
Impaired Wound Healing, Fibrosis, and Cancer: The Paradigm of Recessive Dystrophic Epidermolysis Bullosa


Abstract
:1. Introduction
2. Recessive Dystrophic Epidermolysis Bullosa
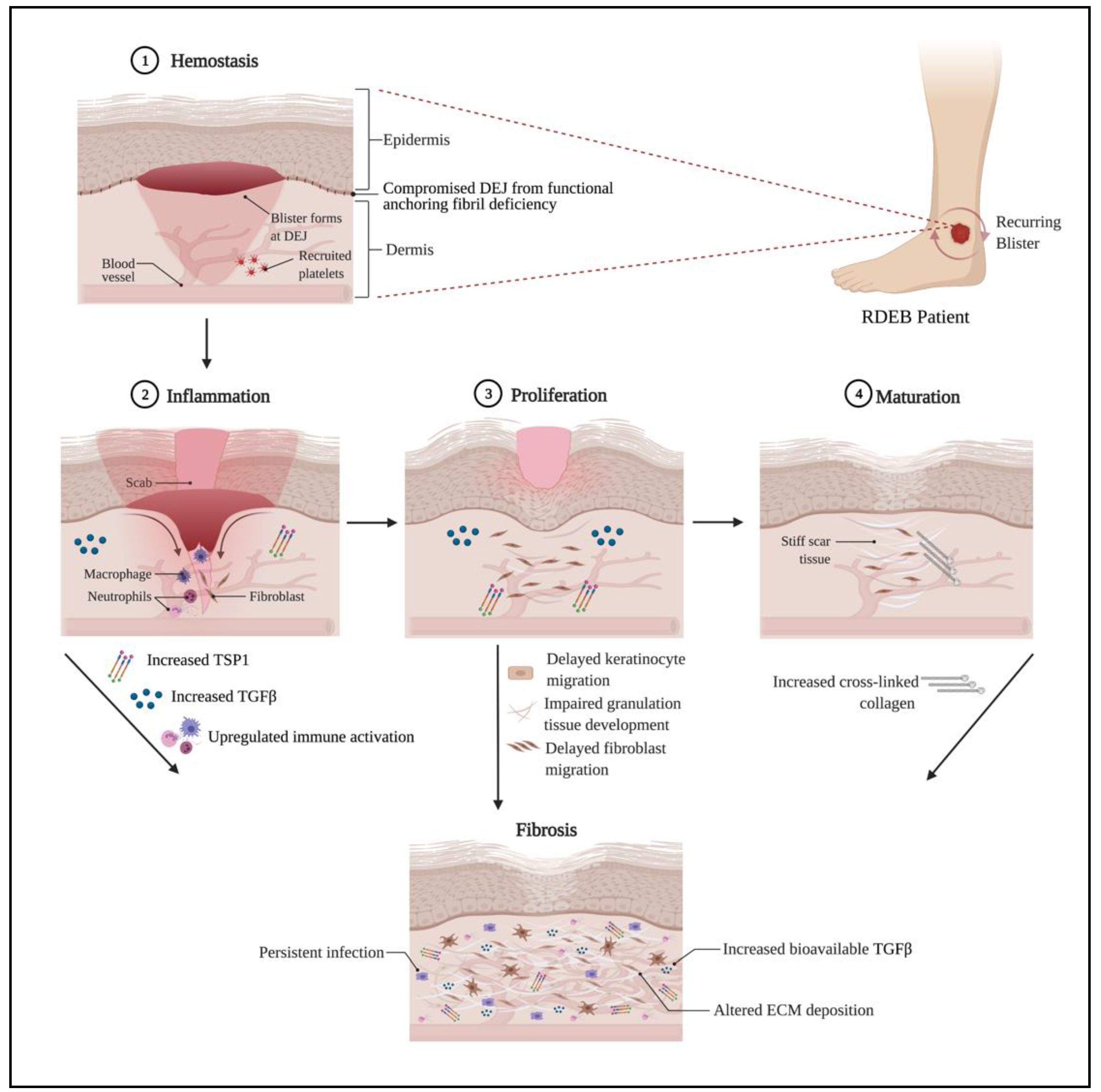
3. Impaired Wound Healing in RDEB
4. TGFβ and Fibrosis in RDEB
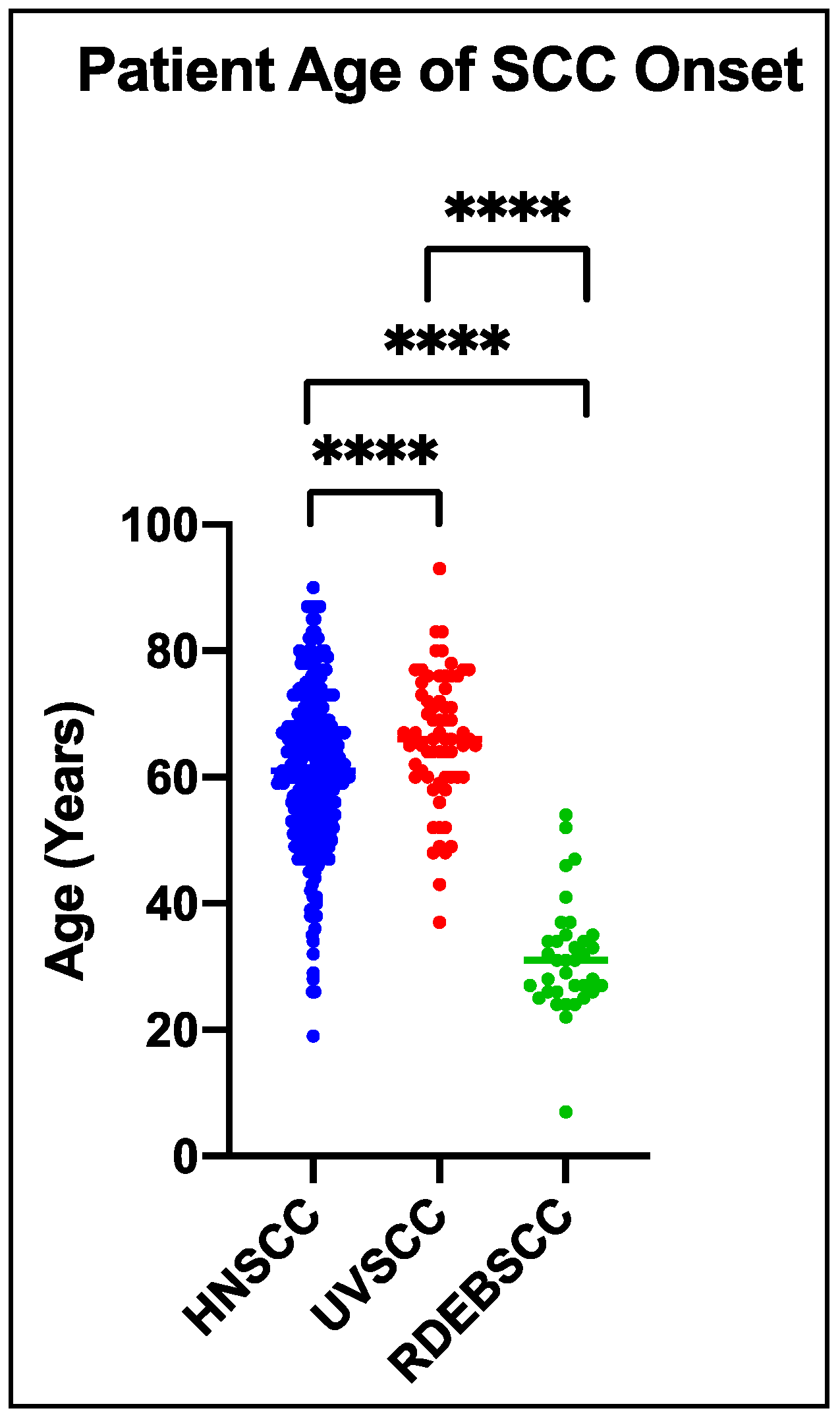
5. Cutaneous Cancer in RDEB
6. Closing Remarks about Parallels Between RDEB and Aging Pathologies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKT | Protein Kinase B |
| C7 | Type VII Collagen |
| CAF | Cancer-Associated Fibroblasts |
| cAMP | Cyclic Adenosine Monophosphate |
| cSCC | Cutaneous Squamous Cell Carcinoma |
| EB | Epidermolysis Bullosa |
| ECM | Extracellular Matrix |
| ERS | Endoplasmic Reticulum Stress |
| HMGB1 | High Mobility Group Box 1 protein |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| IL6 | Interleukin 6 |
| JNK | c-Jun N-terminal Kinase |
| LAP | Latency Associated Peptide |
| MAP | Mitogen-Activated Protein |
| MMP | Matrix Metalloproteinase |
| NETs | Neutrophil Extracellular Traps |
| p-EMT | Partial-Epithelial to Mesenchymal Transition |
| PI3 | Phosphoinositide 3 |
| RDEB | Recessive Dystrophic Epidermolysis Bullosa |
| ROS | Reactive Oxygen Species |
| SCC | Squamous Cell Carcinoma |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TGFβ | Transforming Growth Factor Beta |
| TNFα | Tumor Necrosis Factor Alpha |
| TSP-1 | Thrombospondin-1 |
| UPR | Unfolded Protein Response |
| USSCs | Unrestricted Somatic Stem Cells |
| α-SMA | Alpha-Smooth Muscle Actin |
References
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uitto, J.; Jimenez, S. Fibrotic skin diseases. Clinical presentations, etiologic considerations, and treatment options. Arch. Dermatol. 1990, 126, 661–664. [Google Scholar] [CrossRef]
- Rosenbloom, J.; Macarak, E.; Piera-Velazquez, S.; Jimenez, S.A. Human Fibrotic Diseases: Current Challenges in Fibrosis Research. Methods Mol. Biol. 2017, 1627, 1–23. [Google Scholar] [PubMed]
- Nguyen, A.V.; Soulika, A.M. The Dynamics of the Skin’s Immune System. Int. J. Mol. Sci. 2019, 20, 1811. [Google Scholar] [CrossRef] [Green Version]
- desJardins-Park, H.E.; Foster, D.S.; Longaker, M.T. Fibroblasts and wound healing: An update. Regen. Med. 2018, 13, 491–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taha, I.N.; Naba, A. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019, 63, 417–432. [Google Scholar] [PubMed]
- Montaudié, H.; Chiaverini, C.; Sbidian, E.; Charlesworth, A.; Lacour, J.P. Inherited epidermolysis bullosa and squamous cell carcinoma: A systematic review of 117 cases. Orphanet J. Rare Dis. 2016, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.F.; desJardins-Park, H.E.; Mascharak, S.; Borrelli, M.R.; Longaker, M.T. Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis. Models Mech. 2020, 13, dmm044164. [Google Scholar] [CrossRef] [PubMed]
