SATB1-Mediated Upregulation of the Oncogenic Receptor Tyrosine Kinase HER3 Antagonizes MET Inhibition in Gastric Cancer Cells

 and
and
Abstract
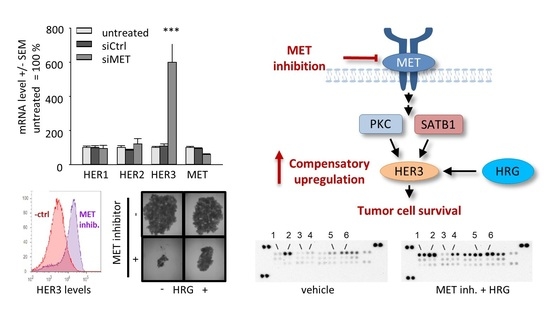
:
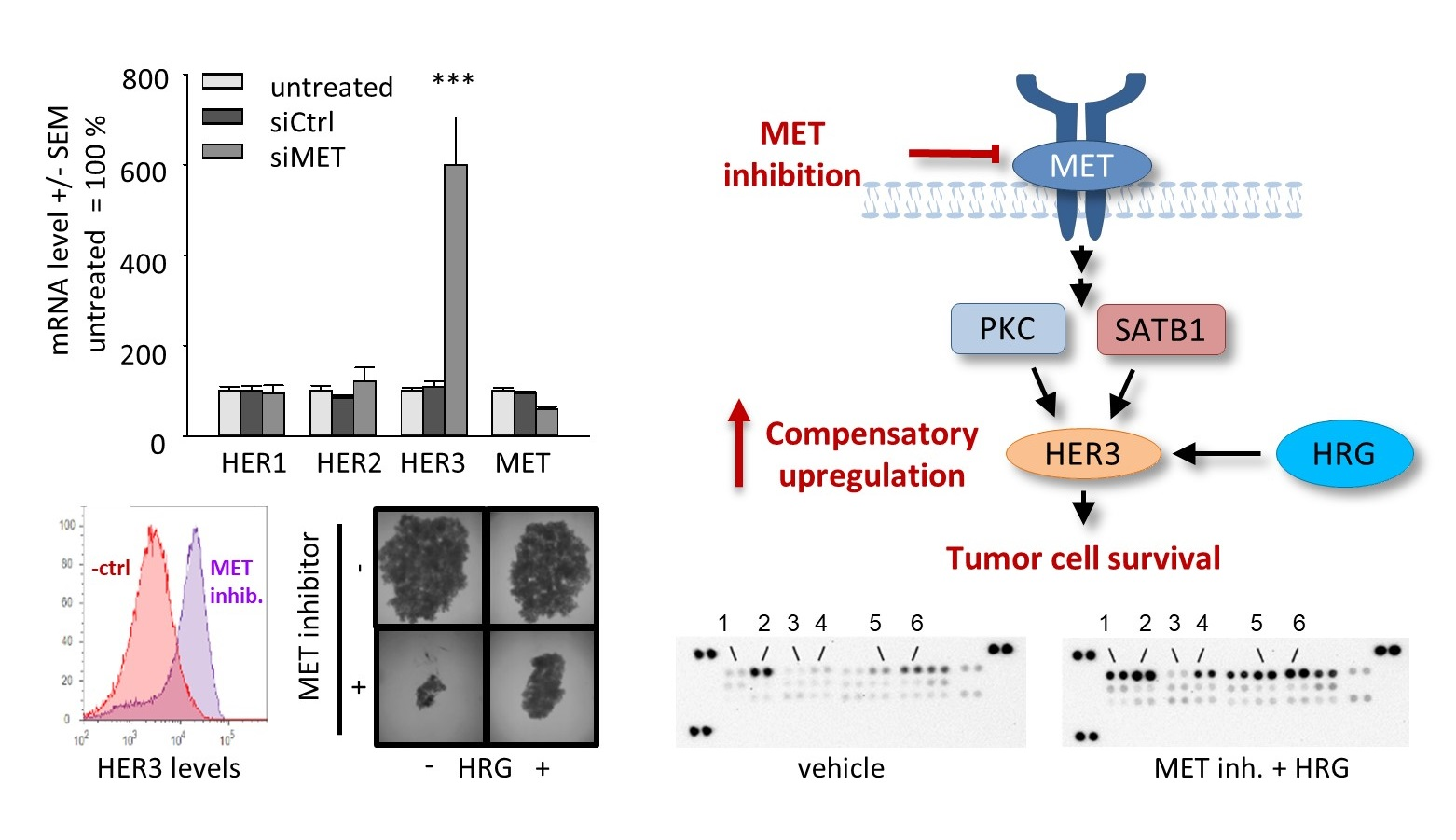{kind=link}
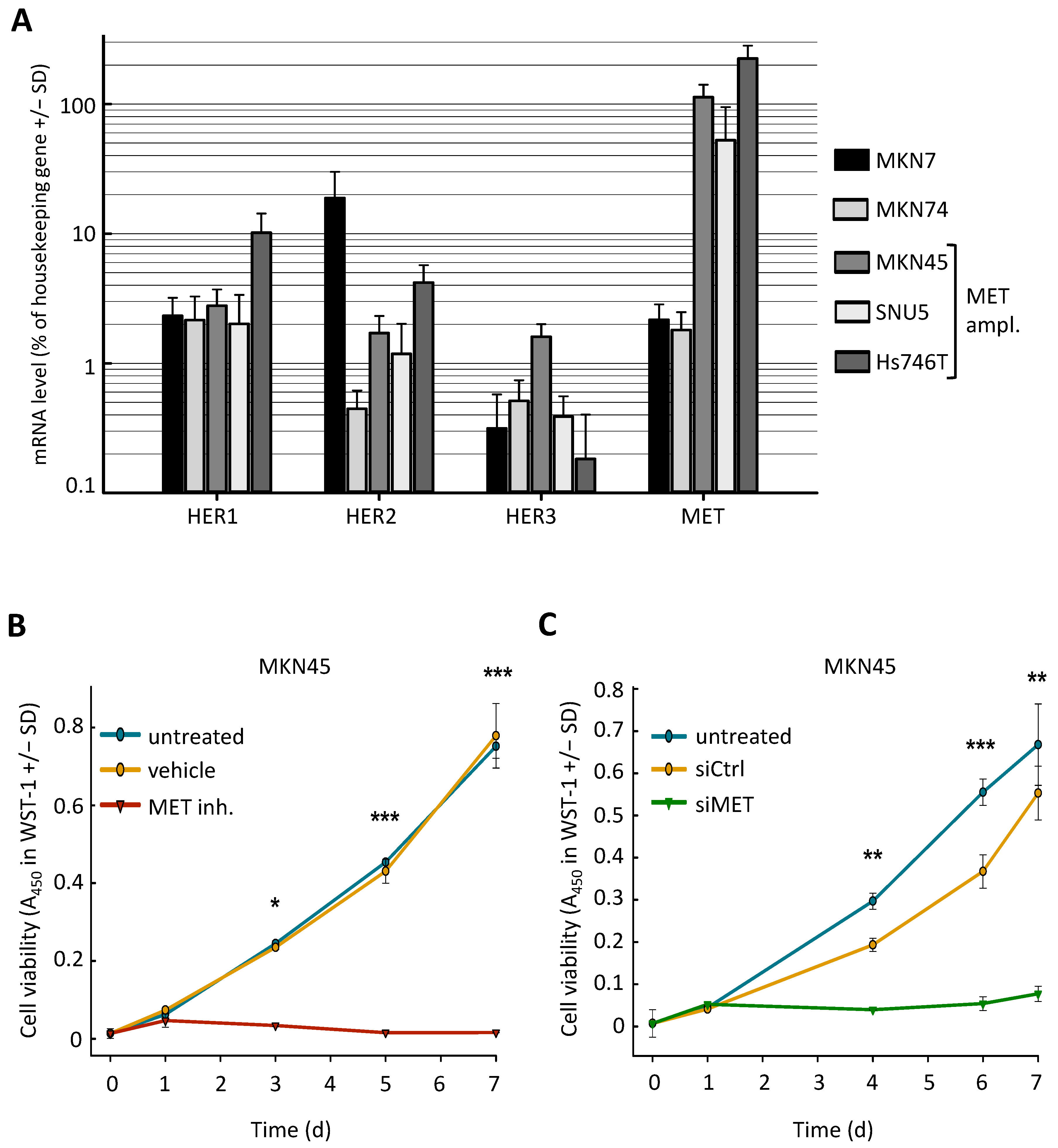{kind=link}
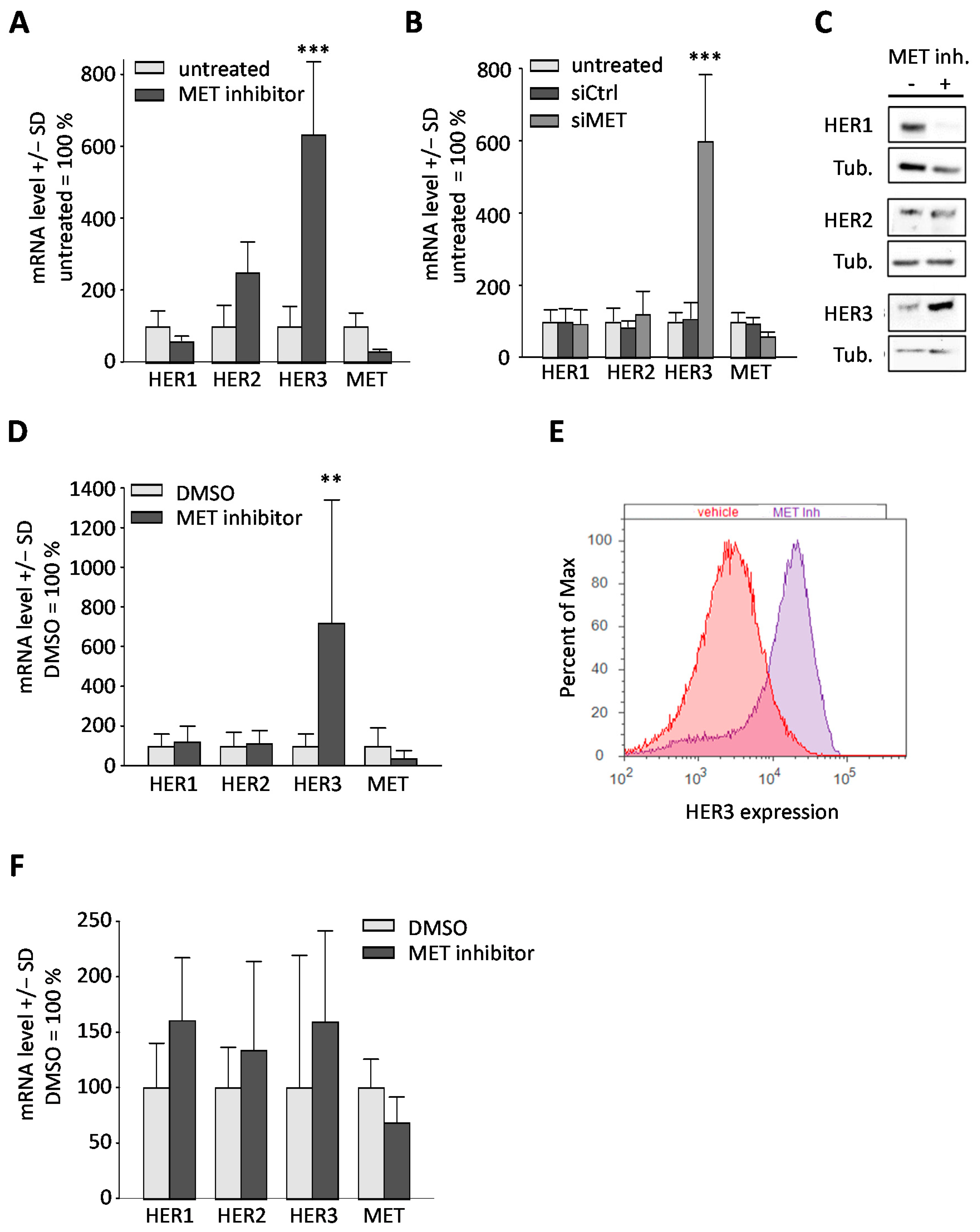{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
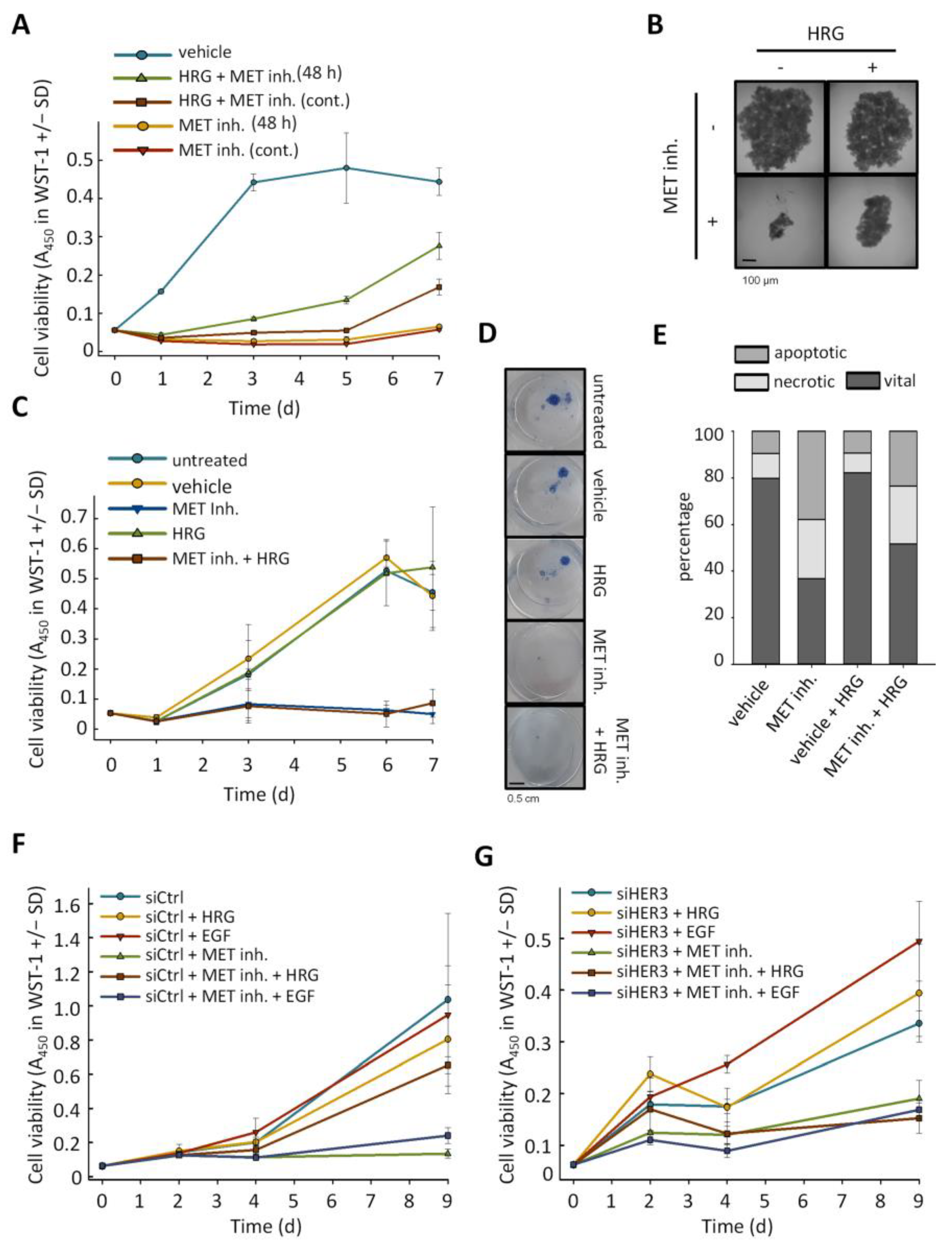
2.1. MET-Amplified Gastric Cancer Cells Are Highly Sensitive to MET Inhibition or siRNA-Mediated MET Knockdown
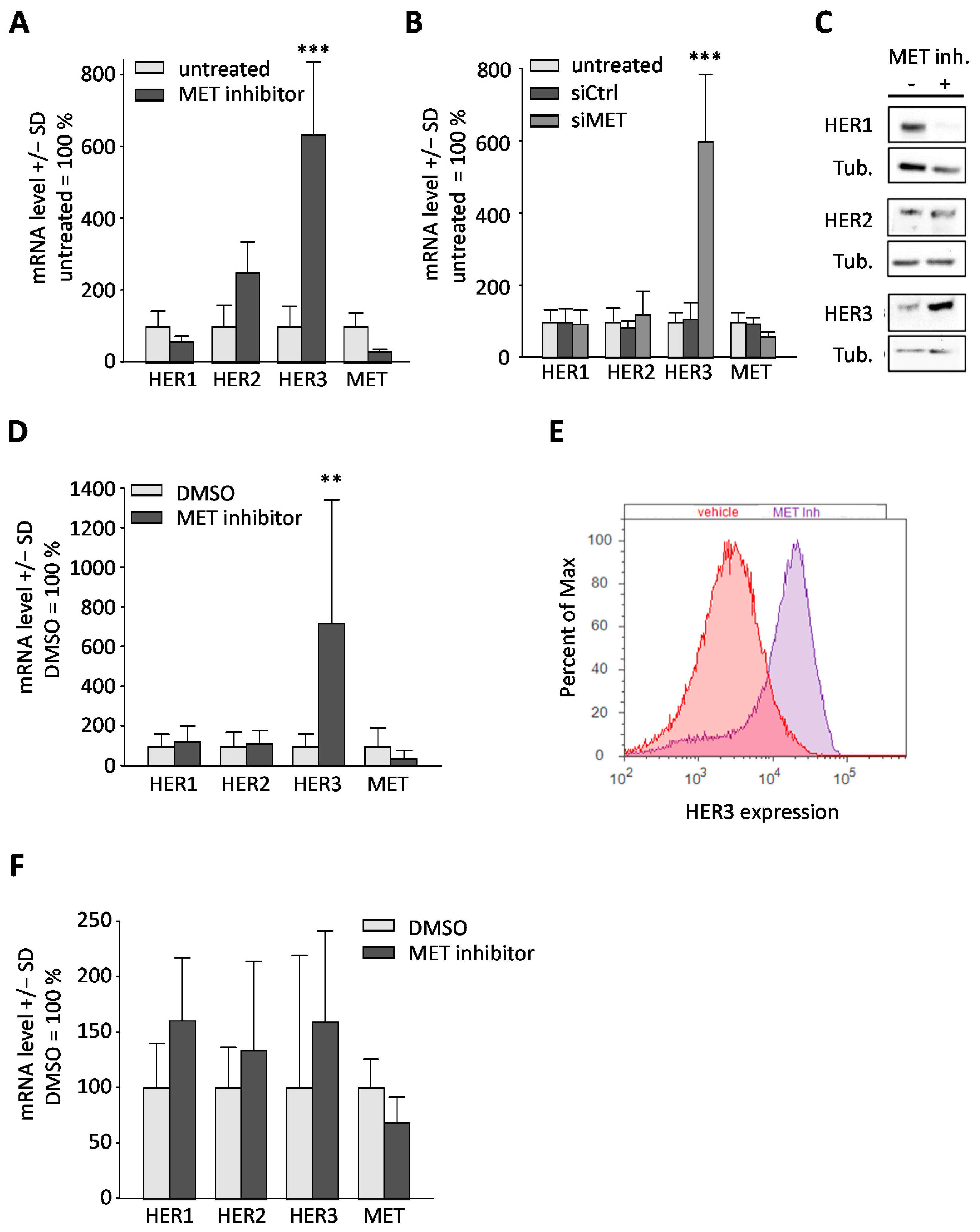
2.2. Downregulation or Inhibition of MET Leads to Upregulation of HER3
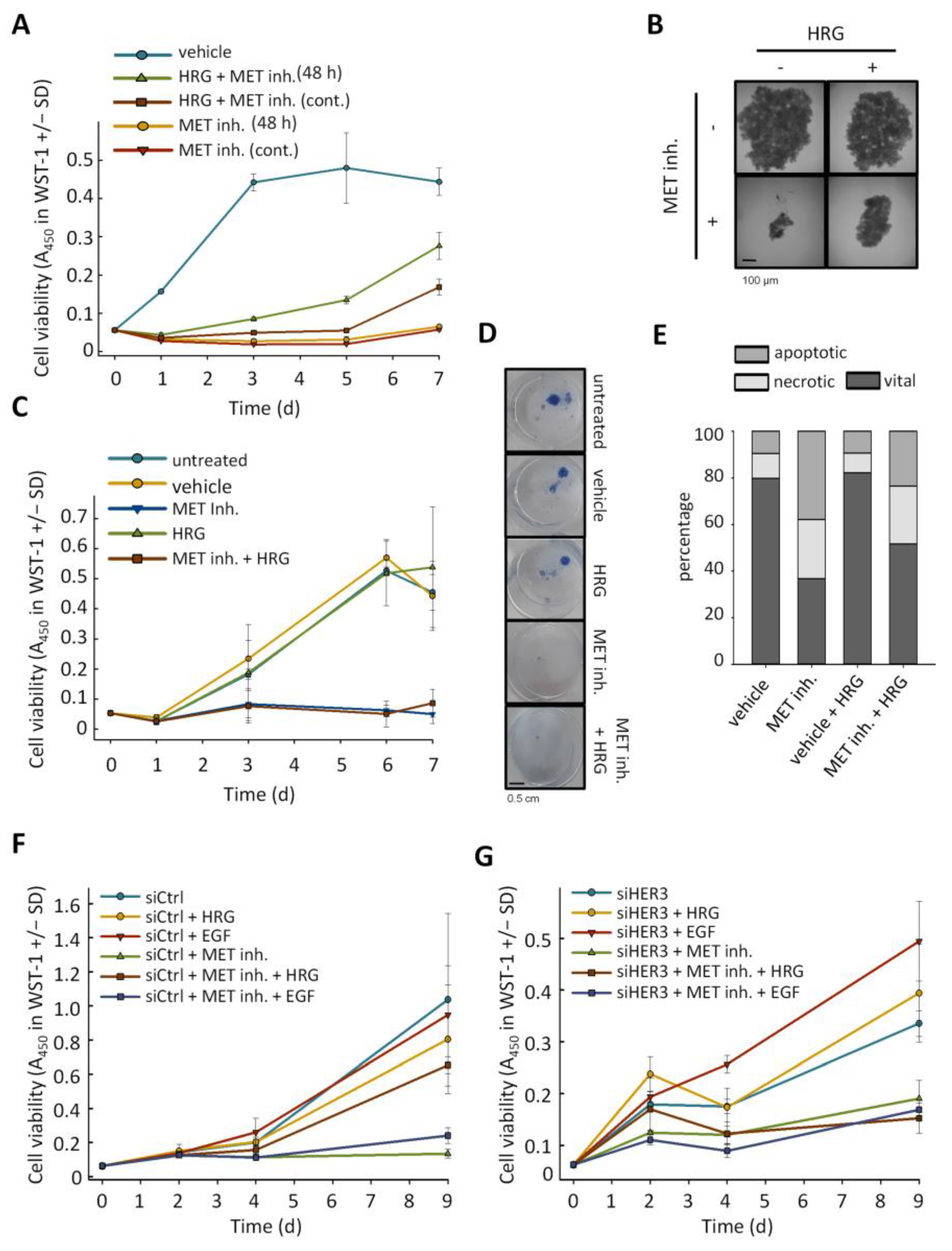
2.3. Anti-Proliferative Effects of MET Inhibition Are Partially Abolished by Treatment with HER3 Activator Heregulin
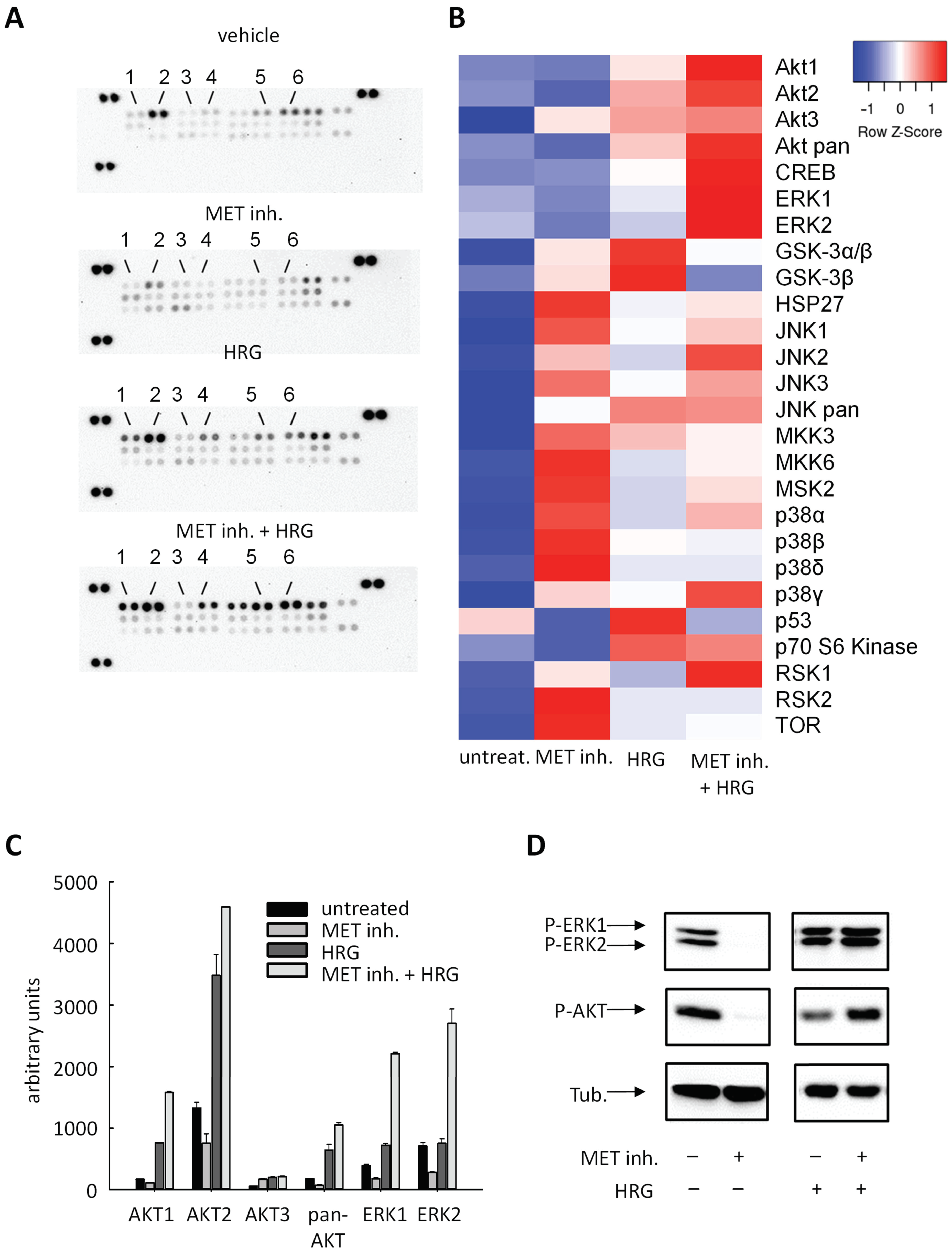
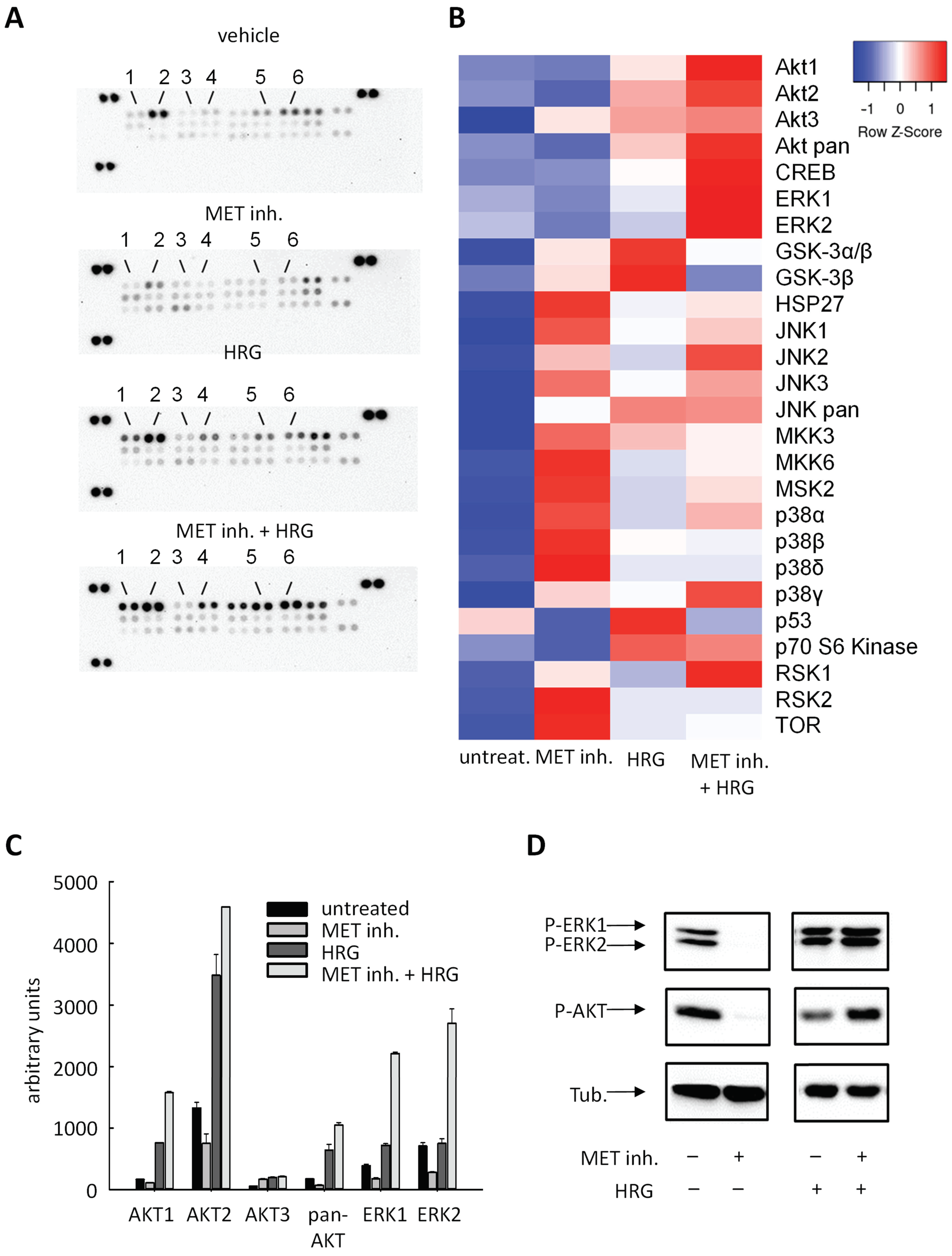
2.4. Cellular Effects Are Mediated by Alterations in MAPK Signaling
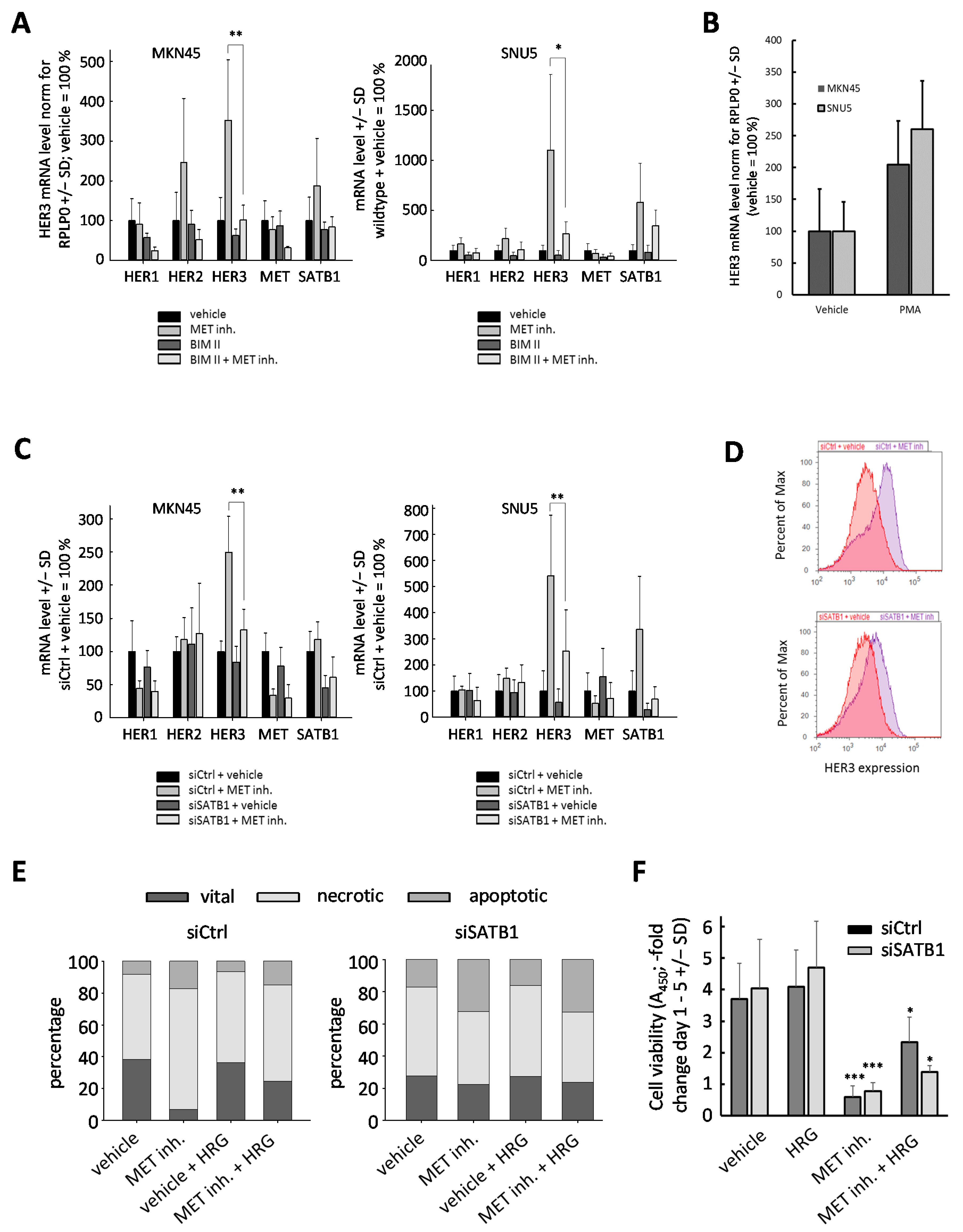
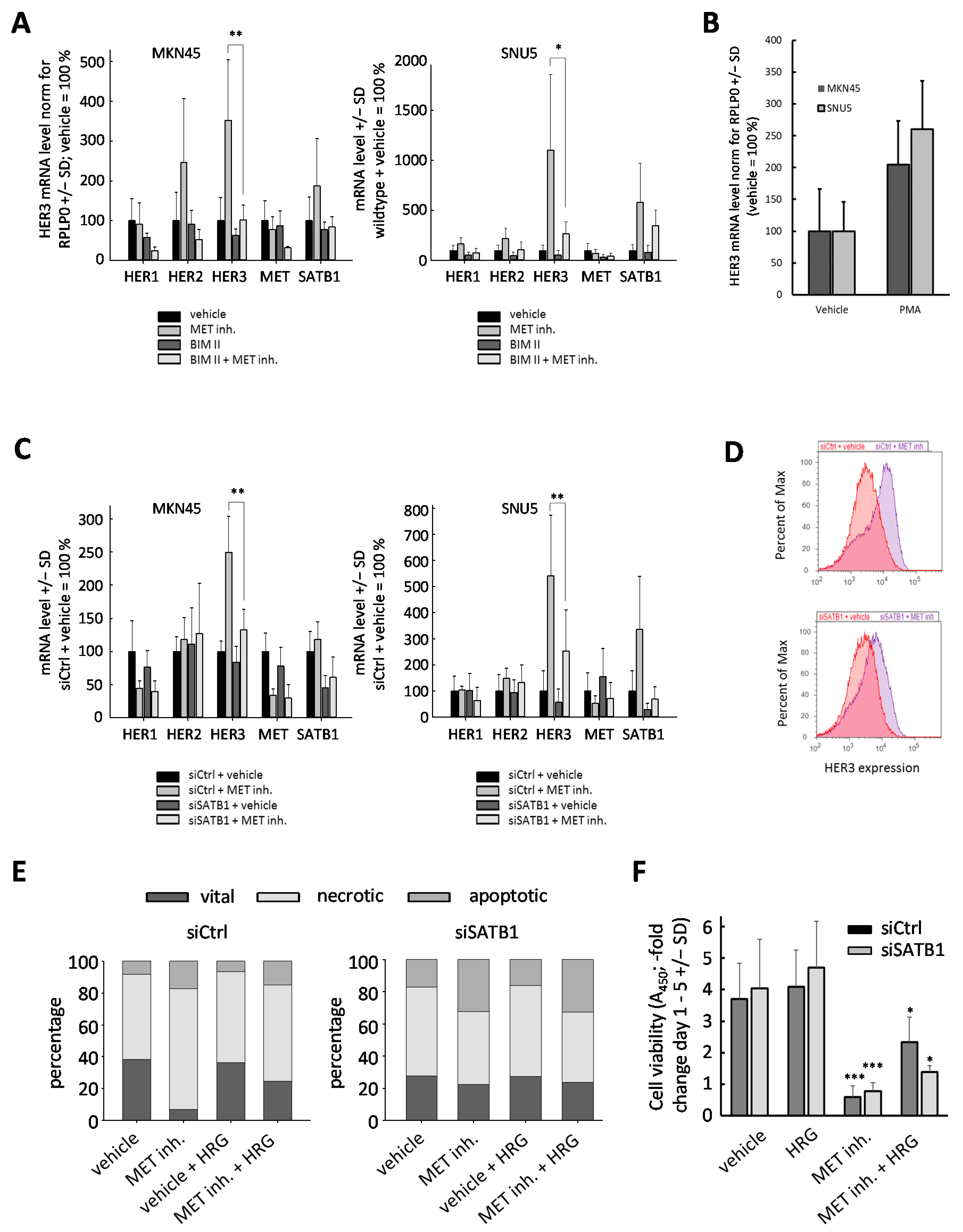
2.5. Upregulation of HER3 upon MET Inhibition Is Dependent on PKC and SATB1
3. Discussion
4. Material and Methods
4.1. Materials
4.2. Methods
4.2.1. Cell Culture
4.2.2. Cell Transfection and Treatment
4.2.3. WST-1 Assay
4.2.4. Colony Forming Assay
4.2.5. RNA Isolation and RT-qPCR
