
Glucose Metabolism in Burns—What Happens?

 ,
,
Abstract
:1. Introduction
2. Glucose Metabolism in Burns
- -
- decreased plasma volume due to extravasation into the burn wound and later into normal tissues because of vasodilation, increased blood flow, increased vascular permeability (produced by histamine, prostaglandin E2, prostacyclin, thromboxane A2, thromboxane B2, bradykinin, serotonin, reactive oxygen species (ROS)) [22,23,24];
- -
- -
- -
- -
- tachycardia and increased blood pressure due to the action of catecholamines and stimulation of the sympathetic nervous system [25];
- -
- peripheral insulin resistance [18];
- -
- increased glycolysis [5];
- -
- augmented glycogenolysis [5];
- -
- accentuated gluconeogenesis [5];
- -
- elevated lipolysis [5];
- -
- persistent proteolysis [5].
- -
- immunodeficiency and increased risk of systemic and/or burn wound infections;
- -
- delayed wound healing;
- -
- poor quality scars;
- -
- complicated graft taking.
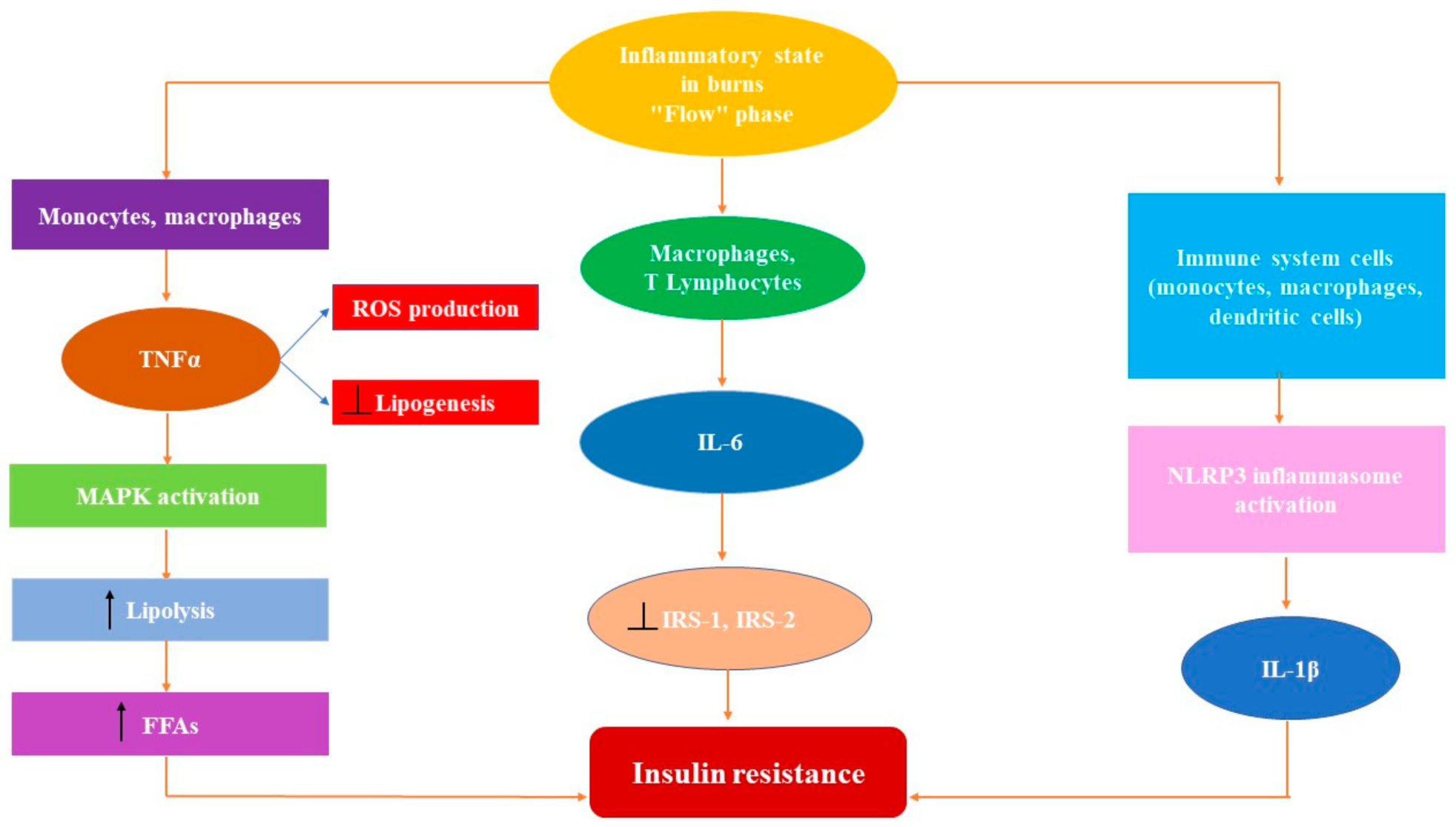
2.1. The Key Role of Proinflammatory Cytokines in Hypermetabolic Response
2.1.1. Tumor Necrosis Factorα
2.1.2. Interleukin 1β (IL-1β)
2.1.3. Interleukin 6 (IL-6)
2.2. Stress Crosstalk from Burns to Glucose Metabolism
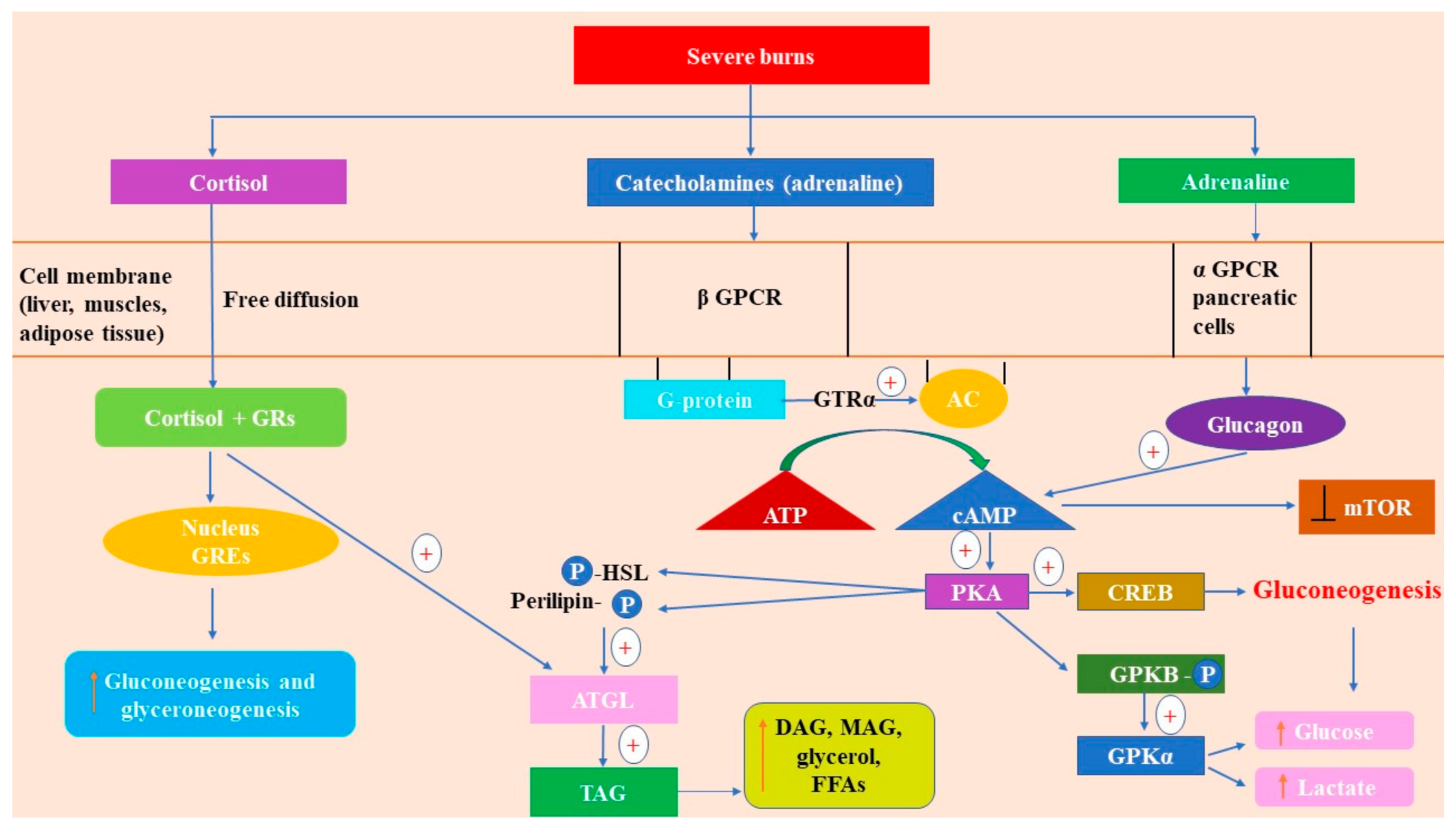
2.2.1. Catecholamines
2.2.2. Cortisol
2.3. How Glucagon and Insulin Modulate Glucose Metabolism in Burns
2.3.1. Glucagon
2.3.2. Insulin
Molecular Mechanisms of Insulin Action
Insulin Resistance—Signaling Pathway
3. Current Clinical Strategies in Managing Metabolism Status/Glucose Metabolism after Severe Burns
3.1. Maintaining the Core Temperature of the Patient
3.2. Sedation and Pain Control
3.3. Nutritional Support
3.4. Early Excision of Burn Wounds
3.5. Physical Exercise
3.6. Psychological Support
4. Pharmacologic Modulation of Glucose Metabolism in Burns
4.1. Insulin Therapy
4.2. Fenofibrate
4.3. Glucagon-Like Peptide-1 (GLP-1) and Analogs
4.4. Metformin
4.5. Sitagliptin
4.6. Recombinant Human Growth Hormone (rhGH)
4.7. Beta Blockers
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peck, M.D.; Kruger, G.E.; van der Merwe, A.E.; Godakumbura, W.; Ahuja, R.B. Burns and fires from non-electric domestic appliances in low and middle income countries Part I. The scope of the problem. Burns 2008, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Forjuoh, S.N. Burns in low-and middle-income contries: A review of available literature on descriptive epidemiology, risk factors, treatment, and prevention. Burns 2006, 32, 529–537. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2008. Available online: www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf (accessed on 2 February 2021).
- Herndon, D.N.; Tompkins, R.G. Support of the metabolic response to burn injury. Lancet 2004, 363, 1895–1902. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Mlcak, R.P.; Finnerty, C.C.; Norbury, W.B.; Gauglitz, G.G.; Kulp, G.A.; Herndon, D.N. Burn size determines the in-flammatory and hypermetabolic response. Crit. Care 2007, 11, R90. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, D.G. Management of Burns. N. Engl. J. Med. 2019, 380, 2349–2359. [Google Scholar] [CrossRef]
- Nielson, C.B.; Duethman, N.C.; Howard, J.M.; Moncure, M.; Wood, J.G. Burns: Pathophysiology of systemic complications and current management. J. Burn. Care Res. 2017, 38, e469–e481. [Google Scholar] [CrossRef]
- Finnerty, C.C.; Herndon, D.N.; Przkora, R.; Pereira, C.T.; Oliveira, H.M.; Queiroz, D.M.; Rocha, A.M.; Jeschke, M.G. Cytokine expression profile over time in severely burned pediatric patients. Shock 2006, 26, 13–19. [Google Scholar] [CrossRef]
- Farina, J.A., Jr.; Rosique, M.J.; Rosique, R.G. Curbing inflammation in burn patients. Int. J. Inflam. 2013, 715645. [Google Scholar] [CrossRef] [Green Version]
- Kallinen, O.; Maisniemi, K.; Böhling, T.; Tukiainen, E.; Koljonen, V. Multiple organ failure as a cause of death in patients with severe burns. J. Burn. Care Res. 2012, 33, 206–211. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Gauglitz, G.G.; Finnerty, C.C.; Kraft, R.; Mlcak, R.P.; Herndon, D.N. Survivors versus nonsurvivors postburn: Differences in inflammatory and hypermetabolic trajectories. Ann. Surg. 2014, 259, 814–823. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, M.G.; Gauglitz, G.G.; Kulp, G.A.; Finnerty, C.C.; Williams, F.N.; Kraft, R.; Suman, O.E.; Mlcak, R.P.; Herndon, D.N. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS ONE 2011, 6, e21245. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, M.G.; Chinkes, D.L.; Finnerty, C.C.; Kulp, G.; Suman, O.E.; Norbury, W.B.; Herndon, D.N. The pathophysiologic response to severe burn injury. Ann. Surg. 2008, 248, 387–401. [Google Scholar] [CrossRef] [Green Version]
- McCowen, K.C.; Malhotra, A.; Bistrian, B.R. Stress-induced hyperglycemia. Crit. Care Clin. 2001, 17, 107–124. [Google Scholar] [CrossRef]
- Herling, A.; König, M.; Bulik, S.; Holzhütter, H.G. Enzymatic features of the glucose metabolism in tumor cells. FEBS J. 2011, 278, 2436–2459. [Google Scholar] [CrossRef]
- Gearhart, M.M.; Parbhoo, S.K. Hyperglycemia in the critically ill patient. AACN Clin. Issues 2006, 17, 50–55. [Google Scholar] [CrossRef]
- Robinson, L.E.; van Soeren, M.H. Insulin resistance and hyperglycemia in critical illness: Role of insulin in glycemic control. AACN Clin. Issues 2004, 15, 45–62. [Google Scholar] [CrossRef]
