
Transcriptome and Metabolomic Analyses Reveal Regulatory Networks Controlling Maize Stomatal Development in Response to Blue Light


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Gene Expression Profile in Light Treatments
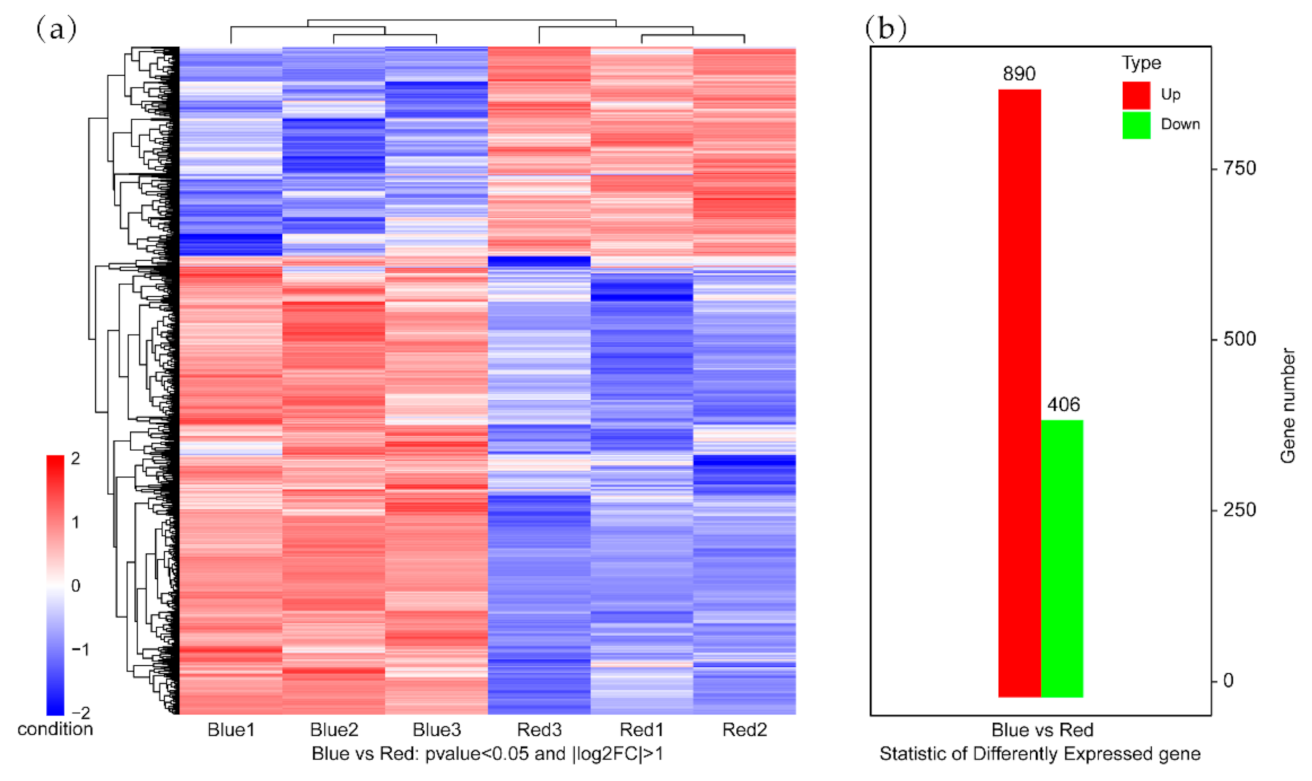
2.2. DEGs in Maize That Respond to Blue Light
2.3. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analyses of the DEGs
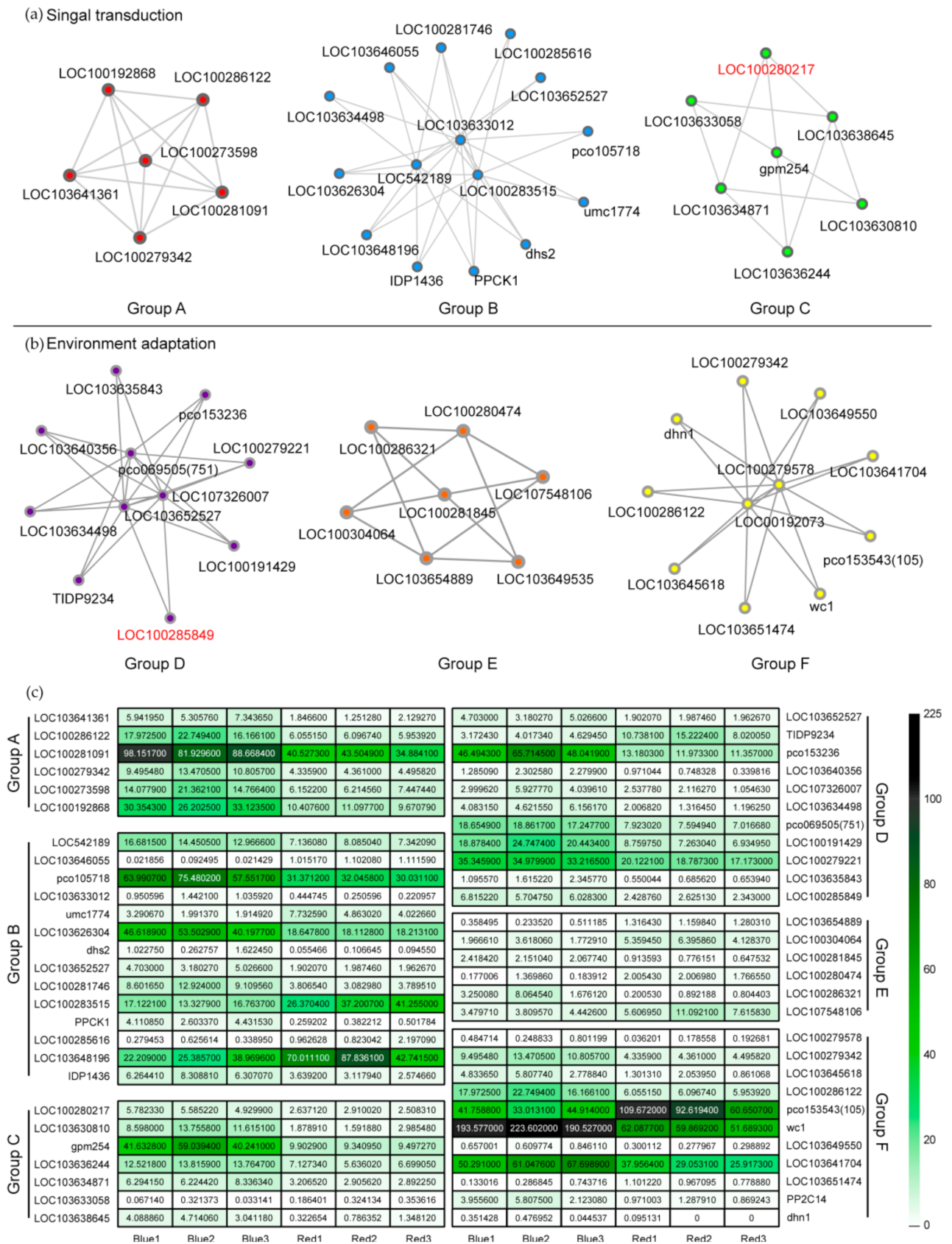
2.4. Protein–Protein Interaction (PPI) Network Analysis of Correlated DEGs and Identification of Potential Functional DEGs
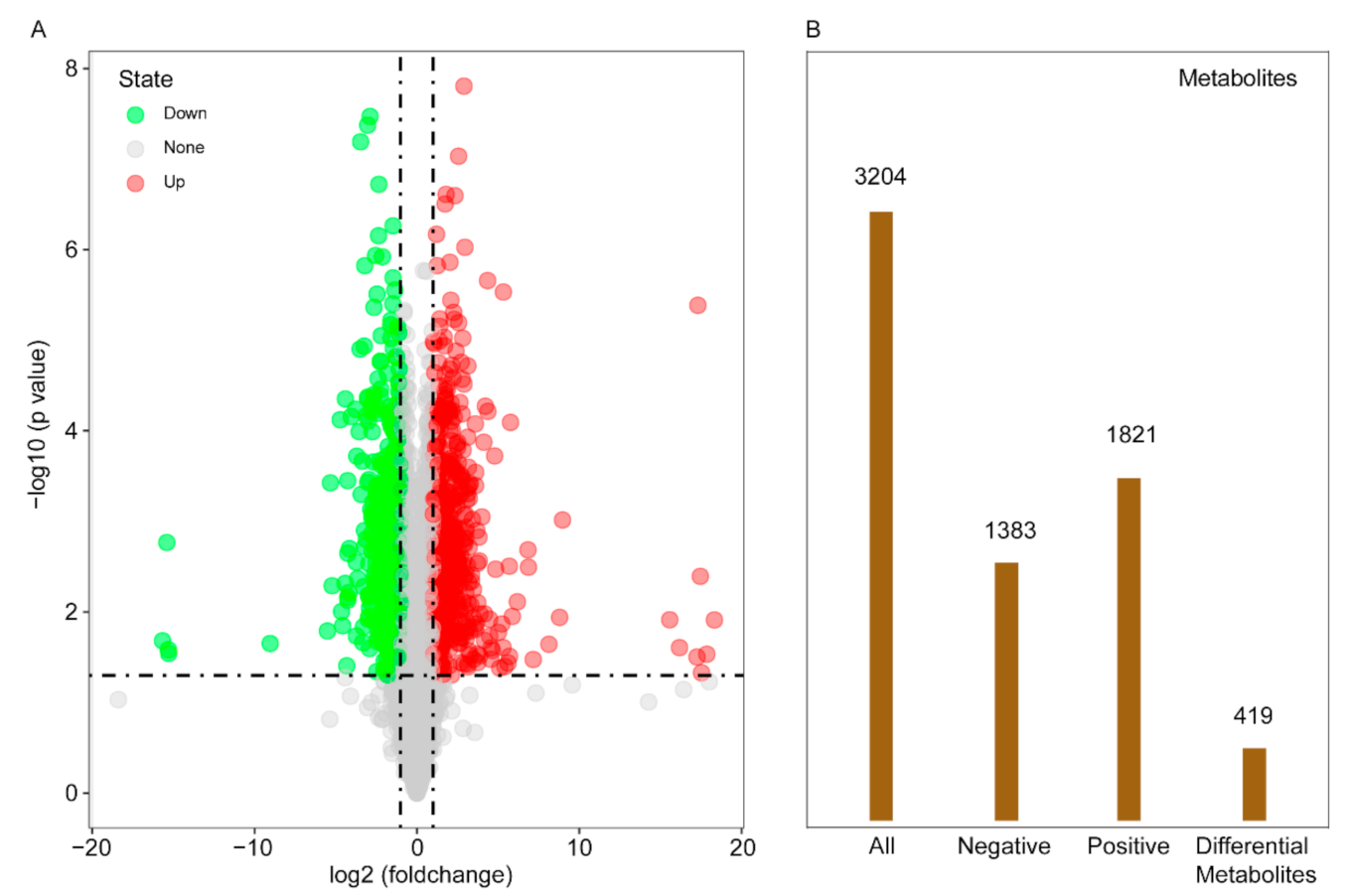
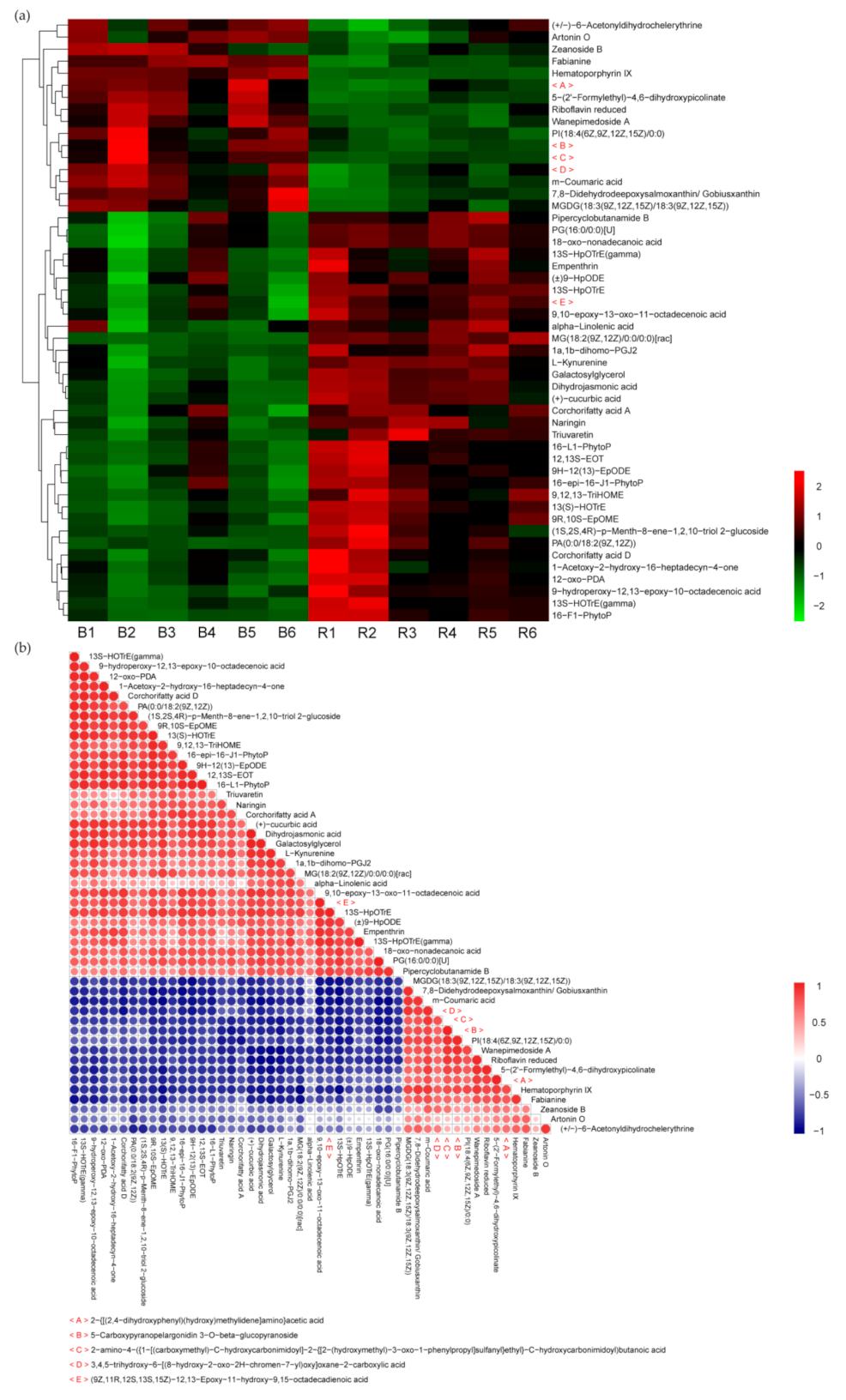
2.5. Metabolite Profile and Screening of Differential Metabolites in Maize in Response to Light Change
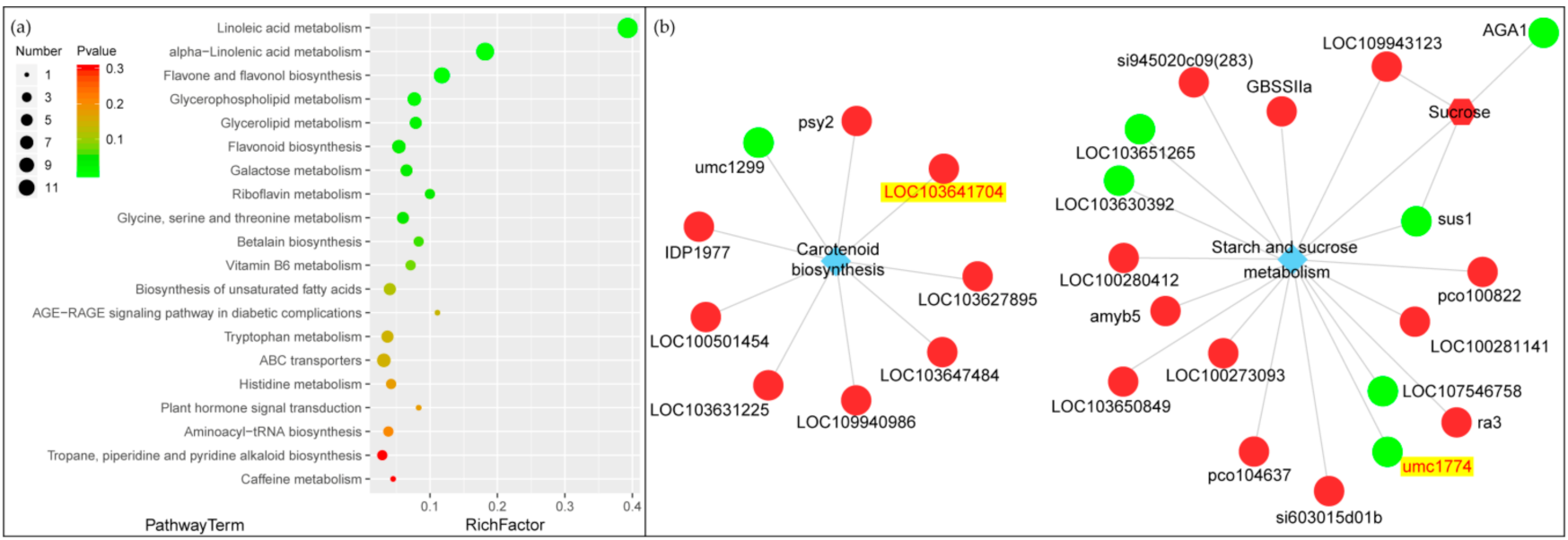
2.6. Effect of Light Change on Metabolic Pathways and Gene Screening in the Joint Analysis of the Transcriptome and Metabolome
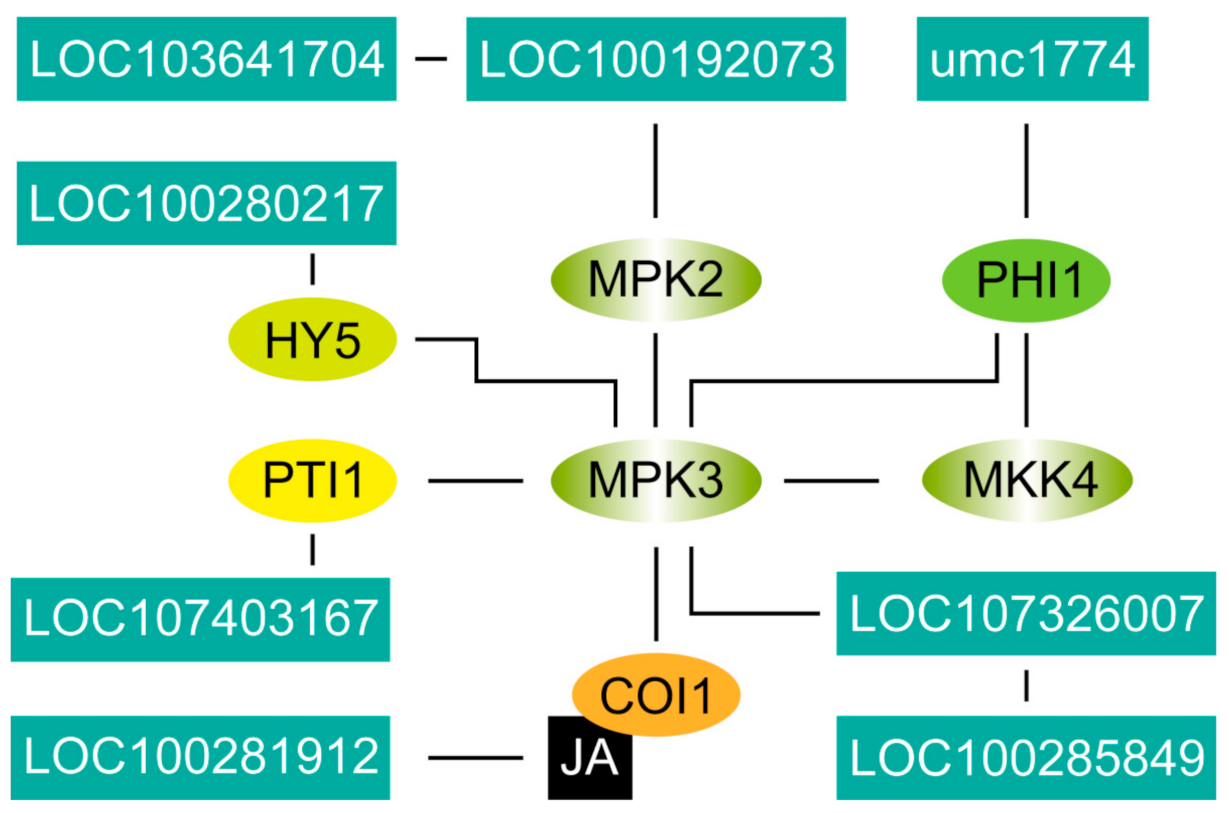