4.2.6. Immunoblot
4.2.7. Flow Cytometry
4.2.8. Spheroid Outgrow and Spheroid Formation
4.2.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Den Hoed, C.M.; Kuipers, E.J. Gastric Cancer: How Can We Reduce the Incidence of This Disease? Curr. Gastroenterol. Rep. 2016, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric Cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Jemal, A.; Center, M.M.; DeSantis, C.; Ward, E.M. Global Patterns of Cancer Incidence and Mortality Rates and Trends. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 1893–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mestier, L.; Lardière-Deguelte, S.; Volet, J.; Kianmanesh, R.; Bouché, O. Recent Insights in the Therapeutic Management of Patients with Gastric Cancer. Dig. Liver Dis. 2016, 48, 984–994. [Google Scholar] [CrossRef]
- Shum, H.; Rajdev, L. Multimodality Management of Resectable Gastric Cancer: A Review. World J. Gastrointest. Oncol. 2014, 6, 393. [Google Scholar] [CrossRef]
- Obermannová, R.; Lordick, F. Management of Metastatic Gastric Cancer. Hematol. Oncol. Clin. N. Am. 2017, 31, 469–483. [Google Scholar] [CrossRef]
- Lordick, F.; Janjigian, Y.Y. Clinical Impact of Tumour Biology in the Management of Gastroesophageal Cancer. Nat. Rev. Clin. Oncol. 2016, 13, 348–360. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in Combination with Chemotherapy versus Chemotherapy Alone for Treatment of HER2-Positive Advanced Gastric or Gastro-Oesophageal Junction Cancer (ToGA): A Phase 3, Open-Label, Randomised Controlled Trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Lee, H.E.; Kim, M.A.; Lee, H.S.; Jung, E.-J.; Yang, H.-K.; Lee, B.L.; Bang, Y.-J.; Kim, W.H. MET in Gastric Carcinomas: Comparison between Protein Expression and Gene Copy Number and Impact on Clinical Outcome. Br. J. Cancer 2012, 107, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; Shen, D.; Yin, X.; Gavine, P.; Zhang, T.; Su, X.; Zhan, P.; Xu, Y.; Lv, J.; Qian, J.; et al. HER2, MET and FGFR2 Oncogenic Driver Alterations Define Distinct Molecular Segments for Targeted Therapies in Gastric Carcinoma. Br. J. Cancer 2014, 110, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C. (Not Giving up) the Marathon Race of Met Targeting Therapy: Are We There Yet? Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erjala, K.; Sundvall, M.; Junttila, T.T.; Zhang, N.; Savisalo, M.; Mali, P.; Kulmala, J.; Pulkkinen, J.; Grenman, R.; Elenius, K. Signaling via ErbB2 and ErbB3 Associates with Resistance and Epidermal Growth Factor Receptor (EGFR) Amplification with Sensitivity to EGFR Inhibitor Gefitinib in Head and Neck Squamous Cell Carcinoma Cells. Clin. Cancer Res. 2006, 12, 4103–4111. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Chang, S.S.; Hsu, J.L.; Hung, M.C. Signaling Cross-Talk in the Resistance to HER Family Receptor Targeted Therapy. Oncogene 2014, 33, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachleitner-Hofmann, T.; Sun, M.Y.; Chen, C.-T.; Tang, L.; Song, L.; Zeng, Z.; Shah, M.; Christensen, J.G.; Rosen, N.; Solit, D.B.; et al. HER Kinase Activation Confers Resistance to MET Tyrosine Kinase Inhibition in MET Oncogene-Addicted Gastric Cancer Cells. Mol. Cancer Ther. 2008, 7, 3499–3508. [Google Scholar] [CrossRef] [Green Version]
- Corso, S.; Ghiso, E.; Cepero, V.; Sierra, J.R.; Migliore, C.; Bertotti, A.; Trusolino, L.; Comoglio, P.M.; Giordano, S. Activation of HER Family Members in Gastric Carcinoma Cells Mediates Resistance to MET Inhibition. Mol. Cancer 2010, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.; Gang, L.; Rongmin, G.; Xu, W.; Xuezhi, M.; Huanqiu, C. Essential Role of Her3 in Two Signaling Transduction Patterns: Her2/Her3 and MET/Her3 in Proliferation of Human Gastric Cancer. Mol. Carcinog. 2015, 54, 1700–1709. [Google Scholar] [CrossRef]
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 Reprogrammes Gene Expression to Promote Breast Tumour Growth and Metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef]