- Auger, C.; Samadi, O.; Jeschke, M.G. The biochemical alterations underlying post-burn hypermetabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2633–2644. [Google Scholar] [CrossRef]
- Brooks, N.C.; Marshall, A.H.; Qa’aty, N.; Hiyama, Y.; Boehning, D.; Jeschke, M.G. XBP-1s is linked to suppressed gluconeogenesis in the Ebb phase of burn injury. Mol. Med. 2013, 19, 72–78. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Gauglitz, G.G.; Song, J.; Kulp, G.A.; Finnerty, C.C.; Cox, R.A.; Barral, J.M.; Herndon, D.N.; Boehning, D. Calcium and ER stress mediate hepatic apoptosis after burn injury. J. Cell Mol. Med. 2009, 13, 1857–1865. [Google Scholar] [CrossRef]
- Jeschke, M.G. The hepatic response to thermal injury: Is the liver important for post-burn outcomes? Mol. Med. 2009, 15, 337–351. [Google Scholar] [CrossRef]
- Aulick, L.H.; Wilmore, D.W.; Mason, A.D., Jr.; Pruitt, B.A., Jr. Influence of the burn wound on peripheral circulation in thermally injured patients. Am. J. Physiol. 1977, 233, H520–H526. [Google Scholar] [CrossRef]
- Baxter, C.R. Fluid volume and electrolyte changes of the early postburn period. Clin. Plast. Surg. 1974, 1, 693–703. [Google Scholar] [CrossRef]
- Lund, T.; Reed, R.K. Acute hemodynamic effects of thermal skin injury in the rat. Circ. Shock 1986, 20, 105–114. [Google Scholar]
- Wolfe, R.R. Review: Acute versus chronic response to burn injury. Circ. Shock 1981, 8, 105–115. [Google Scholar]
- Newsome, T.W.; Mason, A.D., Jr.; Pruitt, B.A., Jr. Weight loss following thermal injury. Ann. Surg. 1973, 178, 215–227. [Google Scholar] [CrossRef]
- Gore, D.C.; Chinkes, D.; Heggers, J.; Herndon, D.N.; Wolf, S.E.; Desai, M. Association of hyperglycemia with increased mortality after severe burn injury. J. Trauma 2001, 51, 540–544. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Yamada, Y.; Endo, S.; Inada, K.; Nakae, H.; Nasu, W.; Taniguchi, S.; Ishikura, H.; Tanaka, T.; Wakabayashi, G.; Taki, K.; et al. Tumor necrosis factor-alpha and tumor necrosis factor receptor I, II levels in patients with severe burns. Burns 2000, 26, 239–244. [Google Scholar] [CrossRef]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryden, M.; Dicker, A.; van Harmelen, V.; Hauner, H.; Brunnberg, M.; Perbeck, L.; Lonnqvist, F.; Arner, P. Mapping of early signaling events in tumor necrosis factor-alpha -mediated lipolysis in human fat cells. J. Biol. Chem. 2002, 277, 1085–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Miles, P.D.; Ladd, C.M.; Ross, K.; Golub, T.R.; Olefsky, J.M.; Lodish, H.F. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-α: Implications for insulin resistance. Diabetes 2002, 51, 3176–3188. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammatory pathways and insulin action. Int. J. Obes. Relat. Metab. Disord. 2003, 27, S53–S55. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Sim, Y.B.; Park, S.H.; Kang, Y.J.; Jung, J.S.; Ryu, O.H.; Choi, M.G.; Choi, S.S.; Suh, H.W. Interleukin-1β (IL-1β) increases pain behavior and the blood glucose level: Possible involvement of glucocorticoid system. Cytokine 2013, 64, 351–356. [Google Scholar] [CrossRef]
- Sirivelu, M.P.; MohanKumar, P.S.; MohanKumar, S.M. Interleukin-1 beta simultaneously affects the stress and reproductive axes by modulating norepinephrine levels in different brain areas. Life Sci. 2012, 91, 878–884. [Google Scholar] [CrossRef] [Green Version]
- Sapolsky, R.; Rivier, C.; Yamamoto, G.; Plotsky, P.; Vale, W. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science 1987, 238, 522–524. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Lin, W.L.; Shen, H.D.; Fang, R.H. Changes in circulating levels of interleukin 6 in burned patients. Burns 1999, 25, 131–136. [Google Scholar] [CrossRef]
- Wolsk, E.; Mygind, H.; Grøndahl, T.S.; Pedersen, B.K.; van Hall, G. IL-6 selectively stimulates fat metabolism in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E832–E840. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Glund, S.; Deshmukh, A.; Long, Y.C.; Moller, T.; Koistinen, H.A.; Caidahl, K.; Zierath, J.R.; Krook, A. Interleukin-6 directly increases glucose metabolism in resting human skeletal muscle. Diabetes 2007, 56, 1630–1637. [Google Scholar] [CrossRef] [Green Version]
- Dokken, B.B.; Sloniger, J.A.; Henriksen, E.J. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1188–E1194. [Google Scholar] [CrossRef]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell Biol. 2004, 24, 5434–5546, Erratum in: Mol. Cell Biol. 2005, 25, 8762. [Google Scholar] [CrossRef] [Green Version]
- Wallenius, V.; Wallenius, K.; Ahrén, B.; Rudling, M.; Carlsten, H.; Dickson, S.L.; Ohlsson, C.; Jansson, J.O. Interleukin-6-deficient mice develop mature-onset obesity. Nat. Med. 2002, 8, 75–79. [Google Scholar] [CrossRef]
- Campbell, I.L.; Erta, M.; Lim, S.L.; Frausto, R.; May, U.; Rose-John, S.; Scheller, J.; Hidalgo, J. Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J. Neurosci. 2014, 34, 2503–2513. [Google Scholar] [CrossRef] [Green Version]
- Mackiewicz, A.; Schooltink, H.; Heinrich, P.C.; Rose-John, S. Complex of soluble human IL-6-receptor/IL-6 up-regulates expression of acute-phase proteins. J. Immunol. 1992, 149, 2021–2027. [Google Scholar]
