
The Emerging Role of Neutrophils in the Pathogenesis of Thrombosis in COVID-19

 and
and 
Abstract
:1. Introduction
2. Neutrophils
3. Induction and Molecular Mechanisms of NET Formation
4. Neutrophils, NETs, and Endothelial Damage
5. Neutrophils, NETs, and Thromboinflammation
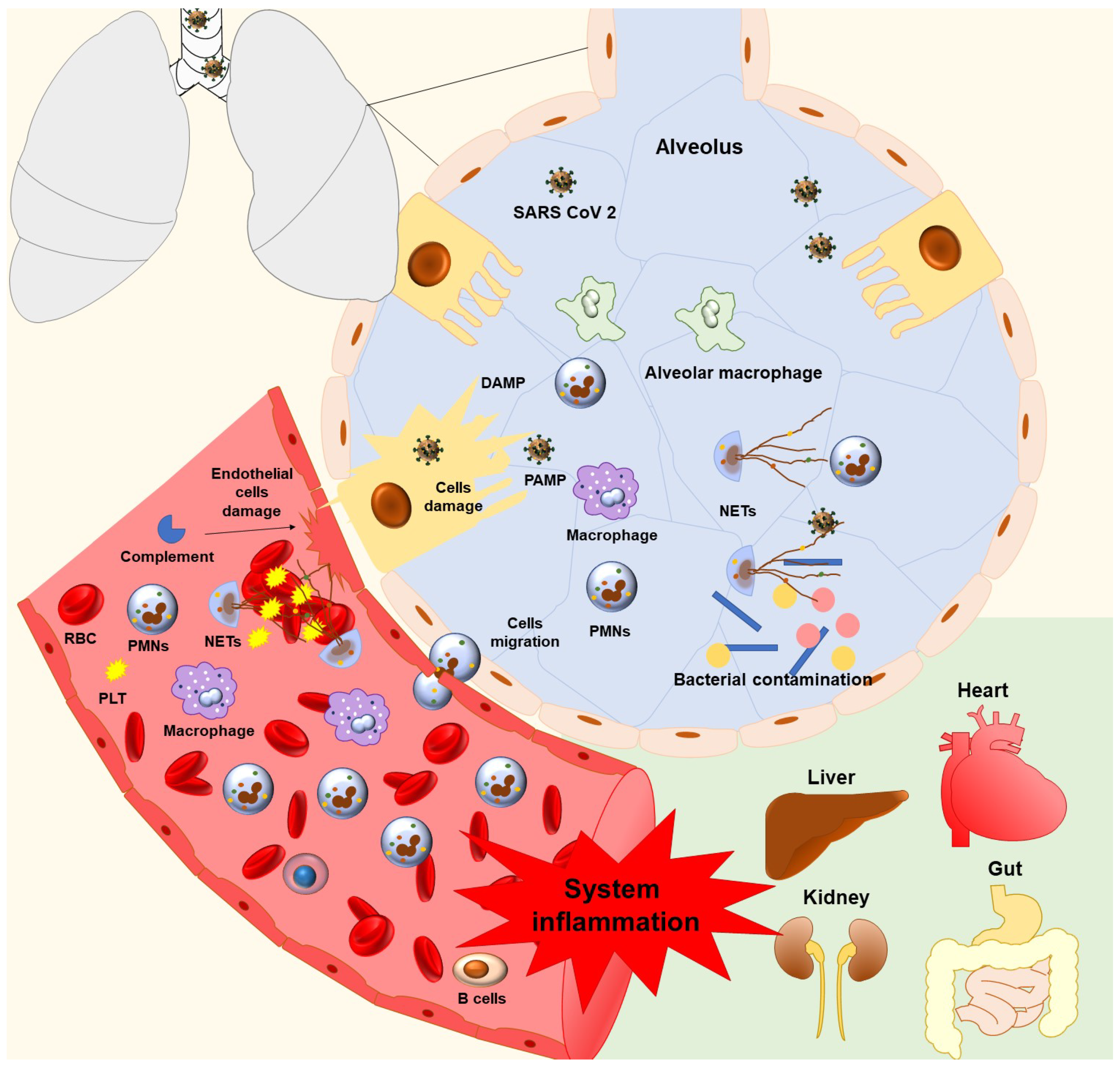
6. Neutrophils, NETs, and Lung Damage in COVID-19
7. Neutrophils, NETs, and Kidney Damage in COVID-19 Thromboinflammation
8. Neutrophils, NETs, and Kawasaki Disease
9. Neutrophils, NETs, and Therapeutical Interventions in COVID-19
9.1. Interleukin Targeting
9.2. Neutrophil Elastase Inhibitors
9.3. DNase Inhibitors
9.4. Colchicine
9.5. Corticosteroids
9.6. Other Therapeutic Intervention That Affects Neutrophils
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 28 March 2021).