- Frömberg, A.; Rabe, M.; Aigner, A. Multiple Effects of the Special AT-Rich Binding Protein 1 (SATB1) in Colon Carcinoma. Int. J. Cancer 2014, 135, 2537–2546. [Google Scholar] [CrossRef]
- Frömberg, A.; Rabe, M.; Oppermann, H.; Gaunitz, F.; Aigner, A. Analysis of Cellular and Molecular Antitumor Effects upon Inhibition of SATB1 in Glioblastoma Cells. BMC Cancer 2017, 17. [Google Scholar] [CrossRef]
- Zhang, S.; Tong, Y.X.; Xu, X.S.; Lin, H.; Chao, T.F. Prognostic Significance of SATB1 in Gastrointestinal Cancer: A Meta-Analysis and Literature Review. Oncotarget 2017, 8, 48410–48423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinomiya, N.; Chong, F.G.; Xie, Q.; Gustafson, M.; Waters, D.J.; Zhang, Y.W.; Vande Woude, G.F. RNA Interference Reveals That Ligand-Independent Met Activity Is Required for Tumor Cell Signaling and Survival. Cancer Res. 2004, 64, 7962–7970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraway, K.L.; Cantley, L.C. A Neu Acquaintance for ErbB3 and ErbB4: A Role for Receptor Heterodimerization in Growth Signaling. Cell 1994, 5–8. [Google Scholar] [CrossRef]