- Herndon, D.N.; Hart, D.W.; Wolf, S.E.; Chinkes, D.L.; Wolfe, R.R. Reversal of catabolism by beta-blockade after severe burns. N. Engl. J. Med. 2001, 345, 1223–1229. [Google Scholar] [CrossRef]
- Vega, G.L.; Baxter, C.R. Induction of hypertriglyceridemia in rabbits by thermal injury: I. Time course of elevated plasma triglyceride levels. J. Burn Care Rehabil. 1988, 9, 266–270. [Google Scholar] [CrossRef]
- Kulp, G.A.; Herndon, D.N.; Lee, J.O.; Suman, O.E.; Jeschke, M.G. Extent and magnitude of catecholamine surge in pediatric burned patients. Shock 2010, 33, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.V.; Accili, D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011, 14, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr 2006, 26, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.R. Pharmacogenetics of the human beta-adrenergic receptors. Pharm. J. 2007, 7, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2014; pp. 832–843. [Google Scholar]
- Stouten, P.F.; Sander, C.; Wittinghofer, A.; Valencia, A. How does the switch II region of G-domains work? FEBS Lett. 1993, 320, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Aiston, S.; Hampson, L.; Gómez-Foix, A.M.; Guinovart, J.J.; Agius, L. Hepatic glycogen synthesis is highly sensitive to phosphorylase activity: Evidence from metabolic control analysis. J. Biol. Chem. 2001, 276, 23858–23866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobilka, B.K. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol. Sci. 2011, 32, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, U.D.L.; Cox, M.M. Lehninger Principles of Biochemistry, 6th ed.; WH Freeman & Co.: New York, NY, USA, 2012. [Google Scholar]
- Debuyser, A.; Drews, G.; Henquin, J.C. Adrenaline inhibition of insulin release: Role of the repolarization of the B cell membrane. Pflug. Arch. 1991, 419, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Gromada, J.; Bokvist, K.; Ding, W.G.; Barg, S.; Buschard, K.; Renström, E.; Rorsman, P. Adrenaline stimulates glucagon secretion in pancreatic A-cells by increasing the Ca2+ current and the number of granules close to the L-type Ca2+ channels. J. Gen. Physiol. 1997, 110, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef] [Green Version]
- Brasaemle, D.L.; Subramanian, V.; Garcia, A.; Marcinkiewicz, A.; Rothenberg, A. Perilipin A and the control of triacylglycerol metabolism. Mol. Cell Biochem. 2009, 326, 15–21. [Google Scholar] [CrossRef]
- Chaves, V.E.; Frasson, D.; Kawashita, N.H. Several agents and pathways regulate lipolysis in adipocytes. Biochimie 2011, 93, 1631–1640. [Google Scholar] [CrossRef]
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J. Biol. Chem. 2006, 281, 40236–40241. [Google Scholar] [CrossRef] [Green Version]
- Lehner, R.; Quiroga, A.D. Fatty Acid Handling in Mammalian Cells. In Biochemistry of Lipids, Lipoproteins and Membranes, 6th ed.; Ridgway, N.D., McLeod, R.S., Eds.; Elsevier: Boston, MA, USA, 2016; pp. 149–184. [Google Scholar] [CrossRef]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. Fat signals-lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Mullins, G.R.; Wang, L.; Raje, V.; Sherwood, S.G.; Grande, R.C.; Boroda, S.; Eaton, J.M.; Blancquaert, S.; Roger, P.P.; Leitinger, N.; et al. Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 17450–17455. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, C.; Radermacher, P.; Wepler, M.; Nußbaum, B. Non-hemodynamic effects of catecholamines. Shock 2017, 48, 390–400. [Google Scholar] [CrossRef]
- Whirledge, S.; DeFranco, D.B. Glucocorticoid signaling in health and disease: Insights from tissue-specific GR knockout mice. Endocrinology 2018, 159, 46–64. [Google Scholar] [CrossRef]
- Almawi, W.Y. Molecular mechanisms of glucocorticoid effects. Mod. Aspects Immunobiol. 2001, 2, 78–82. [Google Scholar]
- Buttgereit, F.; Straub, R.H.; Wehling, M.; Burmester, G.R. Glucocorticoids in the treatment of rheumatic diseases: An update on the mechanisms of action. Arthritis Rheum. 2004, 50, 3408–3417. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Croxtall, J.D.; Choudhury, Q.; Flower, R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000, 130, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Spies, C.M.; Schaumann, D.H.; Berki, T.; Mayer, K.; Jakstadt, M.; Huscher, D.; Wunder, C.; Burmester, G.R.; Radbruch, A.; Lauster, R.; et al. Membrane glucocorticoid receptors are down regulated by glucocorticoids in patients with systemic lupus erythematosus and use a caveolin-1-independent expression pathway. Ann. Rheum. Dis. 2006, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.; McQueen, A.; Chen, T.C.; Wang, J.C. Regulation of glucose homeostasis by glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar] [CrossRef]
- Hanson, R.W.; Reshef, L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu. Rev. Biochem. 1997, 66, 581–611. [Google Scholar] [CrossRef]
- Xu, C.; He, J.; Jiang, H.; Zu, L.; Zhai, W.; Pu, S.; Xu, G. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol. Endocrinol. 2009, 23, 1161–1170. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.; Harris, C.A.; Wang, J.C. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol. Cell Endocrinol. 2013, 380, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Coderre, L.; Srivastava, A.K.; Chiasson, J.L. Role of glucocorticoid in the regulation of glycogen metabolism in skeletal muscle. Am. J. Physiol. 1991, 260(Pt. 1), E927–E932. [Google Scholar] [CrossRef]