- Christiano, A.M.; Greenspan, D.S.; Hoffman, G.G.; Zhang, X.; Tamai, Y.; Lin, A.N.; Dietz, H.C.; Hovnanian, A.; Uitto, J. A missense mutation in type VII collagen in two affected siblings with recessive dystrophic epidermolysis bullosa. Nat. Genet. 1993, 4, 62–66. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [Green Version]
- Flannery, D.; Doyle, C.; Hickey, S.; Aherne, F.; Kennan, A. Direct Costs of Epidermolysis Bullosa by Disease Severity. Acta Derm. Venereol. 2020, 100, adv00116. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; South, A.; Uitto, J. Molecular Therapeutics in Development for Epidermolysis Bullosa: Update 2020. Mol. Diagn. Ther. 2020, 24, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Li, K.P.; Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef]
- Burgeson, R.E. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J. Investig. Dermatol. 1993, 101, 252–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Y.Z.; Pourreyron, C.; Salas-Alanis, J.C.; Dayal, J.H.; Cepeda-Valdes, R.; Yan, W.; Wright, S.; Chen, M.; Fine, J.D.; Hogg, F.J.; et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 2012, 72, 3522–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küttner, V.; Mack, C.; Gretzmeier, C.; Bruckner-Tuderman, L.; Dengjel, J. Loss of collagen VII is associated with reduced transglutaminase 2 abundance and activity. J. Investig. Dermatol. 2014, 134, 2381–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küttner, V.; Mack, C.; Rigbolt, K.T.; Kern, J.S.; Schilling, O.; Busch, H.; Bruckner-Tuderman, L.; Dengjel, J. Global remodelling of cellular microenvironment due to loss of collagen VII. Mol. Syst. Biol. 2013, 9, 657. [Google Scholar] [CrossRef]
- Nyström, A.; Velati, D.; Mittapalli, V.R.; Fritsch, A.; Kern, J.S.; Bruckner-Tuderman, L. Collagen VII plays a dual role in wound healing. J. Clin. Investig. 2013, 123, 3498–3509. [Google Scholar] [CrossRef] [Green Version]
- Nyström, A.; Bornert, O.; Kühl, T.; Gretzmeier, C.; Thriene, K.; Dengjel, J.; Pfister-Wartha, A.; Kiritsi, D.; Bruckner-Tuderman, L. Impaired lymphoid extracellular matrix impedes antibacterial immunity in epidermolysis bullosa. Proc. Natl. Acad. Sci. USA 2018, 115, E705–E714. [Google Scholar] [CrossRef] [Green Version]
- Thriene, K.; Grüning, B.A.; Bornert, O.; Erxleben, A.; Leppert, J.; Athanasiou, I.; Weber, E.; Kiritsi, D.; Nyström, A.; Reinheckel, T.; et al. Combinatorial Omics Analysis Reveals Perturbed Lysosomal Homeostasis in Collagen VII-deficient Keratinocytes. Mol. Cell. Proteom. 2018, 17, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Mittapalli, V.R.; Madl, J.; Löffek, S.; Kiritsi, D.; Kern, J.S.; Römer, W.; Nyström, A.; Bruckner-Tuderman, L. Injury-Driven Stiffening of the Dermis Expedites Skin Carcinoma Progression. Cancer Res. 2016, 76, 940–951. [Google Scholar] [CrossRef] [Green Version]
- Akasaka, E.; Kleiser, S.; Sengle, G.; Bruckner-Tuderman, L.; Nyström, A. Diversity of Mechanisms Underlying Latent TGF-β Activation in Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Russell, R.J.; Webster, T.G.; Cao, Q.; Agarwal, P.; Lim, Y.Z.; Krishnan, S.; Fuentes, I.; Guttmann-Gruber, C.; McGrath, J.A.; et al. Thrombospondin-1 Is a Major Activator of TGF-β Signaling in Recessive Dystrophic Epidermolysis Bullosa Fibroblasts. J. Investig. Dermatol. 2019, 139, 1497–1505.e5. [Google Scholar] [CrossRef] [PubMed]