- Cui, J.J.; McTigue, M.; Nambu, M.; Tran-Dubé, M.; Pairish, M.; Shen, H.; Jia, L.; Cheng, H.; Hoffman, J.; Le, P.; et al. Discovery of a Novel Class of Exquisitely Selective Mesenchymal-Epithelial Transition Factor (c-MET) Protein Kinase Inhibitors and Identification of the Clinical Candidate 2-(4-(1-(Quinolin-6-Ylmethyl)-1H-[1–3]Triazolo[4,5-b]Pyrazin-6-Yl)-1H-Pyraz. J. Med. Chem. 2012, 55, 8091–8109. [Google Scholar] [CrossRef] [PubMed]
- Ocana, A.; Vera-Badillo, F.; Seruga, B.; Templeton, A.; Pandiella, A.; Amir, E. HER3 Overexpression and Survival in Solid Tumors: A Meta-Analysis. J. Natl. Cancer Inst. 2013, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Qu, Y.; Li, X.; Sui, F.; Yao, D.; Yang, Q.; Shi, B.; Ji, M.; Hou, P. Increased Expression of EHF via Gene Amplification Contributes to the Activation of HER Family Signaling and Associates with Poor Survival in Gastric Cancer. Cell Death Dis. 2016. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT Inhibition Relieves Feedback Suppression of Receptor Tyrosine Kinase Expression and Activity. Cancer Cell 2011. [Google Scholar] [CrossRef] [Green Version]
- Pavan Kumar, P.; Purbey, P.K.; Sinha, C.K.; Notani, D.; Limaye, A.; Jayani, R.S.; Galande, S. Phosphorylation of SATB1, a Global Gene Regulator, Acts as a Molecular Switch Regulating Its Transcriptional Activity In Vivo. Mol. Cell 2006, 22, 231–243. [Google Scholar] [CrossRef]