- Morgan, S.A.; Sherlock, M.; Gathercole, L.L.; Lavery, G.G.; Lenaghan, C.; Bujalska, I.J.; Laber, D.; Yu, A.; Convey, G.; Mayers, R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 2009, 58, 2506–2515. [Google Scholar] [CrossRef] [Green Version]
- Dimitriadis, G.; Leighton, B.; Parry-Billings, M.; Sasson, S.; Young, M.; Krause, U.; Bevan, S.; Piva, T.; Wegener, G.; Newsholme, E.A. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochem. J. 1997, 321, 707–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brillon, D.J.; Zheng, B.; Campbell, R.G.; Matthews, D.E. Effect of cortisol on energy expenditure and amino acid metabolism in humans. Am. J. Physiol. 1995, 268, E501–E513. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, R.R.; Herndon, D.N.; Jahoor, F.; Miyoshi, H.; Wolfe, M. Effect of severe burn injury on substrate cycling by glucose and fatty acids. N. Engl. J. Med. 1987, 317, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Harp, J.B.; Yancopoulos, G.D.; Gromada, J. Glucagon orchestrates stress-induced hyperglycaemia. Diabetes Obes. Metab. 2016, 18, 648–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.J.; Thombare, K.; Sarsenbayeva, A.; Kamble, P.G.; Almby, K.; Lundqvist, M.; Eriksson, J.W. Direct effects of glucagon on glucose uptake and lipolysis in human adipocytes. Mol. Cell Endocrinol. 2020, 503, 110696. [Google Scholar] [CrossRef]
- Kraft, G.; Coate, K.C.; Winnick, J.J.; Dardevet, D.; Donahue, E.P.; Cherrington, A.D.; Williams, P.E.; Moore, M.C. Glucagon’s effect on liver protein metabolism in vivo. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E263–E272. [Google Scholar] [CrossRef]
- Davies, A.O.; Lefkowitz, R.J. Regulation of beta-adrenergic receptors by steroid hormones. Annu. Rev. Physiol. 1984, 46, 119–130. [Google Scholar] [CrossRef]
- Taborsky, G.J., Jr. The physiology of glucagon. J. Diabetes Sci. Technol. 2010, 4, 1338–1344. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Runge, S.; Gram, C.; Brauner-Osborne, H.; Madsen, K.; Knudsen, L.B.; Wulff, B.S. Three distinct epitopes on the extracellular face of the glucagon receptor determine specificity for the glucagon amino terminus. J. Biol. Chem. 2003, 278, 28005–28010. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183, Erratum in: Nature 2001, 413, 652. [Google Scholar] [CrossRef]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. Transcriptional regulators of hepatic gluconeogenesis. Arch. Pharm. Res. 2013, 36, 189–200. [Google Scholar] [CrossRef]
- Castaño, J.G.; Nieto, A.; Felíu, J.E. Inactivation of phosphofructokinase by glucagon in rat hepatocytes. J. Biol. Chem. 1979, 254, 5576–5579. [Google Scholar] [CrossRef]
- Pilkis, S.J.; Claus, T.H. Hepatic gluconeogenesis/glycolysis: Regulation and structure/function relationships of substrate cycle enzymes. Annu. Rev. Nutr. 1991, 11, 465–515. [Google Scholar] [CrossRef]
- Gutiérrez-Rodelo, C.; Roura-Guiberna, A.; Olivares-Reyes, J.A. Mecanismos moleculares de la resistencia a la insulina: Una actualización [Molecular mechanisms of insulin resistance: An update]. Gac. Med. Mex. 2017, 153, 214–228. [Google Scholar]
- Saini, V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J. Diabetes 2010, 1, 68–75. [Google Scholar] [CrossRef]
- dos Santos, J.L.; de Araujo, S.S.; dos Santos Estevam, C.; Lima, C.A.; de Oliveira Carvalho, C.R.; Lima, F.B.; Marçal, A.C. Molecular mechanisms of muscle glucose uptake in response to resistance exercise: A review. J. Exer. Physiol. Online 2017, 20, 200–211. [Google Scholar]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef]
- Bunner, A.E.; Chandrasekera, P.C.; Barnard, N.D. Knockout mouse models of insulin signaling: Relevance past and future. World J. Diabetes 2014, 5, 146–159. [Google Scholar] [CrossRef]
- Kitamura, T.; Kahn, C.R.; Accili, D. Insulin receptor knockout mice. Annu. Rev. Physiol. 2003, 65, 313–332. [Google Scholar] [CrossRef]
- Kubota, T.; Kubota, N.; Kadowaki, T. Imbalanced insulin actions in obesity and type 2 diabetes: Key mouse models of insulin signaling pathway. Cell Metab. 2017, 25, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Besse-Patin, A.; Jeromson, S.; Levesque-Damphousse, P.; Secco, B.; Laplante, M.; Estall, J.L. PGC1A regulates the IRS1:IRS2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc. Natl. Acad. Sci. USA 2019, 116, 4285–4290. [Google Scholar] [CrossRef] [Green Version]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.W.; Garofalo, R.S. Positive and negative regulatory role of insulin receptor substrate 1 and 2 (IRS-1 and IRS-2) serine/threonine phosphorylation. Biochemistry 2002, 41, 7082–7091. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N. Effects of burn injury on insulin secretion and on sensitivity to insulin in the rat in vivo. Eur. J. Clin. Investig. 1975, 5, 331–337. [Google Scholar] [CrossRef]
- Cree, M.G.; Wolfe, R.R. Postburn trauma insulin resistance and fat metabolism. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E1–E9. [Google Scholar] [CrossRef]
- Gauglitz, G.G.; Herndon, D.N.; Kulp, G.A.; Meyer III, W.J.; Jeschke, M.G. Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. J. Clin. Endocrinol. Metab. 2009, 94, 1656–1664. [Google Scholar] [CrossRef] [Green Version]