- Chacón-Solano, E.; Aguado, T.; Sánchez-Domínguez, R.; Leon, C.; Carretero, M.; Escamez, M.; Ruiz-Mezcua, B.; Sánchez-Puelles, J.; Larcher, F.; Río, M. 631 Oxidative stress imbalance as contributing factor in the establishment of fibrosis in recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2019, 139, S323. [Google Scholar] [CrossRef] [Green Version]
- Odorisio, T.; Di Salvio, M.; Orecchia, A.; Di Zenzo, G.; Piccinni, E.; Cianfarani, F.; Travaglione, A.; Uva, P.; Bellei, B.; Conti, A.; et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-β signalling in modifying disease severity. Hum. Mol. Genet. 2014, 23, 3907–3922. [Google Scholar] [CrossRef] [Green Version]
- Titeux, M.; Pendaries, V.; Tonasso, L.; Décha, A.; Bodemer, C.; Hovnanian, A. A frequent functional SNP in the MMP1 promoter is associated with higher disease severity in recessive dystrophic epidermolysis bullosa. Hum. Mutat. 2008, 29, 267–276. [Google Scholar] [CrossRef]
- Aburima, A.; Berger, M.; Spurgeon, B.E.; Webb, B.A.; Wraith, K.S.; Febbraio, M.; Poole, A.W.; Naseem, K.M. Thrombospondin-1 promotes haemostasis through modulation of cAMP signalling in blood platelets. Blood 2021, 137, 678–689. [Google Scholar] [CrossRef]
- Choudhary, P.; Gutteridge, A.; Impey, E.; Storer, R.I.; Owen, R.M.; Whiting, P.J.; Bictash, M.; Benn, C.L. Targeting the cAMP and Transforming Growth Factor-β Pathway Increases Proliferation to Promote Re-Epithelialization of Human Stem Cell-Derived Retinal Pigment Epithelium. Stem Cells Transl. Med. 2016, 5, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, S.Q.; Hassid, A.; Ostrom, R.S. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol. Pharmacol. 2006, 70, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, M.; Dennler, S.; Anderegg, U.; Kokot, A.; Simon, J.C.; Luger, T.A.; Mauviel, A.; Böhm, M. Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J. Biol. Chem. 2010, 285, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-β1-Induced Profibrotic Signaling. Cell. Physiol. Biochem. 2015, 36, 1911–1927. [Google Scholar] [CrossRef]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeloffzen, W.W.; Kluin-Nelemans, H.C.; Bosman, L.; de Wolf, J.T. Effects of red blood cells on hemostasis. Transfusion 2010, 50, 1536–1544. [Google Scholar] [CrossRef]
- Hwang, S.J.E.; Daniel, B.S.; Fergie, B.; Davey, J.; Murrell, D.F. Prevalence of anemia in patients with epidermolysis bullosa registered in Australia. Int. J. Womens Dermatol. 2015, 1, 37–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimer, A.; Hess, M.; Schwieger-Briel, A.; Kiritsi, D.; Schauer, F.; Schumann, H.; Bruckner-Tuderman, L.; Has, C. Natural history of growth and anaemia in children with epidermolysis bullosa: A retrospective cohort study. Br. J. Dermatol. 2020, 182, 1437–1448. [Google Scholar] [CrossRef]