- Purbey, P.K.; Singh, S.; Notani, D.; Kumar, P.P.; Limaye, A.S.; Galande, S. Acetylation-Dependent Interaction of SATB1 and CtBP1 Mediates Transcriptional Repression by SATB1. Mol. Cell. Biol. 2009, 29, 1321–1337. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.; J. Pradhan, S.; Galande, S. Chromatin Organizer SATB1 As a Novel Molecular Target for Cancer Therapy. Curr. Drug Targets 2012, 13, 1603–1615. [Google Scholar] [CrossRef]
- Frömberg, A.; Engeland, K.; Aigner, A. The Special AT-Rich Sequence Binding Protein 1 (SATB1) and Its Role in Solid Tumors. Cancer Lett. 2018, 417, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Lu, X.; Li, H.; Peng, Z.; Wu, K.; Wang, G.; Tong, Q. Special AT-Rich Sequence Binding Protein 1 Regulates the Multidrug Resistance and Invasion of Human Gastric Cancer Cells. Oncol. Lett. 2012, 4, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, C.; Fang, E.; Lu, X.; Wang, G.; Tong, Q. Co-Delivery of Doxorubicin and SATB1 ShRNA by Thermosensitive Magnetic Cationic Liposomes for Gastric Cancer Therapy. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Sakamoto, C.; Wada, K.; Akamatsu, T.; Uchida, T.; Tatsuguchi, A.; Matsui, H.; Fukui, H.; Fujimori, T.; Kasuga, M. Expression of Heregulin α, ErbB2, and ErbB3 and Their Influences on Proliferation of Gastric Epithelial Cells. Gastroenterology 1999, 117, 1119–1127. [Google Scholar] [CrossRef]
