, K.; Menendez-Benito, V.; Masucci, M.G.; Dantuma, N.P. A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 2003, 21, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.E.; De Sousa, J.R.; Silva, L.M.; Junior, L.B.D.; Furlaneto, I.P.; Carneiro, F.R.O.; Aarão, T.L.D.S.; Sotto, M.N.; Quaresma, J.A.S. Endoplasmic Reticulum Stress Markers and Their Possible Implications in Leprosy’s Pathogenesis. Dis. Markers 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Elkharaz, J.; Ugun-Klusek, A.; Constantin-Teodosiu, D.; Lawler, K.; Mayer, R.J.; Billett, E.; Lowe, J.; Bedford, L. Implications for oxidative stress and astrocytes following 26S proteasomal depletion in mouse forebrain neurones. Biochim. Biophys. Acta 2013, 1832, 1930–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [Green Version]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Wu, J.; Back, S.-H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR Pathways Combine to Prevent Hepatic Steatosis Caused by ER Stress-Mediated Suppression of Transcriptional Master Regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, J.L.; McCracken, A.A. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 1999, 10, 507–513. [Google Scholar] [CrossRef]
- Bell, M.C.; Meier, S.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the Ubiquitin-Proteasome System by Protein Aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Andre, R.; Tabrizi, S.J. Misfolded PrP and a novel mechanism of proteasome inhibition. Prion 2012, 6, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, Q.; Xu, K.; Gao, C.; Chen, C.; Li, X.-L.; Wang, G.-R.; Tian, C.; Han, J.; Dong, X.-P. Familial CJD Associated PrP Mutants within Transmembrane Region Induced Ctm-PrP Retention in ER and Triggered Apoptosis by ER Stress in SH-SY5Y Cells. PLoS ONE 2011, 6, e14602. [Google Scholar] [CrossRef] [PubMed]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The Unfolded Protein Response and the Role of Protein Disulfide Isomerase in Neurodegeneration. Front. Cell Dev. Biol. 2016, 3, 80. [Google Scholar] [CrossRef] [Green Version]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Eikelenboom, P.; De Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Eikelenboom, P.; Scheper, W. The Unfolded Protein Response Is Activated in Pretangle Neurons in Alzheimer’s Disease Hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Hoozemans, J.J.M.; Van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Scheper, W. Activation of the Unfolded Protein Response Is an Early Event in Alzheimer’s and Parkinson’s Disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Nijholt, D.A.T.; Van Haastert, E.S.; Rozemuller, A.J.M.; Scheper, W.; Hoozemans, J.J.M. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 2012, 226, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.L.; Gilman, S.; Lee, V.M.-Y.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D.; PSP Genetics Study Group. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.P.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nat. Cell Biol. 2012, 485, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, R.; Ryten, M.; Simone, R.; Trabzuni, D.; Nicolaou, N.; Hondhamuni, G.; Ramasamy, A.; Vandrovcova, J.; Weale, M.E.; Lees, A.J.; et al. Assessment of common variability and expression quantitative trait loci for genome-wide associations for progressive supranuclear palsy. Neurobiol. Aging 2014, 35, 1514.e1–1514.e12. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, V.I.; Van Hecke, W.; Scheper, W.; Van Osch, M.A.J.; Hermsen, W.J.M.; Rozemuller, A.J.M.; Hoozemans, J.J.M. Activation of the unfolded protein response and granulovacuolar degeneration are not common features of human prion pathology. Acta Neuropathol. Commun. 2016, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Eroller, C.; Emaddalo, D. The Molecular Chaperone GRP78/BiP in the Development of Chemoresistance: Mechanism and Possible Treatment. Front. Pharmacol. 2013, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-W.; Kim, G.E.; Morales, R.; Moda, F.; Moreno-Gonzalez, I.; Concha-Marambio, L.; Lee, A.S.; Hetz, C.; Soto, C. The Endoplasmic Reticulum Chaperone GRP78/BiP Modulates Prion Propagation in vitro and in vivo. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Azzu, V.; Valencak, T.G. Energy Metabolism and Ageing in the Mouse: A Mini-Review. Gerontology 2017, 63, 327–336. [Google Scholar] [CrossRef]
- Hetz, C.; Lee, A.-H.; Gonzalez-Romero, D.; Thielen, P.; Castilla, J.; Soto, C.; Glimcher, L.H. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Steele, A.D.; Hetz, C.; Yi, C.H.; Jackson, W.S.; Borkowski, A.W.; Yuan, J.; Wollmann, R.H.; Lindquist, S. Prion Pathogenesis is Independent of Caspase-12. Prion 2007, 1, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middeldorp, J.; Kamphuis, W.; Sluijs, J.A.; Achoui, D.; Leenaars, C.H.C.; Feenstra, M.G.P.; Van Tijn, P.; Fischer, D.F.; Berkers, C.; Ovaa, H.; et al. Intermediate filament transcription in astrocytes is repressed by proteasome inhibition. FASEB J. 2009, 23, 2710–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldbaum, O.; Riedel, M.; Stahnke, T.; Richter-Landsberg, C. The small heat shock protein HSP25 protects astrocytes against stress induced by proteasomal inhibition. Glia 2009, 57, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Titler, A.M.; Posimo, J.M.; Leak, R.K. Astrocyte plasticity revealed by adaptations to severe proteotoxic stress. Cell Tissue Res. 2013, 352, 427–443. [Google Scholar] [CrossRef]
- Jansen, A.H.P.; Reits, E.A.; Hol, E.M. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front. Mol. Neurosci. 2014, 7, 73. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, J.; Xu, Y.; Ren, K.; Xie, W.-L.; Yan, Y.-E.; Zhang, B.-Y.; Shi, Q.; Liu, Y.; Dong, X.-P. Abnormally Upregulated αB-crystallin Was Highly Coincidental with the Astrogliosis in the Brains of Scrapie-Infected Hamsters and Human Patients with Prion Diseases. J. Mol. Neurosci. 2013, 51, 734–748. [Google Scholar] [CrossRef]
- Lopez Salon, M.; Pasquini, L.; Besio Moreno, M.; Pasquini, J.M.; Soto, E. Relationship between beta-amyloid degradation and the 26S proteasome in neural cells. Exp. Neurol. 2003, 180, 131–143. [Google Scholar] [CrossRef]
- Makarava, N.; Chang, J.C.-Y.; Kushwaha, R.; Baskakov, I.V. Region-Specific Response of Astrocytes to Prion Infection. Front. Neurosci. 2019, 13, 1048. [Google Scholar] [CrossRef]
- Ashok, A.; Hegde, R.S. Selective Processing and Metabolism of Disease-Causing Mutant Prion Proteins. PLoS Pathog. 2009, 5, e1000479. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Borges, N.; Di Bari, M.A.; Eraña, H.; Sanchez-Martin, M.A.; Pirisinu, L.; Parra, B.; Elezgarai, S.R.; Vanni, I.; López-Moreno, R.; Vaccari, G.; et al. Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol. 2017, 135, 179–199. [Google Scholar] [CrossRef]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Possible involvement of proteasome inhibition in aging: Implications for oxidative stress. Mech. Ageing Dev. 2000, 113, 61–70. [Google Scholar] [CrossRef]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monleón, E.; Monzón, M.; Hortells, P.; Vargas, A.; Acín, C.; Badiola, J.J. Detection of PrPscon Lymphoid Tissues from Naturally Affected Scrapie Animals: Comparison of Three Visualization Systems. J. Histochem. Cytochem. 2004, 52, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, Z.; Díaz-Hernández, M.; Maynard, C.J.; Hernández, F.; Dantuma, N.P.; Lucas, J.J. Acute Polyglutamine Expression in Inducible Mouse Model Unravels Ubiquitin/Proteasome System Impairment and Permanent Recovery Attributable to Aggregate Formation. J. Neurosci. 2010, 30, 3675–3688. [Google Scholar] [CrossRef]
- Sarasa, R.; Martínez, A.; Monleón, E.; Bolea, R.; Vargas, A.; Badiola, J.J.; Monzón, M. Involvement of astrocytes in transmissible spongiform encephalopathies: A confocal microscopy study. Cell Tissue Res. 2012, 350, 127–134. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ubiquitin | PDI | PERK | BiP | Spongiosis | PrPSc | GFAP | ||
|---|---|---|---|---|---|---|---|---|
| Preclinical stage (Preclinical +controls) | Ubiquitin | ---- | 0.2676 * | 0.1249 n.s. | 0.1950 n.s. | 0.4701 **** | 0.6190 **** | 0.6020 **** |
| PDI | 0.2676 * | ---- | −0.2014 n.s. | 0.4944 **** | 0.4183 *** | 0.5211 **** | 0.4302 *** | |
| PERK | 0.1249 n.s. | −0.2014 n.s. | ---- | 0.1496 n.s. | 0.2894 * | 0.2041 n.s. | 0.2518 * | |
| BiP | 0.1950 n.s. | 0.4944 **** | 0.1496 n.s. | ---- | 0.4008 *** | 0.5105 **** | 0.5214 **** | |
| Spongiosis | 0.4701 **** | 0.4183 *** | 0.2894 * | 0.4008 *** | ---- | 0.7603 **** | 0.6152 **** | |
| PrPSc | 0.6190 **** | 0.5211 **** | 0.2041 n.s. | 0.5105 **** | 0.7603 **** | ---- | 0.8550 **** | |
| GFAP | 0.6020 **** | 0.4302 *** | 0.2518 * | 0.5214 **** | 0.6152 **** | 0.8550 **** | ---- | |
| Clinical stage (Clinical +controls) | Ubiquitin | ---- | 0.3131 *** | 0.1124 n.s. | 0.1606 n.s. | 0.4616 **** | 0.5947 **** | 0.5059 **** |
| PDI | 0.3131 *** | ---- | 0.1962 n.s. | 0.4457 **** | 0.4007 **** | 0.3172 ** | 0.4170 **** | |
| PERK | 0.1124 n.s. | 0.1962 n.s. | ---- | 0.2819 ** | 0.2554 * | 0.3182 ** | 0.2663 * | |
| BiP | 0.1606 n.s. | 0.4457 **** | 0.2819 ** | ---- | 0.2573 * | 0.3391 *** | 0.5395 **** | |
| Spongiosis | 0.4616 **** | 0.4007 **** | 0.2554 * | 0.2573 * | ---- | 0.8507 **** | 0.6619 **** | |
| PrPSc | 0.5947 **** | 0.3172 ** | 0.3182 ** | 0.3391 *** | 0.8507 **** | ---- | 0.7958 **** | |
| GFAP | 0.5059 **** | 0.4170 **** | 0.2663 * | 0.5395 **** | 0.6619 **** | 0.7958 **** | ---- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otero, A.; Betancor, M.; Eraña, H.; Fernández Borges, N.; Lucas, J.J.; Badiola, J.J.; Castilla, J.; Bolea, R. Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease. Int. J. Mol. Sci. 2021, 22, 465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010465
Otero A, Betancor M, Eraña H, Fernández Borges N, Lucas JJ, Badiola JJ, Castilla J, Bolea R. Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease. International Journal of Molecular Sciences. 2021; 22(1):465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010465
Chicago/Turabian StyleOtero, Alicia, Marina Betancor, Hasier Eraña, Natalia Fernández Borges, José J. Lucas, Juan José Badiola, Joaquín Castilla, and Rosa Bolea. 2021. "Prion-Associated Neurodegeneration Causes Both Endoplasmic Reticulum Stress and Proteasome Impairment in a Murine Model of Spontaneous Disease" International Journal of Molecular Sciences 22, no. 1: 465. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010465