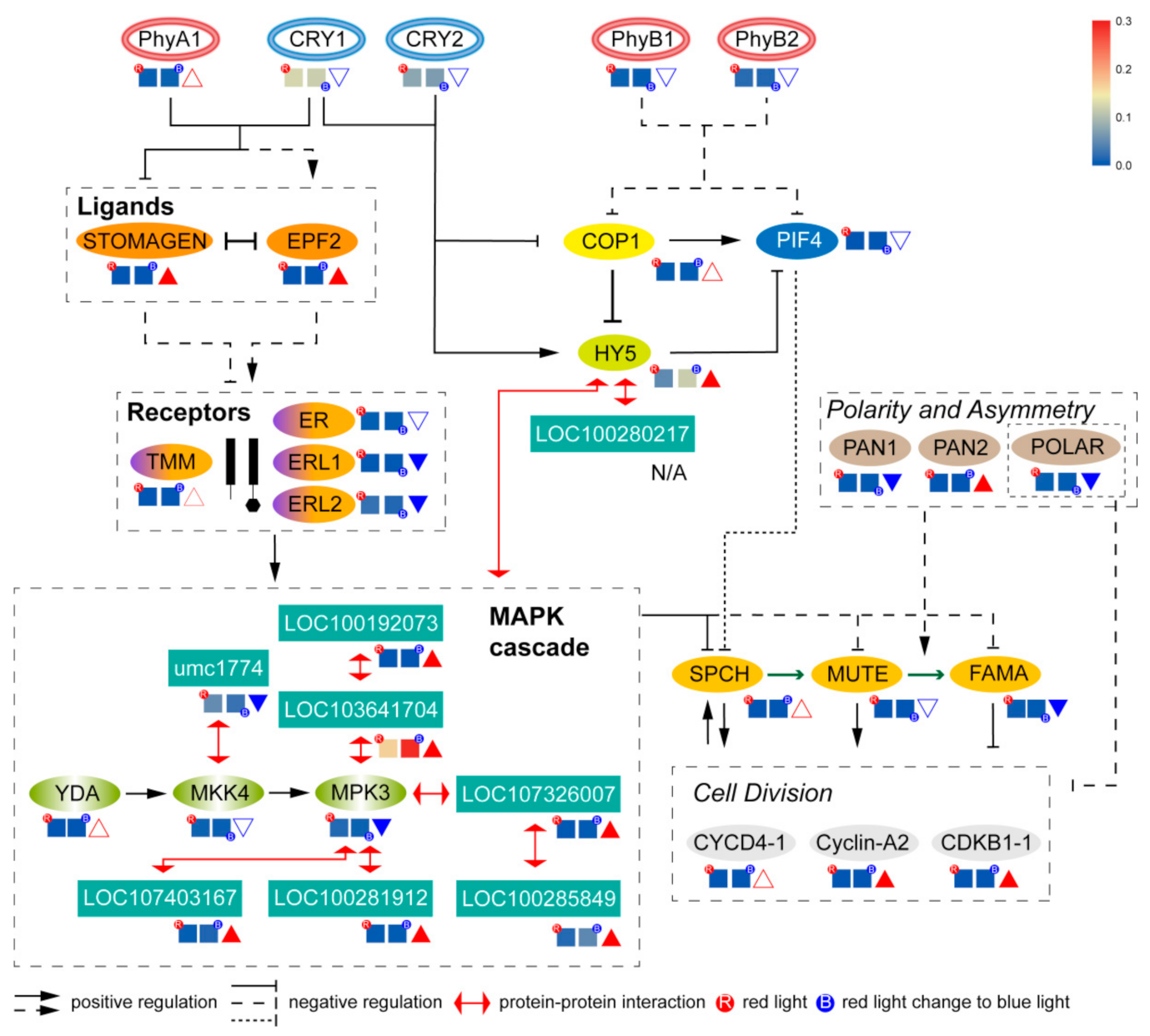
2.7. Location of Key Genes and Their Expression in the Network of Stomatal Development
3. Discussion
3.1. Effects of Blue Light on Transcription Expression and Metabolite Formation in Maize Leaves
3.2. Complement Network of Stomatal Development in Maize by PPI and KGML
3.3. Regulation of the Key Nodes of HY5 and MAPKs by Blue Light Was the Determining Factor for the Differences in Stomatal Development
3.4. Speculation of the Signaling Pathway of Blue Light Affecting the Stomatal Development of Maize
4. Materials and Methods
4.1. Plant Material and Growth Conditions
4.2. RNA Sequencing and Transcriptome Analysis
4.3. Metabolite Extraction and LC-MS Analysis
4.4. Data Processing and Differentially Accumulated Metabolite Identification
4.5. Gene Excavation and Location and RT-PCR Expression Analysis