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The Origin, Transmission and Clinical Therapies on Coronavirus Disease 2019 (COVID-19) Outbreak-an Update on the Status. Mil Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [Green Version]
- Ibañez, C.; Perdomo, J.; Calvo, A.; Ferrando, C.; Reverter, J.C.; Tassies, D.; Blasi, A. High D Dimers and Low Global Fibrinolysis Coexist in COVID19 Patients: What Is Going on in There? J. Thromb. Thrombolysis 2020, 51, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Arachchillage, D.R.J.; Laffan, M. Abnormal Coagulation Parameters Are Associated with Poor Prognosis in Patients with Novel Coronavirus Pneumonia. J. Thromb. Haemost. 2020, 18, 1233–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal Coagulation Parameters Are Associated with Poor Prognosis in Patients with Novel Coronavirus Pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Słomka, A.; Kowalewski, M.; Żekanowska, E. Coronavirus Disease 2019 (COVID–19): A Short Review on Hematological Manifestations. Pathogens 2020, 9, 493. [Google Scholar] [CrossRef]
- Eljilany, I.; Elzouki, A.-N. D-Dimer, Fibrinogen, and IL-6 in COVID-19 Patients with Suspected Venous Thromboembolism: A Narrative Review. VHRM 2020, 16, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of Natural Anticoagulant, Coagulant Factor and Anti-Phospholipid Antibody in Critically Ill COVID-19 Patients. J. Thromb. Thrombolysis 2020, 50, 580–586. [Google Scholar] [CrossRef]
- Gazzaruso, C.; Paolozzi, E.; Valenti, C.; Brocchetta, M.; Naldani, D.; Grignani, C.; Salvucci, F.; Marino, F.; Coppola, A.; Gallotti, P. Association between Antithrombin and Mortality in Patients with COVID-19. A Possible Link with Obesity. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1914–1919. [Google Scholar] [CrossRef] [PubMed]
- Gazzaruso, C.; Valenti, C.; Coppola, A.; Gallotti, P. Impact of Convalescent and Nonimmune Plasma on Mortality of Patients with COVID-19: A Potential Role for Antithrombin. Clin. Microbiol. Infect. 2020, 27, 637–638. [Google Scholar] [CrossRef]
- Lerman, Y.V.; Kim, M. Neutrophil Migration under Normal and Sepsis Conditions. Cardiovasc. Hematol. Disord. Drug Targets 2015, 15, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schymeinsky, J.; Mócsai, A.; Walzog, B. Neutrophil Activation via Beta2 Integrins (CD11/CD18): Molecular Mechanisms and Clinical Implications. Thromb. Haemost. 2007, 98, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Beneficial Suicide: Why Neutrophils Die to Make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, T.; Kambas, K.; Mitsios, A.; Panopoulou, M.; Tsironidou, V.; Dellaporta, E.; Kouklakis, G.; Arampatzioglou, A.; Angelidou, I.; Mitroulis, I.; et al. Immunomodulatory Role of Clarithromycin in Acinetobacter Baumannii Infection via Formation of Neutrophil Extracellular Traps. Antimicrob. Agents Chemother. 2016, 60, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Halverson, T.W.R.; Wilton, M.; Poon, K.K.H.; Petri, B.; Lewenza, S. DNA Is an Antimicrobial Component of Neutrophil Extracellular Traps. PLoS Pathog. 2015, 11, e1004593. [Google Scholar] [CrossRef] [Green Version]
- Martinod, K.; Deppermann, C. Immunothrombosis and Thromboinflammation in Host Defense and Disease. Platelets 2020, 32, 314–324. [Google Scholar] [CrossRef]
- Hidalgo, A. A NET-Thrombosis Axis in COVID-19. Blood 2020, 136, 1118–1119. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Liberale, L.; Carbone, F.; Vecchié, A.; Diaz-Cañestro, C.; Camici, G.G.; Montecucco, F.; Dallegri, F. The Pathophysiological Role of Neutrophil Extracellular Traps in Inflammatory Diseases. Thromb. Haemost. 2018, 118, 6–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, Z.; Long, Q.; Huang, J.; Hong, T.; Liu, W.; Lin, J. Insights Into Immunothrombosis: The Interplay Among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front. Immunol. 2020, 11, 610696. [Google Scholar] [CrossRef] [PubMed]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [Green Version]