- Wright, J.A.; Richards, T.; Srai, S.K. The role of iron in the skin and cutaneous wound healing. Front. Pharmacol. 2014, 5, 156. [Google Scholar] [CrossRef] [Green Version]
- Dovi, J.V.; He, L.K.; DiPietro, L.A. Accelerated wound closure in neutrophil-depleted mice. J. Leukoc. Biol. 2003, 73, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, I.; Guttmann-Gruber, C.; Tockner, B.; Diem, A.; Klausegger, A.; Cofré-Araneda, G.; Figuera, O.; Hidalgo, Y.; Morandé, P.; Palisson, F.; et al. Cells from discarded dressings differentiate chronic from acute wounds in patients with Epidermolysis Bullosa. Sci. Rep. 2020, 10, 15064. [Google Scholar] [CrossRef]
- Goren, I.; Allmann, N.; Yogev, N.; Schürmann, C.; Linke, A.; Holdener, M.; Waisman, A.; Pfeilschifter, J.; Frank, S. A transgenic mouse model of inducible macrophage depletion: Effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am. J. Pathol. 2009, 175, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Mori, R.; Kondo, T.; Nishie, T.; Ohshima, T.; Asano, M. Impairment of skin wound healing in beta-1,4-galactosyltransferase-deficient mice with reduced leukocyte recruitment. Am. J. Pathol. 2004, 164, 1303–1314. [Google Scholar] [CrossRef]
- Tanno, H.; Kawakami, K.; Kanno, E.; Suzuki, A.; Takagi, N.; Yamamoto, H.; Ishii, K.; Imai, Y.; Maruyama, R.; Tachi, M. Invariant NKT cells promote skin wound healing by preventing a prolonged neutrophilic inflammatory response. Wound Repair Regen. 2017, 25, 805–815. [Google Scholar] [CrossRef]
- DiPietro, L.A.; Wilgus, T.A.; Koh, T.J. Macrophages in Healing Wounds: Paradoxes and Paradigms. Int. J. Mol. Sci. 2021, 22, 950. [Google Scholar] [CrossRef]
- Fuentes, I.; Guttmann-Gruber, C.; Tay, A.S.L.; Piñón Hofbauer, J.; Denil, S.; Reichelt, J.; Palisson, F.; Common, J.E.A.; South, A.P. Reduced Microbial Diversity Is a Feature of Recessive Dystrophic Epidermolysis Bullosa-Involved Skin and Wounds. J. Investig. Dermatol. 2018, 138, 2492–2495. [Google Scholar] [CrossRef] [Green Version]
- Levin, L.E.; Shayegan, L.H.; Lucky, A.W.; Hook, K.P.; Bruckner, A.L.; Feinstein, J.A.; Whittier, S.; Lauren, C.T.; Pope, E.; Lara-Corrales, I.; et al. Characterization of wound microbes in epidermolysis bullosa: Results from the epidermolysis bullosa clinical characterization and outcomes database. Pediatr. Dermatol. 2021, 38, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Granick, M.S.; Tomaselli, N.L. Wound Dressings and Comparative Effectiveness Data. Adv. Wound Care 2014, 3, 511–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, D.C.; Gorell, E.S.; Teng, C.; Barriga, M.; Nazaroff, J.; Li, S.; Subica, A.; Lu, Y.; Marinkovich, M.P.; Tang, J.Y. Clinical characteristics associated with increased wound size in patients with recessive dystrophic epidermolysis bullosa. Pediatr. Dermatol. 2021. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, I.B.; Moseley, R.; Baird, D.M.; Kipling, D.; Giles, P.; Laffafian, I.; Price, P.E.; Thomas, D.W.; Stephens, P. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J. Investig. Dermatol. 2008, 128, 2526–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willyard, C. Unlocking the secrets of scar-free skin healing. Nature 2018, 563, S86–S88. [Google Scholar] [CrossRef]