4.6. Drawing and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qu, X.; Peterson, K.M.; Torii, K.U. Stomatal development in time: The past and the future. Curr. Opin. Genet. Dev. 2017, 45, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Kong, D.; Yang, J.; Wang, H. Light Regulation of Stomatal Development and Patterning: Shifting the Paradigm from Arabidopsis to Grasses. Plant Commun. 2020, 1, 100030. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, Q.; Deng, W.; Wang, X.; Piao, M.; Cai, D.; Li, Y.; Barshop, W.D.; Yu, X.; Zhou, T.; et al. Molecular basis for blue light-dependent phosphorylation of Arabidopsis cryptochrome 2. Nat. Commun. 2017, 8, 15234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Q.; Han, Y.J.; Liu, Q.; Gu, L.F.; Yang, Z.H.; Su, J.; Liu, B.B.; Zuo, Z.C.; He, W.J.; et al. A CRY-BIC negative-feedback circuitry regulating blue light sensitivity of Arabidopsis. Plant J. 2017, 92, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Martínez, C.; Nieto, C.; Prat, S. Convergent regulation of PIFs and the E3 ligase COP1/SPA1 mediates thermosensory hypocotyl elongation by plant phytochromes. Curr. Opin. Plant Biol. 2018, 45, 188–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.D.; Zhang, X.W.; Xu, Y. The effect of light quality on the expression of stomatal development genes of maize. Photosynthetica 2019, 57, 556–563. [Google Scholar] [CrossRef]
- Torii, K.U. Mix-and-match: Ligand–receptor pairs in stomatal development and beyond. Trends Plant Sci. 2012, 17, 711–719. [Google Scholar] [CrossRef]
- Sugano, S.S.; Shimada, T.; Imai, Y.; Okawa, K.; Tamai, A.; Mori, M.; Hara-Nishimura, I. Stomagen positively regulates stomatal density in Arabidopsis. Nature 2010, 463, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-K.; Torii, K.U. Linking cell cycle to stomatal differentiation. Curr. Opin. Plant Biol. 2019, 51, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.D.; Zhang, X.W.; Xu, Y. Influence of red light on the expression of genes on stomatal formation in maize seedlings. Can. J. Plant Sci. 2020, 100, 296–303. [Google Scholar] [CrossRef]