- Waller, A.P.; Kohler, K.; Burns, T.A.; Mudge, M.C.; Belknap, J.K.; Lacombe, V.A. Naturally occurring compensated insulin resistance selectively alters glucose transporters in visceral and subcutaneous adipose tissues without change in AS160 activation. Biochim. Biophys. Acta 2011, 1812, 1098–1103. [Google Scholar] [CrossRef]
- Biolo, G.; Fleming, R.Y.; Maggi, S.P.; Nguyen, T.T.; Herndon, D.N.; Wolfe, R.R. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J. Clin. Endocrinol. Metab. 2002, 87, 3378–3384. [Google Scholar] [CrossRef]
- Cree, M.G.; Aarsland, A.; Herndon, D.N.; Wolfe, R.R. Role of fat metabolism in burn trauma-induced skeletal muscle insulin resistance. Crit. Care Med. 2007, 35, S476–S483. [Google Scholar] [CrossRef]
- Jeschke, M.G.; Boehning, D.F.; Finnerty, C.C.; Herndon, D.N. Effect of insulin on the inflammatory and acute phase response after burn injury. Crit. Care Med. 2007, 35, S519–S523. [Google Scholar] [CrossRef]
- Gauglitz, G.G.; Herndon, D.N.; Jeschke, M.G. Insulin resistance postburn: Underlying mechanisms and current therapeutic strategies. J. Burn Care Res. 2008, 29, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Furnary, A.P.; Wu, Y.; Bookin, S.O. Effect of hyperglycemia and continuous intravenous insulin infusions on outcomes of cardiac surgical procedures: The Portland Diabetic Project. Endocr. Pract. 2004, 10, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Aronson, D. Hyperglycemia and the pathobiology of diabetic complications. Adv. Cardiol. 2008, 45, 1–16. [Google Scholar] [CrossRef]
- Southern, C.; Schulster, D.; Green, I.C. Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an L-arginine-dependent nitric oxide generating mechanism. FEBS Lett. 1990, 276, 42–44. [Google Scholar] [CrossRef] [Green Version]
- Gremlich, S.; Bonny, C.; Waeber, G.; Thorens, B. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J. Biol. Chem. 1997, 272, 30261–30269. [Google Scholar] [CrossRef] [Green Version]
- Reimer, M.K.; Ahrén, B. Altered beta-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57BL/6J mice. Diabetes 2002, 51, S138–S143. [Google Scholar] [CrossRef] [Green Version]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Investig. 1995, 96, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflug. Arch. 2020, 472, 1249–1272. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Chen, M.Z.; Olefsky, J.M.; Marth, J.D. Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat. Med. 2011, 17, 1067–1075. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Le Roith, D.; Zick, Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 2001, 24, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Krycer, J.R.; Kearney, A.L.; Hocking, S.L.; James, D.E. Muscle and adipose tissue insulin resistance: Malady without mechanism? J. Lipid Res. 2019, 60, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Yazıcı, D.; Sezer, H. Insulin resistance, obesity and lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Matulewicz, N.; Karczewska-Kupczewska, M. Insulin resistance and chronic inflammation. Postepy. Hig. Med. Dosw. 2016, 70, 1245–1258. [Google Scholar]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Engin, A.B. What is lipotoxicity? Adv. Exp. Med. Biol. 2017, 96, 197–220. [Google Scholar] [CrossRef]
- Szendroedi, J.; Frossard, M.; Klein, N.; Bieglmayer, C.; Wagner, O.; Pacini, G.; Decker, J.; Nowotny, P.; Müller, M.; Roden, M. Lipid-induced insulin resistance is not mediated by impaired transcapillary transport of insulin and glucose in humans. Diabetes 2012, 61, 3176–3180. [Google Scholar] [CrossRef] [Green Version]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- El-Sonbaty, Y.A.; Suddek, G.M.; Megahed, N.; Gameil, N.M. Protocatechuic acid exhibits hepatoprotective, vasculoprotective, antioxidant and insulin-like effects in dexamethasone-induced insulin-resistant rats. Biochimie 2019, 167, 119–134. [Google Scholar] [CrossRef]
- Mutlur Krishnamoorthy, R.; Carani Venkatraman, A. Polyphenols activate energy sensing network in insulin resistant models. Chem. Biol. Interact. 2017, 275, 95–107. [Google Scholar] [CrossRef]
- Lima, M.H.; Ueno, M.; Thirone, A.C.; Rocha, E.M.; Carvalho, C.R.; Saad, M.J. Regulation of IRS-1/SHP2 interaction and AKT phosphorylation in animal models of insulin resistance. Endocrine 2002, 18, 1–12. [Google Scholar] [CrossRef]
- Zhang, H.; Ta, N.; Chen, P.; Wang, H. Erchen decoction and linguizhugan decoction ameliorate hepatic insulin resistance by inhibiting IRS-1Ser307 phosphorylation in vivo and in vitro. Evid. Based Complement. Alternat. Med. 2017, 2017, 1589871. [Google Scholar] [CrossRef]
- Wang, H.; Du, L.; Wu, T.; Yang, G.; Hu, W.; Wang, H.; Yang, M.; Liu, D.; Gu, H.F.; Zhu, Z.; et al. Circulating betatrophin is associated with insulin resistance in humans: Cross-sectional and interventional studies in vivo and in vitro. Oncotarget 2017, 8, 96604–96614. [Google Scholar] [CrossRef] [Green Version]
- Russo, B.; Picconi, F.; Malandrucco, I.; Frontoni, S. Flavonoids and insulin-resistance: From molecular evidences to clinical trials. Int. J. Mol. Sci. 2019, 20, 2061. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Yu, H.; Shi, X.; Warren, C.R.; Lotta, L.A.; Friesen, M.; Meissner, T.B.; Langenberg, C.; Wabitsch, M.; Wareham, N.; et al. Functional screening of candidate causal genes for insulin resistance in human preadipocytes and adipocytes. Circ. Res. 2020, 126, 330–346. [Google Scholar] [CrossRef]