- Breitenbach, J.S.; Rinnerthaler, M.; Trost, A.; Weber, M.; Klausegger, A.; Gruber, C.; Bruckner, D.; Reitsamer, H.A.; Bauer, J.W.; Breitenbach, M. Transcriptome and ultrastructural changes in dystrophic Epidermolysis bullosa resemble skin aging. Aging 2015, 7, 389–411. [Google Scholar] [CrossRef] [Green Version]
- Eng, V.A.; Solis, D.C.; Gorell, E.S.; Choi, S.; Nazaroff, J.; Li, S.; de Souza, M.P.; Murrell, D.F.; Marinkovich, M.P.; Tang, J.Y. Patient-reported outcomes and quality of life in recessive dystrophic epidermolysis bullosa: A global cross-sectional survey. J. Am. Acad. Dermatol. 2020. [Google Scholar] [CrossRef]
- Condorelli, A.G.; Dellambra, E.; Logli, E.; Zambruno, G.; Castiglia, D. Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives. Int. J. Mol. Sci. 2019, 20, 5707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoste, E.; Arwert, E.N.; Lal, R.; South, A.P.; Salas-Alanis, J.C.; Murrell, D.F.; Donati, G.; Watt, F.M. Innate sensing of microbial products promotes wound-induced skin cancer. Nat. Commun. 2015, 6, 5932. [Google Scholar] [CrossRef] [Green Version]
- Tamai, K.; Yamazaki, T.; Chino, T.; Ishii, M.; Otsuru, S.; Kikuchi, Y.; Iinuma, S.; Saga, K.; Nimura, K.; Shimbo, T.; et al. PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc. Natl. Acad. Sci. USA 2011, 108, 6609–6614. [Google Scholar] [CrossRef] [Green Version]
- Hoste, E.; Maueröder, C.; van Hove, L.; Catrysse, L.; Vikkula, H.K.; Sze, M.; Maes, B.; Karjosukarso, D.; Martens, L.; Gonçalves, A.; et al. Epithelial HMGB1 Delays Skin Wound Healing and Drives Tumor Initiation by Priming Neutrophils for NET Formation. Cell Rep. 2019, 29, 2689–2701.e4. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116 Pt 2, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Costanza, B.; Umelo, I.A.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal Modulators of TGF-β in Cancer. J. Clin. Med. 2017, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Cheng, D.S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Cunnington, R.H.; Gupta, S.; Yeganeh, B.; Filomeno, K.L.; Freed, D.H.; Chen, S.; Klonisch, T.; Halayko, A.J.; Ambrose, E.; et al. Autophagy is a regulator of TGF-β1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 2015, 6, e1696. [Google Scholar] [CrossRef] [Green Version]
- Lenna, S.; Trojanowska, M. The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr. Opin. Rheumatol. 2012, 24, 663–668. [Google Scholar] [CrossRef]
- Owusu, B.Y.; Zimmerman, K.A.; Murphy-Ullrich, J.E. The role of the endoplasmic reticulum protein calreticulin in mediating TGF-β-stimulated extracellular matrix production in fibrotic disease. J. Cell Commun. Signal. 2018, 12, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef]