- González, C.V.; Prieto, J.A.; Mazza, C.; Jeréz, D.N.; Biruk, L.N.; Jofré, M.F.; Giordano, C.V. Grapevine morphological shade acclimation is mediated by light quality whereas hydraulic shade acclimation is mediated by light intensity. Plant Sci. 2021, 307, 110893. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhu, Z. Temporal and Spatial View of Jasmonate Signaling. Trends Plant Sci. 2017, 22, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Que, F.; Li, T.; Zhang, R.-R.; Khadr, A.; Xu, Z.-S.; Tian, Y.-S.; Xiong, A.-S. DcABF3, an ABF transcription factor from carrot, alters stomatal density and reduces ABA sensitivity in transgenic Arabidopsis. Plant Sci. 2021, 302, 110699. [Google Scholar] [CrossRef]
- Simmons, A.R.; Bergmann, D.C. Transcriptional control of cell fate in the stomatal lineage. Curr. Opin. Plant Biol. 2016, 29, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rasouli, F.; Kiani-Pouya, A.; Tahir, A.; Shabala, L.; Chen, Z.; Shabala, S. A comparative analysis of stomatal traits and photosynthetic responses in closely related halophytic and glycophytic species under saline conditions. Environ. Exp. Bot. 2021, 181, 104300. [Google Scholar] [CrossRef]
- Alakus, T.B.; Turkoglu, I. A novel entropy-based mapping method for determining the protein-protein interactions in viral genomes by using coevolution analysis. Biomed. Signal Process. Control 2021, 65, 102359. [Google Scholar] [CrossRef]
- Zhang, K.; Duan, L.; Lin, Z.; Sung, K.; Osakada, Y.; Cui, B. Light-Controlled Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway in Live Cells. Biophys. J. 2013, 104, 679a. [Google Scholar] [CrossRef] [Green Version]