- Lepretti, M.; Martucciello, S.; Burgos Aceves, M.A.; Putti, R.; Lionetti, L. Omega-3 fatty acids and insulin resistance: Focus on the regulation of mitochondria and endoplasmic reticulum stress. Nutrients 2018, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Biobaku, F.; Ghanim, H.; Batra, M.; Dandona, P. Macronutrient-mediated inflammation and oxidative stress: Relevance to insulin resistance, obesity, and atherogenesis. J. Clin. Endocrinol. Metab. 2019, 104, 6118–6128. [Google Scholar] [CrossRef]
- Cha, H.N.; Park, S.; Dan, Y.; Kim, J.R.; Park, S.Y. Peroxiredoxin2 deficiency aggravates aging-induced insulin resistance and delines muscle strength. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 147–154. [Google Scholar] [CrossRef]
- Xia, Q.S.; Lu, F.E.; Wu, F.; Huang, Z.Y.; Dong, H.; Xu, L.J.; Gong, J. New role for ceramide in hypoxia and insulin resistance. World J. Gastroenterol. 2020, 26, 2177–2186. [Google Scholar] [CrossRef]
- Corallo, J.P.; King, B.; Pizano, L.R.; Namias, N.; Schulman, C.I. Core warming of a burn pa-tient during excision to prevent hypothermia. Burns 2008, 34, 418–420. [Google Scholar] [CrossRef]
- Kelemen, J.J., III; Cioffi, W.G., Jr.; Mason, A.D., Jr.; Mozingo, D.W.; McManus, W.F.; Pruitt, B.A., Jr. Effect of ambient temperature on metabolic rate after thermal injury. Ann. Surg. 1996, 223, 406–412. [Google Scholar] [CrossRef]
- Griggs, C.; Goverman, J.; Bittner, E.A.; Levi, B. Sedation and pain management in burn patients. Clin. Plast. Surg. 2017, 44, 535–540. [Google Scholar] [CrossRef]
- Summer, G.J.; Puntillo, K.A.; Miaskowski, C.; Green, P.G.; Levine, J.D. Burn injury pain: The continuing challenge. J. Pain 2007, 8, 533–548. [Google Scholar] [CrossRef]
- Patterson, D.R.; Hofland, H.W.; Espey, K.; Sharar, S.; Nursing Committee of the International Society for Burn Injuries. Pain management. Burns 2004, 30, A10-5, Erratum in: Burns 2005, 31, 537. [Google Scholar] [CrossRef]
- Walker, J.; Maccallum, M.; Fischer, C.; Kopcha, R.; Saylors, R.; McCall, J. Sedation using dexmedetomidine in pediatric burn patients. J. Burn Care Res. 2006, 27, 206–210. [Google Scholar] [CrossRef]
- Faucher, L.; Furukawa, K. Practice guidelines for the management of pain. J. Burn Care Res. 2006, 27, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.; Williams, B.; Cramond, T. Successful use of gabapentin in acute pain management following burn injury: A case series. Pain Med. 2008, 9, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.K.; Hromatka, K.A.; Miller, K.R. Energy Expenditure and Protein Requirements Following Burn Injury. Nutr. Clin. Pract. 2019, 34, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.; Imran, J.; Madni, T.; Wolf, S.E. Nutrition and metabolism in burn patients. Burns Trauma 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- McClave, S.A.; Snider, H.L. Use of indirect calorimetry in clinical nutrition. Nutr. Clin. Pract. 1992, 7, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Trocki, O.; Dominioni, L.; Brackett, K.A.; Joffe, S.N.; Alexander, J.W. Mechanism of prevention of postburn hypermetabolism and catabolism by early enteral feeding. Ann. Surg. 1984, 200, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Garrel, D.R.; Razi, M.; Larivière, F.; Jobin, N.; Naman., N.; Emptoz-Bonneton, A.; Pugeat, M.M. Improved clinical status and length of care with low-fat nutrition support in burn patients. J. Parenter. Enteral. Nutr. 1995, 19, 482–491. [Google Scholar] [CrossRef]
- Hall, K.L.; Shahrokhi, S.; Jeschke, M.G. Enteral nutrition support in burn care: A review of current recommendations as instituted in the Ross Tilley Burn Centre. Nutrients 2012, 4, 1554–1565. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, A.F.; Losser, M.R.; Ichai, C.; Berger, M.M. ESPEN endorsed recommendations: Nutritional therapy in major burns. Clin. Nutr. 2013, 32, 497–502, Erratum in: Clin. Nutr. 2013, 32, 1083. [Google Scholar] [CrossRef]
- Vinha, P.P.; Martinez, E.Z.; Vannucchi, H.; Marchini, J.S.; Farina, J.A., Jr.; Jordao, A.A., Jr.; Cunha, S.F. Effect of acute thermal injury in status of serum vitamins, inflammatory markers, and oxidative stress markers: Preliminary data. J. Burn Care Res. 2013, 34, e87–e91. [Google Scholar] [CrossRef]
- Nordlund, M.J.; Pham, T.N.; Gibran, N.S. Micronutrients after burn injury: A review. J. Burn Care Res. 2014, 35, 121–133. [Google Scholar] [CrossRef]
- Berger, M.M.; Baines, M.; Raffoul, W.; Benathan, M.; Chiolero, R.L.; Reeves, C.; Revelly, J.P.; Cayeux, M.C.; Sénéchaud, I.; Shenkin, A. Trace element supplementation after major burns modulates antioxidant status and clinical course by way of increased tissue trace element concentrations. Am. J. Clin. Nutr. 2007, 85, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Hart, D.W.; Wolf, S.E.; Chinkes, D.L.; Beauford, R.B.; Mlcak, R.P.; Heggers, J.P.; Wolfe, R.R.; Herndon, D.N. Effects of early excision and aggressive enteral feeding on hypermetabolism, catabolism, and sepsis after severe burn. J. Trauma 2003, 54, 755–764. [Google Scholar] [CrossRef]
- Horton, J.W.; Sanders, B.; White, D.J.; Maass, D.L. The effects of early excision and grafting on myocardial inflammation and function after burn injury. J. Trauma 2006, 61, 1069–1077. [Google Scholar] [CrossRef]