- Yoo, D.; Floyd, M.; Winn, M.; Moskowitz, S.M.; Rada, B. NET Formation Induced by Pseudomonas Aeruginosa Cystic Fibrosis Isolates Measured as Release of Myeloperoxidase-DNA and Neutrophil Elastase-DNA Complexes. Immunol. Lett. 2014, 160, 186–194. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil Extracellular Traps Capture and Kill Candida Albicans Yeast and Hyphal Forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Al-Ghoul, W.M.; Kim, M.S.; Fazal, N.; Azim, A.C.; Ali, A. Evidence for Simvastatin Anti-Inflammatory Actions Based on Quantitative Analyses of NETosis and Other Inflammation/Oxidation Markers. Results Immunol. 2014, 4, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Jhunjhunwala, S.; Aresta-DaSilva, S.; Tang, K.; Alvarez, D.; Webber, M.J.; Tang, B.C.; Lavin, D.M.; Veiseh, O.; Doloff, J.C.; Bose, S.; et al. Neutrophil Responses to Sterile Implant Materials. PLoS ONE 2015, 10, e0137550. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET Formation by Gene Therapy in CGD Controls Aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Hagiwara, T.; Yamada, M. Nuclear Localization of Peptidylarginine Deiminase V and Histone Deimination in Granulocytes. J. Biol. Chem. 2002, 277, 49562–49568. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 Is Essential for Antibacterial Innate Immunity Mediated by Neutrophil Extracellular Traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil Extracellular Trap Formation Is Associated with IL-1β and Autophagy-Related Signaling in Gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Jin, C.; Ding, Z.; Zhu, Y.; He, Q.; Zhang, X.; Ai, R.; Yin, Y.; He, Y. TLR4 Regulates ROS and Autophagy to Control Neutrophil Extracellular Traps Formation against Streptococcus Pneumoniae in Acute Otitis Media. Pediatr. Res. 2020, 89, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhang, Y.; Yin, S.-W.; Gao, X.-J.; Shi, W.-W.; Wang, Y.; Huang, X.; Wang, L.; Zou, L.-Y.; Zhao, J.-H.; et al. Neutrophil Extracellular Trap Formation Is Associated with Autophagy-Related Signalling in ANCA-Associated Vasculitis. Clin. Exp. Immunol. 2015, 180, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Mazzoleni, V.; Zimmermann-Meisse, G.; Smirnova, A.; Tarassov, I.; Prévost, G. Staphylococcus Aureus Panton-Valentine Leukocidin Triggers an Alternative NETosis Process Targeting Mitochondria. FASEB J. 2020, 35, e21167. [Google Scholar] [CrossRef]
- Esmon, C.T.; Esmon, N.L. The Link between Vascular Features and Thrombosis. Annu. Rev. Physiol. 2011, 73, 503–514. [Google Scholar] [CrossRef]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial Dysfunction Contributes to COVID-19-Associated Vascular Inflammation and Coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.-P.; et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Iba, T.; Connors, J.M.; Levy, J.H. The Coagulopathy, Endotheliopathy, and Vasculitis of COVID-19. Inflamm. Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zuo, Y.; Gandhi, A.A.; Sule, G.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Wang, J.; Zuo, M.; Shi, Y.; et al. Endothelial Cell-Activating Antibodies in COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Evert, K.; Dienemann, T.; Brochhausen, C.; Lunz, D.; Lubnow, M.; Ritzka, M.; Keil, F.; Trummer, M.; Scheiter, A.; Salzberger, B.; et al. Autopsy Findings after Long-Term Treatment of COVID-19 Patients with Microbiological Correlation. Virchows Arch. 2021, 1–12. [Google Scholar] [CrossRef]
- Philippe, A.; Chocron, R.; Gendron, N.; Bory, O.; Beauvais, A.; Peron, N.; Khider, L.; Guerin, C.L.; Goudot, G.; Levasseur, F.; et al. Circulating Von Willebrand Factor and High Molecular Weight Multimers as Markers of Endothelial Injury Predict COVID-19 in-Hospital Mortality. Angiogenesis 2021, 1–13. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Pires da Silva, B.G.P.; Fonseca, B.A.L.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 Inhibition in Severe COVID-19: Early Clinical Findings Reveal Differential Biological Efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef]
- Egorina, E.M.; Sovershaev, M.A.; Olsen, J.O.; Østerud, B. Granulocytes Do Not Express but Acquire Monocyte-Derived Tissue Factor in Whole Blood: Evidence for a Direct Transfer. Blood 2008, 111, 1208–1216. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil Extracellular Traps Promote Deep Vein Thrombosis in Mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Kambas, K.; Markiewski, M.M.; Pneumatikos, I.A.; Rafail, S.S.; Theodorou, V.; Konstantonis, D.; Kourtzelis, I.; Doumas, M.N.; Magotti, P.; Deangelis, R.A.; et al. C5a and TNF-Alpha up-Regulate the Expression of Tissue Factor in Intra-Alveolar Neutrophils of Patients with the Acute Respiratory Distress Syndrome. J. Immunol. 2008, 180, 7368–7375. [Google Scholar] [CrossRef] [Green Version]