- Jobling, R.; D’Souza, R.; Baker, N.; Lara-Corrales, I.; Mendoza-Londono, R.; Dupuis, L.; Savarirayan, R.; Ala-Kokko, L.; Kannu, P. The collagenopathies: Review of clinical phenotypes and molecular correlations. Curr. Rheumatol. Rep. 2014, 16, 394. [Google Scholar] [CrossRef] [PubMed]
- Omari, S.; Makareeva, E.; Roberts-Pilgrim, A.; Mirigian, L.; Jarnik, M.; Ott, C.; Lippincott-Schwartz, J.; Leikin, S. Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E10099–E10108. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, H.; Patel, S.B.; Pastar, I. The Role of TGFβ Signaling in Wound Epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal-Ponce, A.; Tiruneh, M.W.; Lee, J.; Guerrero-Juarez, C.F.; Kuhn, J.; David, J.A.; Dammeyer, K.; Mc Kell, R.; Kwong, J.; Rabbani, P.S.; et al. Keratinocyte-Macrophage Crosstalk by the Nrf2/Ccl2/EGF Signaling Axis Orchestrates Tissue Repair. Cell Rep. 2020, 33, 108417. [Google Scholar] [CrossRef] [PubMed]
- Chacón-Solano, E.; León, C.; Díaz, F.; García-García, F.; García, M.; Escámez, M.J.; Guerrero-Aspizua, S.; Conti, C.J.; Mencía, Á.; Martínez-Santamaría, L.; et al. Fibroblast activation and abnormal extracellular matrix remodelling as common hallmarks in three cancer-prone genodermatoses. Br. J. Dermatol. 2019, 181, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Cianfarani, F.; De Domenico, E.; Nyström, A.; Mastroeni, S.; Abeni, D.; Baldini, E.; Ulisse, S.; Uva, P.; Bruckner-Tuderman, L.; Zambruno, G.; et al. Decorin counteracts disease progression in mice with recessive dystrophic epidermolysis bullosa. Matrix Biol. 2019, 81, 3–16. [Google Scholar] [CrossRef]
- Suto, M.J.; Gupta, V.; Mathew, B.; Zhang, W.; Pallero, M.A.; Murphy-Ullrich, J.E. Identification of Inhibitors of Thrombospondin 1 Activation of TGF-β. ACS Med. Chem. Lett. 2020, 11, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [Green Version]
- Nyström, A.; Thriene, K.; Mittapalli, V.; Kern, J.S.; Kiritsi, D.; Dengjel, J.; Bruckner-Tuderman, L. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 2015, 7, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Aspizua, S.; González-Masa, A.; Conti, C.J.; García, M.; Chacón-Solano, E.; Larcher, F.; Del Río, M. Humanization of Tumor Stroma by Tissue Engineering as a Tool to Improve Squamous Cell Carcinoma Xenograft. Int. J. Mol. Sci. 2020, 21, 1951. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [CrossRef] [Green Version]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Shain, A.H. The landscape of driver mutations in cutaneous squamous cell carcinoma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Mandal, R.; Şenbabaoğlu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Parikh, A.S.; Tirosh, I. Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol. Cell. Oncol. 2018, 5, e1448244. [Google Scholar] [CrossRef]