- Wurzer, P.; Keil, H.; Branski, L.K.; Parvizi, D.; Clayton, R.P.; Finnerty, C.C.; Herndon, D.N.; Kamolz, L.P. The use of skin substitutes and burn care—A survey. J. Surg. Res. 2016, 201, 293–298. [Google Scholar] [CrossRef]
- Jackson, D.; Topley, E.; Cason, J.S.; Lowbury, E.J. Primary excision and grafting of large burns. Ann. Surg. 1960, 152, 167–189. [Google Scholar] [CrossRef]
- Flores, O.; Tyack, Z.; Stockton, K.; Ware, R.; Paratz, J.D. Exercise training for improving outcomes post-burns: A systematic review and meta-analysis. Clin. Rehabil. 2018, 32, 734–746. [Google Scholar] [CrossRef]
- Suman, O.E.; Mlcak, R.P.; Herndon, D.N. Effect of exercise training on pulmonary function in children with thermal injury. J. Burn Care Rehabil. 2002, 23, 288–293, Discussion 287. [Google Scholar] [CrossRef]
- Suman, O.E.; Spies, R.J.; Celis, M.M.; Mlcak, R.P.; Herndon, D.N. Effects of a 12-wk resistance exercise program on skeletal muscle strength in children with burn injuries. J. Appl. Physiol. 2001, 91, 1168–1175. [Google Scholar] [CrossRef]
- Porro, L.J.; Al-Mousawi, A.M.; Williams, F.; Herndon, D.N.; Mlcak, R.P.; Suman, O.E. Effects of propranolol and exercise training in children with severe burns. J. Pediatr. 2013, 62, 799–803.e1. [Google Scholar] [CrossRef] [Green Version]
- Cleary, M.; Visentin, D.C.; West, S.; Kornhaber, R. The importance of mental health considerations for critical care burns patients. J. Adv. Nurs. 2018, 74, 1233–1235. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, M.G. Clinical review: Glucose control in severely burned patients—Current best practice. Crit. Care 2013, 17, 232. [Google Scholar] [CrossRef] [Green Version]
- Ferrando, A.A.; Chinkes, D.L.; Wolf, S.E.; Matin, S.; Herndon, D.N.; Wolfe, R.R. A submaximal dose of insulin promotes net skeletal muscle protein synthesis in patients with severe burns. Ann. Surg. 1999, 229, 11–18. [Google Scholar] [CrossRef]
- Stanojcic, M.; Finnerty, C.C.; Jeschke, M.G. Anabolic and anticatabolic agents in critical care. Curr. Opin. Crit. Care 2016, 22, 325–331. [Google Scholar] [CrossRef]
- NICE-SUGAR Study Investigators; Finfer, S.; Chittock, D.R.; Su, S.Y.; Blair, D.; Foster, D.; Dhingra, V.; Bellomo, R.; Cook, D.; Dodek, P.; et al. Intensive versus conventional glucose control in critically ill patients. N. Engl. J. Med. 2009, 360, 1283–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.M.; Adanichkin, N.; Kurmis, R.; Munn, Z. Intensive insulin therapy, insulin sensitisers and insulin secretagogues for burns: A systematic review of effectiveness and safety. Burns 2018, 44, 1377–1394. [Google Scholar] [CrossRef]
- Nisr, R.B.; Affourtit, C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta 2014, 1837, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecott, G.A.; Al-Mousawi, A.M.; Gauglitz, G.G.; Herndon, D.N.; Jeschke, M.G. The role of hyperglycemia in burned patients: Evidence-based studies. Shock 2010, 33, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, F.N.; Herndon, D.N.; Jeschke, M.G. The hypermetabolic response to burn injury and interventions to modify this response. Clin. Plast. Surg. 2009, 36, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.E. The cytochrome P450 4 (CYP4) family. Gen. Pharmacol. 1997, 28, 351–359. [Google Scholar] [CrossRef]
- Cree, M.G.; Zwetsloot, J.J.; Herndon, D.N.; Qian, T.; Morio, B.; Fram, R.; Sanford, A.P.; Aarsland, A.; Wolfe, R.R. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann. Surg. 2007, 245, 214–221. [Google Scholar] [CrossRef]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Mecott, G.A.; Herndon, D.N.; Kulp, G.A.; Brooks, N.C.; Al-Mousawi, A.M.; Kraft, R.; Rivero, H.G.; Williams, F.N.; Branski, L.K.; Jeschke, M.G. The use of exenatide in severely burned pediatric patients. Crit. Care 2010, 14, R153. [Google Scholar] [CrossRef] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Gore, D.C.; Wolf, S.E.; Sanford, A.; Herndon, D.N.; Wolfe, R.R. Influence of metformin on glucose intolerance and muscle catabolism following severe burn injury. Ann. Surg. 2005, 241, 334–342. [Google Scholar] [CrossRef]
- Diaz, E.C.; Herndon, D.N.; Porter, C.; Sidossis, L.S.; Suman, O.E.; Børsheim, E. Effects of pharmacological interventions on muscle protein synthesis and breakdown in recovery from burns. Burns 2015, 41, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, M.G.; Abdullahi, A.; Burnett, M.; Rehou, S.; Stanojcic, M. Glucose control in severely burned patients using metformin: An interim safety and efficacy analysis of a phase II randomized controlled trial. Ann. Surg. 2016, 264, 518–527. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef]
- Wang, D.S.; Kusuhara, H.; Kato, Y.; Jonker, J.W.; Schinkel, A.H.; Sugiyama, Y. Involvement of organic cation transporter 1 in the lactic acidosis caused by metformin. Mol. Pharmacol. 2003, 63, 844–848. [Google Scholar] [CrossRef]
- Scott, L.J. Sitagliptin: A review in type 2 diabetes. Drugs 2017, 77, 209–224. [Google Scholar] [CrossRef]
- Pruskowski, K.A.; Shields, B.A.; Ainsworth, C.R.; Cancio, L.C. Evaluation of the use of sitagliptin for insulin resistance in burn patients. Int. J. Burns Trauma 2020, 10, 237–245. [Google Scholar]
- Aili Low, J.F.; Barrow, R.E.; Mittendorfer, B.; Jeschke, M.G.; Chinkes, D.L.; Herndon, D.N. The effect of short-term growth hormone treatment on growth and energy expenditure in burned children. Burns 2001, 27, 447–452. [Google Scholar] [CrossRef]