- Nagata, K.; Wada, K.; Tatsuguchi, A.; Futagami, S.; Gudis, K.; Miyake, K.; Tsukui, T.; Sakamoto, C. Heregulin-α and Heregulin-β Expression Is Linked to a COX-2-PGE2 Pathway in Human Gastric Fibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, 1243–1251. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jenke, R.; Holzhäuser-Rein, M.; Mueller-Wilke, S.; Lordick, F.; Aigner, A.; Büch, T. SATB1-Mediated Upregulation of the Oncogenic Receptor Tyrosine Kinase HER3 Antagonizes MET Inhibition in Gastric Cancer Cells. Int. J. Mol. Sci. 2021, 22, 82. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010082
Jenke R, Holzhäuser-Rein M, Mueller-Wilke S, Lordick F, Aigner A, Büch T. SATB1-Mediated Upregulation of the Oncogenic Receptor Tyrosine Kinase HER3 Antagonizes MET Inhibition in Gastric Cancer Cells. International Journal of Molecular Sciences. 2021; 22(1):82. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010082
Chicago/Turabian StyleJenke, Robert, Miriam Holzhäuser-Rein, Stefanie Mueller-Wilke, Florian Lordick, Achim Aigner, and Thomas Büch. 2021. "SATB1-Mediated Upregulation of the Oncogenic Receptor Tyrosine Kinase HER3 Antagonizes MET Inhibition in Gastric Cancer Cells" International Journal of Molecular Sciences 22, no. 1: 82. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010082