- Dayal, J.H.S.; Mason, S.M.; Salas-Alanis, J.C.; McGrath, J.A.; Taylor, R.G.; Mellerio, J.E.; Blyth, K.; South, A.P.; Inman, G.J. Heterogeneous addiction to transforming growth factor-beta signalling in recessive dystrophic epidermolysis bullosa-associated cutaneous squamous cell carcinoma. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFβ receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [Green Version]
- Twaroski, K.; Chen, W.; Pickett-Leonard, M.; Tolar, J. Role of transforming growth factor-β1 in recessive dystrophic epidermolysis bullosa squamous cell carcinoma. Exp. Dermatol. 2021, 30, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Diociaiuti, A.; Steinke, H.; Nyström, A.; Schwieger-Briel, A.; Meiss, F.; Pfannenberg, C.; Bruckner-Tuderman, L.; Ruf, J.; De Vito, R.; El Hachem, M.; et al. EGFR inhibition for metastasized cutaneous squamous cell carcinoma in dystrophic epidermolysis bullosa. Orphanet J. Rare Dis. 2019, 14, 278. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Gorell, E.S.; Dehdashti, F.; Tang, J.Y.; Ansstas, G. Induced Remission of Metastatic Squamous Cell Carcinoma with an Immune Checkpoint Inhibitor in a Patient with Recessive Dystrophic Epidermolysis Bullosa. Case Rep. Oncol. 2020, 13, 911–915. [Google Scholar] [CrossRef]
- Chapman, C.M.; Sun, X.; Roschewski, M.; Aue, G.; Farooqui, M.; Stennett, L.; Gibellini, F.; Arthur, D.; Pérez-Galán, P.; Wiestner, A. ON 01910.Na is selectively cytotoxic for chronic lymphocytic leukemia cells through a dual mechanism of action involving PI3K/AKT inhibition and induction of oxidative stress. Clin. Cancer. Res. 2012, 18, 1979–1991. [Google Scholar] [CrossRef] [Green Version]
- Atanasova, V.S.; Pourreyron, C.; Farshchian, M.; Lawler, M.; Brown, C.A.t.; Watt, S.A.; Wright, S.; Warkala, M.; Guttmann-Gruber, C.; Hofbauer, J.P.; et al. Identification of Rigosertib for the Treatment of Recessive Dystrophic Epidermolysis Bullosa-Associated Squamous Cell Carcinoma. Clin. Cancer Res. 2019, 25, 3384–3391. [Google Scholar] [CrossRef] [Green Version]
- Jacków, J.; Rami, A.; Hayashi, R.; Hansen, C.; Guo, Z.; DeLorenzo, D.; Pappalardo, A.; Alvarez Cespedes, D.; Kim, A.L.; Perez-Lorenzo, R.; et al. Targeting the Jak/Signal Transducer and Activator of Transcription 3 Pathway with Ruxolitinib in a Mouse Model of Recessive Dystrophic Epidermolysis Bullosa-Squamous Cell Carcinoma. J. Investig. Dermatol. 2021, 141, 942–946. [Google Scholar] [CrossRef]
- Mittapalli, V.R.; Kühl, T.; Kuzet, S.E.; Gretzmeier, C.; Kiritsi, D.; Gaggioli, C.; Bruckner-Tuderman, L.; Nyström, A. STAT3 targeting in dystrophic epidermolysis bullosa. Br. J. Dermatol. 2020, 182, 1279–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Ivanova, L.; Zhu, H.; Plumer, T.; Hamby, C.; Mehta, B.; Gevertz, A.; Christiano, A.M.; McGrath, J.A.; Cairo, M.S. Cord Blood-Derived Stem Cells Suppress Fibrosis and May Prevent Malignant Progression in Recessive Dystrophic Epidermolysis Bullosa. Stem Cells 2018, 36, 1839–1850. [Google Scholar] [CrossRef] [Green Version]