- Przkora, R.; Herndon, D.N.; Suman, O.E.; Jeschke, M.G.; Meyer, W.J.; Chinkes, D.L.; Mlcak, R.P.; Huang, T.; Barrow, R.E. Beneficial effects of extended growth hormone treatment after hospital discharge in pediatric burn patients. Ann. Surg. 2006, 243, 796–803. [Google Scholar] [CrossRef]
- Keane, J.; Tajouri, L.; Gray, B. The effect of growth hormone administration on the regulation of mitochondrial apoptosis in-vivo. Int. J. Mol. Sci. 2015, 16, 12753–12772. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, R.J.; Wolf, S.E.; Barrow, R.E.; Herndon, D.N. Growth hormone treatment in pediatric burns: A safe therapeutic approach. Ann. Surg. 1998, 228, 439–448. [Google Scholar] [CrossRef]
- Singh, K.P.; Prasad, R.; Chari, P.S.; Dash, R.J. Effect of growth hormone therapy in burn patients on conservative treatment. Burns 1998, 24, 733–738. [Google Scholar] [CrossRef]
- Takala, J.; Ruokonen, E.; Webster, N.R.; Nielsen, M.S.; Zandstra, D.F.; Vundelinckx, G.; Hinds, C.J. Increased mortality associated with growth hormone treatment in critically ill adults. N. Engl. J. Med. 1999, 341, 785–792. [Google Scholar] [CrossRef]
- Herndon, D.N.; Rodriguez, N.A.; Diaz, E.C.; Hegde, S.; Jennings, K.; Mlcak, R.P.; Suri, J.S.; Lee, J.O.; Williams, F.N.; Meyer, W.; et al. Long-term propranolol use in severely burned pediatric patients: A randomized controlled study. Ann. Surg. 2012, 256, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauglitz, G.G.; Williams, F.N.; Herndon, D.N.; Jeschke, M.G. Burns: Where are we standing with propranolol, oxandrolone, recombinant human growth hormone, and the new incretin analogs? Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barret, J.P.; Jeschke, M.G.; Herndon, D.N. Fatty infiltration of the liver in severely burned pediatric patients: Autopsy findings and clinical implications. J. Trauma 2001, 51, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.C.; Song, J.; Boehning, D.; Kraft, R.; Finnerty, C.C.; Herndon, D.N.; Jeschke, M.G. Propranolol improves impaired hepatic phosphatidylinositol 3-kinase/AKT signaling after burn injury. Mol. Med. 2012, 18, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.; Marchionni, N.; Pahor, M. Beta-blockers after acute myocardial infarction in elderly patients with diabetes mellitus: Time to reassess. Drugs Aging 2003, 20, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.A. Effects of beta-blockers on glucose and lipid metabolism. Curr. Med. Res. Opin. 2010, 26, 615–629. [Google Scholar] [CrossRef]
- Damnjanovic, I.; Velickovic-Radovanovic, R.; Kocić, R.; Zlatković-Guberinić, S.; Sokolović, D.; Đinđić, N. Influence of beta-blockers on insulin resistance in patients with diabetes mellitus type 2. Acta Med. Median. 2011, 50, 23–28. [Google Scholar] [CrossRef]
- Toda, N. Vasodilating beta-adrenoceptor blockers as cardiovascular therapeutics. Pharmacol. Ther. 2003, 100, 215–234. [Google Scholar] [CrossRef]
 ” shows an increase; “
” shows an increase; “  ” indicates an inhibitory effect.
” shows an increase; “ ” indicates an inhibitory effect.
” indicates an inhibitory effect.
” shows an increase; “ ” indicates an inhibitory effect.
 ” marks an increase; “
” marks an increase; “  ” shows an inhibitory effect; “
” shows an inhibitory effect; “  ” indicates stimulatory effects.
” marks an increase; “ ” shows an inhibitory effect; “ ” indicates stimulatory effects.
” indicates stimulatory effects.
” marks an increase; “ ” shows an inhibitory effect; “ ” indicates stimulatory effects.
 ” shows an inhibitory effect.
” shows an inhibitory effect.
” shows an inhibitory effect.
” shows an inhibitory effect.

{kind=link}
{kind=link}
{kind=link}
| “Ebb Phase” | “Flow Phase” | |
|---|---|---|
| Plasma volume | decreased | increased |
| Vascular resistance | increased | increased |
| Renal filtration | decreased | decreased |
| Cardiac output | decreased | increased |
| Tissue perfusion | decreased | decreased |
| Metabolism | hypometabolism | hypermetabolism |
| Mitochondrial dysfunction | initiated | accentuated |
| Endoplasmic reticulum stress | initiated | accentuated |
| Glycolysis | ecreased | increased |
| Proteolysis | decreased | increased |
| Lipolysis | decreased | increased |
| Thermogenesis | decreased | increased |
| Resting energy expenditure | decreased | increased |
| Insulin resistance | no | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badoiu, S.C.; Miricescu, D.; Stanescu-Spinu, I.-I.; Ripszky Totan, A.; Badoiu, S.E.; Costagliola, M.; Greabu, M. Glucose Metabolism in Burns—What Happens? Int. J. Mol. Sci. 2021, 22, 5159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105159
Badoiu SC, Miricescu D, Stanescu-Spinu I-I, Ripszky Totan A, Badoiu SE, Costagliola M, Greabu M. Glucose Metabolism in Burns—What Happens? International Journal of Molecular Sciences. 2021; 22(10):5159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105159
Chicago/Turabian StyleBadoiu, Silviu Constantin, Daniela Miricescu, Iulia-Ioana Stanescu-Spinu, Alexandra Ripszky Totan, Silvia Elena Badoiu, Michel Costagliola, and Maria Greabu. 2021. "Glucose Metabolism in Burns—What Happens?" International Journal of Molecular Sciences 22, no. 10: 5159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105159