- Kambas, K.; Mitroulis, I.; Apostolidou, E.; Girod, A.; Chrysanthopoulou, A.; Pneumatikos, I.; Skendros, P.; Kourtzelis, I.; Koffa, M.; Kotsianidis, I.; et al. Autophagy Mediates the Delivery of Thrombogenic Tissue Factor to Neutrophil Extracellular Traps in Human Sepsis. PLoS ONE 2012, 7, e45427. [Google Scholar] [CrossRef]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu-Bercu, A.; Grassi, L.; Frontini, M.; Salles-Crawley, I.I.; Woollard, K.; Crawley, J.T. Activated AIIbβ3 on Platelets Mediates Flow-Dependent NETosis via SLC44A2. Elife 2020, 9, e53353. [Google Scholar] [CrossRef] [Green Version]
- Mitsios, A.; Chrysanthopoulou, A.; Arampatzioglou, A.; Angelidou, I.; Vidali, V.; Ritis, K.; Skendros, P.; Stakos, D. Ticagrelor Exerts Immune-Modulatory Effect by Attenuating Neutrophil Extracellular Traps. Int. J. Mol. Sci. 2020, 21, 3625. [Google Scholar] [CrossRef]
- Choi, S.H.; Smith, S.A.; Morrissey, J.H. Polyphosphate Is a Cofactor for the Activation of Factor XI by Thrombin. Blood 2011, 118, 6963–6970. [Google Scholar] [CrossRef] [Green Version]
- Sperling, C.; Fischer, M.; Maitz, M.F.; Werner, C. Neutrophil Extracellular Trap Formation upon Exposure of Hydrophobic Materials to Human Whole Blood Causes Thrombogenic Reactions. Biomater. Sci. 2017, 5, 1998–2008. [Google Scholar] [CrossRef]
- Winnersbach, P.; Rossaint, J.; Buhl, E.M.; Singh, S.; Lölsberg, J.; Wessling, M.; Rossaint, R.; Bleilevens, C. Platelet Count Reduction during in Vitro Membrane Oxygenation Affects Platelet Activation, Neutrophil Extracellular Trap Formation and Clot Stability, but Does Not Prevent Clotting. Perfusion 2021, 267659121989231. [Google Scholar] [CrossRef]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of Functional Tissue Factor by Neutrophil Extracellular Traps in Culprit Artery of Acute Myocardial Infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Kim, S.-W.; Lee, H.; Lee, H.-K.; Kim, I.-D.; Lee, J.-K. Neutrophil Extracellular Trap Induced by HMGB1 Exacerbates Damages in the Ischemic Brain. Acta Neuropathol. Commun. 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Li, T.; Jin, J.; Liu, Y.; Li, B.; Sun, Q.; Tian, J.; Zhao, H.; Liu, Z.; Ma, S.; et al. Interactions between Neutrophil Extracellular Traps and Activated Platelets Enhance Procoagulant Activity in Acute Stroke Patients with ICA Occlusion. EBioMedicine 2020, 53, 102671. [Google Scholar] [CrossRef] [PubMed]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/Autophagy Pathway Promotes Thromboinflammation and Fibrosis in Human Systemic Lupus Erythematosus (SLE) through NETs Decorated with Tissue Factor (TF) and Interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef]
- Gorham, J.; Moreau, A.; Corazza, F.; Peluso, L.; Ponthieux, F.; Talamonti, M.; Izzi, A.; Nagant, C.; Ndieugnou Djangang, N.; Garufi, A.; et al. Interleukine-6 in Critically Ill COVID-19 Patients: A Retrospective Analysis. PLoS ONE 2020, 15, e0244628. [Google Scholar] [CrossRef] [PubMed]
- Didangelos, A. COVID-19 Hyperinflammation: What about Neutrophils? mSphere 2020, 5, e00367. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and Treatment of Cytokine Storm in COVID-19. Theranostics 2021, 11, 316–329. [Google Scholar] [CrossRef]
- Borczuk, A.C. Pulmonary Pathology of COVID-19: A Review of Autopsy Studies. Curr. Opin. Pulm. Med. 2021, 27, 184–192. [Google Scholar] [CrossRef]
- Pandey, P.; Agarwal, S.R. Lung Pathology in COVID-19: A Systematic Review. Int. J. Appl. Basic Med. Res. 2020, 10, 226–233. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Ng, H.; Havervall, S.; Rosell, A.; Aguilera, K.; Parv, K.; von Meijenfeldt, F.A.; Lisman, T.; Mackman, N.; Thalin, C.; Phillipson, M. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients With COVID-19. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 988–994. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11, 2063. [Google Scholar] [CrossRef]
- Duarte-Neto, A.N.; Monteiro, R.A.A.; da Silva, L.F.F.; Malheiros, D.M.A.C.; de Oliveira, E.P.; Theodoro-Filho, J.; Pinho, J.R.R.; Gomes-Gouvêa, M.S.; Salles, A.P.M.; de Oliveira, I.R.S.; et al. Pulmonary and Systemic Involvement in COVID-19 Patients Assessed with Ultrasound-Guided Minimally Invasive Autopsy. Histopathology 2020, 77, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.; Hirschbühl, K.; Burkhardt, K.; Braun, G.; Trepel, M.; Märkl, B.; Claus, R. Postmortem Examination of Patients With COVID-19. JAMA 2020, 323, 2518–2520. [Google Scholar] [CrossRef] [PubMed]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; de Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral Presence and Immunopathology in Patients with Lethal COVID-19: A Prospective Autopsy Cohort Study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Sinha, P.; Calfee, C.S.; Cherian, S.; Brealey, D.; Cutler, S.; King, C.; Killick, C.; Richards, O.; Cheema, Y.; Bailey, C.; et al. Prevalence of Phenotypes of Acute Respiratory Distress Syndrome in Critically Ill Patients with COVID-19: A Prospective Observational Study. Lancet Respir. Med. 2020, 8, 1209–1218. [Google Scholar] [CrossRef]