- Nissinen, L.; Farshchian, M.; Riihilä, P.; Kähäri, V.M. New perspectives on role of tumor microenvironment in progression of cutaneous squamous cell carcinoma. Cell Tissue Res. 2016, 365, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Nissinen, L.; Kähäri, V.M. Matrix metalloproteinases in keratinocyte carcinomas. Exp. Dermatol. 2021, 30, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Hata, H.; Abe, R.; Suto, A.; Homma, E.; Fujita, Y.; Aoyagi, S.; Shimizu, H. MMP13 can be a useful differentiating marker between squamous cell carcinoma and benign hyperkeratotic lesions in recessive dystrophic epidermolysis bullosa. Br. J. Dermatol. 2015, 172, 769–773. [Google Scholar] [CrossRef]
- Ala-aho, R.; Ahonen, M.; George, S.J.; Heikkilä, J.; Grénman, R.; Kallajoki, M.; Kähäri, V.M. Targeted inhibition of human collagenase-3 (MMP-13) expression inhibits squamous cell carcinoma growth in vivo. Oncogene 2004, 23, 5111–5123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meides, A.; Gutschalk, C.M.; Devel, L.; Beau, F.; Czarny, B.; Hensler, S.; Neugebauer, J.; Dive, V.; Angel, P.; Mueller, M.M. Effects of selective MMP-13 inhibition in squamous cell carcinoma depend on estrogen. Int. J. Cancer 2014, 135, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Garcovich, S.; Colloca, G.; Sollena, P.; Andrea, B.; Balducci, L.; Cho, W.C.; Bernabei, R.; Peris, K. Skin Cancer Epidemics in the Elderly as An Emerging Issue in Geriatric Oncology. Aging Dis. 2017, 8, 643–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Potential Target | Underlying Mechanisms |
|---|---|
| TGFβ | Excessive TGFβ is the main driver of fibrosis via activating myofibroblasts, producing aberrant ECM and inhibiting ECM degradation |
| Decorin | Decorin potentially reduces TGFβ and fibrosis and is downregulated in RDEB skin |
| Thrombospondin-1 (TSP1) | TSP1 is an activator of TGFβ pathway and upregulated in RDEB fibroblasts |
| HMGB1 | HMGB1 is upregulated in RDEB, and inhibiting HMGB1 reduces inflammation |
| STAT3 | STAT3 is constitutively activated in RDEB-derived keratinocytes |
| Matrix Metalloproteinase (MMP) | Increased MMP expression is detected in RDEB skin leading to imbalanced synthesis-degradation in the ECM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tartaglia, G.; Cao, Q.; Padron, Z.M.; South, A.P. Impaired Wound Healing, Fibrosis, and Cancer: The Paradigm of Recessive Dystrophic Epidermolysis Bullosa. Int. J. Mol. Sci. 2021, 22, 5104. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105104
Tartaglia G, Cao Q, Padron ZM, South AP. Impaired Wound Healing, Fibrosis, and Cancer: The Paradigm of Recessive Dystrophic Epidermolysis Bullosa. International Journal of Molecular Sciences. 2021; 22(10):5104. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105104
Chicago/Turabian StyleTartaglia, Grace, Qingqing Cao, Zachary M. Padron, and Andrew P. South. 2021. "Impaired Wound Healing, Fibrosis, and Cancer: The Paradigm of Recessive Dystrophic Epidermolysis Bullosa" International Journal of Molecular Sciences 22, no. 10: 5104. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105104