- Calfee, C.S.; Janz, D.R.; Bernard, G.R.; May, A.K.; Kangelaris, K.N.; Matthay, M.A.; Ware, L.B. Distinct Molecular Phenotypes of Direct vs Indirect ARDS in Single-Center and Multicenter Studies. Chest 2015, 147, 1539–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, G.; Zhang, Z.; Peng, J.; Liu, L.; Zhang, C.; Yu, C.; Ma, Z.; Huang, Y.; Liu, W.; Yao, Y.; et al. Renal Involvement and Early Prognosis in Patients with COVID-19 Pneumonia. J. Am. Soc. Nephrol. 2020, 31, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Jing, H.; Wang, C.; Zhao, X.; Zhang, J.; Zhang, S.; Liu, H.; Xie, R.; Shi, J. COVID-19 Associated Thromboinflammation of Renal Capillary: Potential Mechanisms and Treatment. Am. J. Transl. Res. 2020, 12, 7640–7656. [Google Scholar]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney Disease Is Associated with In-Hospital Death of Patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A.B.; Nie, X.; et al. Renal Histopathological Analysis of 26 Postmortem Findings of Patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Pfister, F.; Vonbrunn, E.; Ries, T.; Jäck, H.-M.; Überla, K.; Lochnit, G.; Sheriff, A.; Herrmann, M.; Büttner-Herold, M.; Amann, K.; et al. Complement Activation in Kidneys of Patients With COVID-19. Front. Immunol. 2020, 11, 594849. [Google Scholar] [CrossRef]
- Kawasaki, T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi 1967, 16, 178–222. [Google Scholar] [PubMed]
- Ishii, M.; Ebato, T.; Kato, H. History and Future of Treatment for Acute Stage Kawasaki Disease. Korean Circ. J. 2020, 50, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Imada, Y.; Kawase, M.; Nakagaki, K.; Matsuyama, S.; Taguchi, F. Possible Involvement of Infection with Human Coronavirus 229E, but Not NL63, in Kawasaki Disease. J. Med. Virol. 2014, 86, 2146–2153. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Matsumoto, Y.; Ohya, M.; Harada, K.; Kanno, H. Histologic and Immunohistochemical Evaluation of Infiltrating Inflammatory Cells in Kawasaki Disease Arteritis Lesions. Appl. Immunohistochem. Mol. Morphol. 2021, 29, 62–67. [Google Scholar] [CrossRef]
- Armaroli, G.; Verweyen, E.; Pretzer, C.; Kessel, K.; Hirono, K.; Ichida, F.; Okabe, M.; Cabral, D.A.; Foell, D.; Brown, K.L.; et al. Monocyte-Derived Interleukin-1β As the Driver of S100A12-Induced Sterile Inflammatory Activation of Human Coronary Artery Endothelial Cells: Implications for the Pathogenesis of Kawasaki Disease. Arthritis Rheumatol. 2019, 71, 792–804. [Google Scholar] [CrossRef]
- Yamashita, K.; Takaori-Kondo, A.; Mizugishi, K. Exaggerated Neutrophil Extracellular Trap Formation in Kawasaki Disease: A Key Phenomenon behind the Outbreak in Western Countries? Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Pouletty, M.; Borocco, C.; Ouldali, N.; Caseris, M.; Basmaci, R.; Lachaume, N.; Bensaid, P.; Pichard, S.; Kouider, H.; Morelle, G.; et al. Paediatric Multisystem Inflammatory Syndrome Temporally Associated with SARS-CoV-2 Mimicking Kawasaki Disease (Kawa-COVID-19): A Multicentre Cohort. Ann. Rheum. Dis. 2020, 79, 999–1006. [Google Scholar] [CrossRef]
- Jing, Y.; Ding, M.; Fu, J.; Xiao, Y.; Chen, X.; Zhang, Q. Neutrophil Extracellular Trap from Kawasaki Disease Alter the Biologic Responses of PBMC. Biosci. Rep. 2020, 40, 40. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting Potential Drivers of COVID-19: Neutrophil Extracellular Traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Godement, M.; Zhu, J.; Cerf, C.; Vieillard-Baron, A.; Maillon, A.; Zuber, B.; Bardet, V.; Geri, G. Neutrophil Extracellular Traps in SARS-CoV2 Related Pneumonia in ICU Patients: The NETCOV2 Study. Front. Med. 2021, 8, 615984. [Google Scholar] [CrossRef]
- Li, Y.; Wan, D.; Luo, X.; Song, T.; Wang, Y.; Yu, Q.; Jiang, L.; Liao, R.; Zhao, W.; Su, B. Circulating Histones in Sepsis: Potential Outcome Predictors and Therapeutic Targets. Front Immunol. 2021, 12, 650184. [Google Scholar] [CrossRef]
- Komorowicz, E.; Balázs, N.; Tanka-Salamon, A.; Varga, Z.; Szabó, L.; Bóta, A.; Longstaff, C.; Kolev, K. Size- and Charge-Dependent Modulation of the Lytic Susceptibility and Mechanical Stability of Fibrin-Histone Clots by Heparin and Polyphosphate Variants. J. Thromb. Haemost. 2021, 19, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The Pro-Inflammatory Cytokines in COVID-19 Pathogenesis: What Goes Wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, G.; de Mast, Q.; Markou, N.; Theodorakopoulou, M.; Komnos, A.; Mouktaroudi, M.; Netea, M.G.; Spyridopoulos, T.; Verheggen, R.J.; Hoogerwerf, J.; et al. Favorable Anakinra Responses in Severe Covid-19 Patients with Secondary Hemophagocytic Lymphohistiocytosis. Cell Host Microbe 2020, 28, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Pasin, L.; Cavalli, G.; Navalesi, P.; Sella, N.; Landoni, G.; Yavorovskiy, A.G.; Likhvantsev, V.V.; Zangrillo, A.; Dagna, L.; Monti, G. Anakinra for Patients with COVID-19: A Meta-Analysis of Non-Randomized Cohort Studies. Eur. J. Intern. Med. 2021, 86, 34–40. [Google Scholar] [CrossRef]
- CORIMUNO-19 Collaborative group Effect of Anakinra versus Usual Care in Adults in Hospital with COVID-19 and Mild-to-Moderate Pneumonia (CORIMUNO-ANA-1): A Randomised Controlled Trial. Lancet Respir. Med. 2021, 9, 295–304. [CrossRef]
- De la Calle, C.; López-Medrano, F.; Pablos, J.L.; Lora-Tamayo, J.; Calle, G.M.; Sánchez-Fernández, M.; Fernández-Ruiz, M.; Asín, M.A.P.-J.; Caro-Teller, J.M.; García-García, R.; et al. Effectiveness of Anakinra for Tocilizumab-Refractory Severe COVID-19. A Single Centre Retrospective Comparative Study. Int. J. Infect. Dis. 2021, 105, 319–325. [Google Scholar] [CrossRef]
- Guaraldi, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in Patients with Severe COVID-19: A Retrospective Cohort Study. Lancet Rheumatol. 2020, 2, e474–e484. [Google Scholar] [CrossRef]
- Huang, E.; Isonaka, S.; Yang, H.; Salce, E.; Rosales, E.; Jordan, S.C. Tocilizumab Treatment in Critically Ill Patients with COVID-19: A Retrospective Observational Study. Int. J. Infect. Dis. 2021, 105, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Frigault, M.J.; Serling-Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 2020, 383, 2333–2344. [Google Scholar] [CrossRef]
- Mareev, V.Y.; Orlova, Y.A.; Pavlikova, E.P.; Akopyan, Z.A.; Matskeplishvili, S.T.; Plisyk, A.G.; Seredenina, E.M.; Potapenko, A.V.; Malakhov, P.S.; Samokhodskaya, L.M.; et al. [Proactive anti-inflammatory and anticoagulant therapy in the treatment of advanced stages of novel coronavirus infection (COVID-19). Case Series and Study Design: COLchicine versus ruxolitinib and secukinumab in open prospective randomIzed trial (COLORIT)]. Kardiologiia 2020, 60, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Mugheddu, C.; Sanna, S.; Atzori, L.; Rongioletti, F. Safety of Secukinumab Treatment in COVID-19 Affected Psoriatic Patients. Dermatol. Ther. 2020, 34, e14710. [Google Scholar] [CrossRef]
- Chiang, C.-C.; Korinek, M.; Cheng, W.-J.; Hwang, T.-L. Targeting Neutrophils to Treat Acute Respiratory Distress Syndrome in Coronavirus Disease. Front. Pharmacol. 2020, 11, 572009. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R. Anti-Protease Treatments Targeting Plasmin(Ogen) and Neutrophil Elastase May Be Beneficial in Fighting COVID-19. Physiol. Rev. 2020, 100, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Maki, C.; Inoue, Y.; Ishihara, T.; Hirano, Y.; Kondo, Y.; Sueyoshi, K.; Okamoto, K.; Tanaka, H. Evaluation of Appropriate Indications for the Use of Sivelestat Sodium in Acute Respiratory Distress Syndrome: A Retrospective Cohort Study. Acute Med. Surg. 2020, 7, e471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabata, K.; Hagio, T.; Matsuoka, S. The Role of Neutrophil Elastase in Acute Lung Injury. Eur. J. Pharmacol. 2002, 451, 1–10. [Google Scholar] [CrossRef]
- Ogura, Y.; Tajiri, K.; Murakoshi, N.; Xu, D.; Yonebayashi, S.; Li, S.; Okabe, Y.; Feng, D.; Shimoda, Y.; Song, Z.; et al. Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice. Int. J. Mol. Sci. 2021, 22, 722. [Google Scholar] [CrossRef]
- Sahebnasagh, A.; Saghafi, F.; Safdari, M.; Khataminia, M.; Sadremomtaz, A.; Talaei, Z.; Rezai Ghaleno, H.; Bagheri, M.; Habtemariam, S.; Avan, R. Neutrophil Elastase Inhibitor (Sivelestat) May Be a Promising Therapeutic Option for Management of Acute Lung Injury/Acute Respiratory Distress Syndrome or Disseminated Intravascular Coagulation in COVID-19. J. Clin. Pharm. Ther. 2020, 45, 1515–1519. [Google Scholar] [CrossRef]
- Zeiher, B.G.; Artigas, A.; Vincent, J.-L.; Dmitrienko, A.; Jackson, K.; Thompson, B.T.; Bernard, G.; STRIVE Study Group. Neutrophil Elastase Inhibition in Acute Lung Injury: Results of the STRIVE Study. Crit. Care Med. 2004, 32, 1695–1702. [Google Scholar] [CrossRef]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive Role of Neutrophil Extracellular Traps in Pathogen-Induced Lung Injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Avondt, K.V.; Maegdefessel, L.; Soehnlein, O. Therapeutic Targeting of Neutrophil Extracellular Traps in Atherogenic Inflammation. Thromb. Haemost. 2019, 119, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhongir, R.K.V.; Kasetty, G.; Papareddy, P.; Mörgelin, M.; Herwald, H.; Egesten, A. DNA-Fragmentation Is a Source of Bactericidal Activity against Pseudomonas Aeruginosa. Biochem. J. 2017, 474, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.G.; Chau, A.S.; Egeblad, M.; Barnes, B.J.; Janowitz, T. Nebulized In-Line Endotracheal Dornase Alfa and Albuterol Administered to Mechanically Ventilated COVID-19 Patients: A Case Series. Mol. Med. 2020, 26, 91. [Google Scholar] [CrossRef]
- Desilles, J.P.; Gregoire, C.; Le Cossec, C.; Lambert, J.; Mophawe, O.; Losser, M.R.; Lambiotte, F.; Le Tacon, S.; Cantier, M.; Engrand, N.; et al. Efficacy and Safety of Aerosolized Intra-Tracheal Dornase Alfa Administration in Patients with SARS-CoV-2-Induced Acute Respiratory Distress Syndrome (ARDS): A Structured Summary of a Study Protocol for a Randomised Controlled Trial. Trials 2020, 21, 548. [Google Scholar] [CrossRef]
- Mitroulis, I.; Papadopoulos, V.P.; Konstantinidis, T.; Ritis, K. Anakinra Suppresses Familial Mediterranean Fever Crises in a Colchicine-Resistant Patient. Neth. J. Med. 2008, 66, 489–491. [Google Scholar] [PubMed]
- Slobodnick, A.; Shah, B.; Krasnokutsky, S.; Pillinger, M.H. Update on Colchicine, 2017. Rheumatology 2018, 57, i4–i11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurup, R.; Galougahi, K.K.; Figtree, G.; Misra, A.; Patel, S. The Role of Colchicine in Atherosclerotic Cardiovascular Disease. Hear. Lung Circ. 2021, 30, 795–806. [Google Scholar] [CrossRef]
- Andreu, J.M.; Timasheff, S.N. Tubulin Bound to Colchicine Forms Polymers Different from Microtubules. Proc. Natl. Acad. Sci. USA 1982, 79, 6753–6756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, K.; Tucker, B.; Kurup, R.; Khandkar, C.; Pandzic, E.; Barraclough, J.; Machet, J.; Misra, A.; Kavurma, M.; Martinez, G.; et al. Colchicine Inhibits Neutrophil Extracellular Trap Formation in Patients with Acute Coronary Syndrome After Percutaneous Coronary Intervention. J. Am. Heart Assoc. 2021, 10, e018993. [Google Scholar] [CrossRef]
- Misra, D.P.; Gasparyan, A.Y.; Zimba, O. Benefits and Adverse Effects of Hydroxychloroquine, Methotrexate and Colchicine: Searching for Repurposable Drug Candidates. Rheumatol. Int. 2020, 40, 1741–1751. [Google Scholar] [CrossRef]
- Kow, C.S.; Hasan, S.S. Colchicine as an Adjunct to Heparin for Prophylaxis of Venous Thromboembolism in Patients with COVID-19. Rheumatol. Int. 2021, 41, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.G.; Giannopoulos, G.; Vrachatis, D.A.; Siasos, G.D.; Giotaki, S.G.; Gargalianos, P.; Metallidis, S.; Sianos, G.; Baltagiannis, S.; Panagopoulos, P.; et al. Effect of Colchicine vs Standard Care on Cardiac and Inflammatory Biomarkers and Clinical Outcomes in Patients Hospitalized with Coronavirus Disease 2019. JAMA Netw. Open 2020, 3, e2013136. [Google Scholar] [CrossRef]
- Scarsi, M.; Piantoni, S.; Colombo, E.; Airó, P.; Richini, D.; Miclini, M.; Bertasi, V.; Bianchi, M.; Bottone, D.; Civelli, P.; et al. Association between Treatment with Colchicine and Improved Survival in a Single-Centre Cohort of Adult Hospitalised Patients with COVID-19 Pneumonia and Acute Respiratory Distress Syndrome. Ann. Rheum. Dis. 2020, 79, 1286–1289. [Google Scholar] [CrossRef]
- Lopes, M.I.; Bonjorno, L.P.; Giannini, M.C.; Amaral, N.B.; Menezes, P.I.; Dib, S.M.; Gigante, S.L.; Benatti, M.N.; Rezek, U.C.; Emrich-Filho, L.L.; et al. Beneficial Effects of Colchicine for Moderate to Severe COVID-19: A Randomised, Double-Blinded, Placebo-Controlled Clinical Trial. RMD Open 2021, 7, e001455. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Bridges, L.; Shih, M.-C.; Marik, P.E.; Siemieniuk, R.A.C.; Kocak, M. Prolonged Glucocorticoid Treatment Is Associated with Improved ARDS Outcomes: Analysis of Individual Patients’ Data from Four Randomized Trials and Trial-Level Meta-Analysis of the Updated Literature. Intensive Care Med. 2016, 42, 829–840. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Mandourah, Y.; Al-Hameed, F.; Sindi, A.A.; Almekhlafi, G.A.; Hussein, M.A.; Jose, J.; Pinto, R.; Al-Omari, A.; Kharaba, A.; et al. Corticosteroid Therapy for Critically Ill Patients with Middle East Respiratory Syndrome. Am. J. Respir. Crit. Care Med. 2018, 197, 757–767. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.V.A.O.; et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19: The CoDEX Randomized Clinical Trial. JAMA 2020, 324, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Darbandi, A.; Asadi, A.; Ghanavati, R.; Afifirad, R.; Emamie, A.D.; Kakanj, M.; Talebi, M. The Effect of Probiotics on Respiratory Tract Infection with Special Emphasis on COVID-19: A Periodic Review during 2010-2020. Int. J. Infect. Dis. 2021, 105, 91–104. [Google Scholar] [CrossRef]
- Vong, L.; Lorentz, R.J.; Assa, A.; Glogauer, M.; Sherman, P.M. Probiotic Lactobacillus Rhamnosus Inhibits the Formation of Neutrophil Extracellular Traps. J. Immunol. 2014, 192, 1870–1877. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, K.; Rosołowski, M.; Kaniewska, M.; Kucha, P.; Meler, A.; Wierzba, W.; Rydzewska, G. Clostridioides Difficile Infection in Coronavirus Disease 2019 (COVID-19): An Underestimated Problem? Pol. Arch. Intern. Med. 2020, 131, 121–127. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Microorganisms | Cytokines/Chemokines |
|---|---|
| Bacteria | IL-8 |
| Escherichia coli | TNFα |
| Enterococcus faecalis | IFN-γ |
| Haemophilus influenzae | IFN-α |
| Shigella flexneri | GM-CSF |
| Staphylococcus aureus | GM-CSF + LPS |
| Streptococcus pyogenes | C5a |
| Streptococcus pneumoniae | Other inducers |
| Serratia marcescens | Activated Platelets |
| Pseudomonas aeruginosa | Drugs |
| Intracellular bacteria | Statins |
| Listeria monocytogenes | Antibiotics |
| Mycobacterium tuberculosis | MSU monosodium urate crystals |
| Fungi yeast | PMA |
| Aspergillus fumigatus | Sterile Implant Materials |
| Candida albicans | |
| Cryptococcus neoformans | |
| Parasites | |
| Toxoplasma gondii Leismania amazonensis | |
| Trypanosoma cruzi | |
| Viruses | |
| HIV-1 | |
| RSV—(respiratory syncytial virus) | |
| Influenza A | |
| SARS-CoV-2 |
| Model | Target | Authors | |
|---|---|---|---|
| Proteins | NET Detection Method | ||
| DVT (Animal) | MPO, TF | Immunofluorescence ELISA | Bril et al. [49] |
| STEMY (Human) | MPO, NE, TF | Immunofluorescence WB, ELISA | Stakos et al. [60] |
| Sepsis (Human) | MPO, NE, TF | Immunofluorescence ELISA | Kambas et al. [51] |
| Ischemic stroke (Animal) | Cit H3 HMGB-1 | Immunofluorescence WB | Kim et al. [61] |
| COVID-19 (Human) | Cit H3 | IHC, Immunofluorescence, WB | Leppkes et al. [52] |
| Ischemic stroke (Human) | Cit H3, MPO, NE, TF | Immunofluorescence ELISA | Zhou et al. [62] |
| SLE (Human) | REDD-1, MPO, NE, TF | Immunofluorescence ELISA | Frangou et al. [63] |
| Mechanisms | Target | Drug | |
|---|---|---|---|
| Proteins | Action | ||
| Inhibition of NET formation | PAD-4 | Inhibition of histone citrullination | PAD-4 inhibitor |
| NE | Inhibition of proteas activity | Sivelestat | |
| NF-κB | NF-κB signaling pathway inhibition | Aspirin | |
| HMGB-1 | HMGB-1-targeting | HMGB-1 inhibitors | |
| NETs dissolution | DNA | NET degradation | DNase Dornase alfa |
| DNA–Histone complex | NET degradation | Heparin | |
| NETs protein blocking | IL-1b | IL-1b receptor antagonist | Anakinra |
| Anti-IL-1b Abs | Canakinumab | ||
| Inhibition of IL-1b secretion | Colchicine | ||
| IL-17 | Anti-IL-17 Abs | Secukinumab | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iliadi, V.; Konstantinidou, I.; Aftzoglou, K.; Iliadis, S.; Konstantinidis, T.G.; Tsigalou, C. The Emerging Role of Neutrophils in the Pathogenesis of Thrombosis in COVID-19. Int. J. Mol. Sci. 2021, 22, 5368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105368
Iliadi V, Konstantinidou I, Aftzoglou K, Iliadis S, Konstantinidis TG, Tsigalou C. The Emerging Role of Neutrophils in the Pathogenesis of Thrombosis in COVID-19. International Journal of Molecular Sciences. 2021; 22(10):5368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105368
Chicago/Turabian StyleIliadi, Valeria, Ina Konstantinidou, Konstantina Aftzoglou, Sergios Iliadis, Theocharis G. Konstantinidis, and Christina Tsigalou. 2021. "The Emerging Role of Neutrophils in the Pathogenesis of Thrombosis in COVID-19" International Journal of Molecular Sciences 22, no. 10: 5368. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105368