- Janssens, V. Serine/Threonine Protein Phosphatases. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Duguay, J.; Jamal, S.; Liu, Z.; Wang, T.-W.; Thompson, J.E. Leaf-specific suppression of deoxyhypusine synthase in Arabidopsis thaliana enhances growth without negative pleiotropic effects. J. Plant Physiol. 2007, 164, 408–420. [Google Scholar] [CrossRef]
- Gaid, M.M.; Scharnhop, H.; Ramadan, H.; Beuerle, T.; Beerhues, L. 4-Coumarate:CoA ligase family members from elicitor-treated Sorbus aucuparia cell cultures. J. Plant Physiol. 2011, 168, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Balcerowicz, M.; Kerner, K.; Schenkel, C.; Hoecker, U. SPA Proteins Affect the Subcellular Localization of COP1 in the COP1/SPA Ubiquitin Ligase Complex during Photomorphogenesis. Plant Physiol. 2017, 174, 1314–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santella, L.; Kyozuka, K.; De Riso, L.; Carafoli, E. Calcium, protease action, and the regulation of the cell cycle. Cell Calcium 1998, 23, 123–130. [Google Scholar] [CrossRef]
- Assmann, S.M.; Jegla, T. Guard cell sensory systems: Recent insights on stomatal responses to light, abscisic acid, and CO2. Curr. Opin. Plant Biol. 2016, 33, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorente, B.; Martinez-Garcia, J.F.; Stange, C.; Rodriguez-Concepcion, M. Illuminating colors: Regulation of carotenoid biosynthesis and accumulation by light. Curr. Opin. Plant Biol. 2017, 37, 49–55. [Google Scholar] [CrossRef]
- Toledo-Ortiz, G.; Huq, E.; Rodríguez-Concepción, M. Direct regulation of phytoene synthase gene expression and carotenoid biosynthesis by phytochrome-interacting factors. Proc. Natl. Acad. Sci. USA 2010, 107, 11626–11631. [Google Scholar] [CrossRef] [Green Version]
- Stauder, R.; Welsch, R.; Camagna, M.; Kohlen, W.; Balcke, G.U.; Tissier, A.; Walter, M.H. Strigolactone Levels in Dicot Roots Are Determined by an Ancestral Symbiosis-Regulated Clade of the PHYTOENE SYNTHASE Gene Family. Front. Plant Sci. 2018, 9, 255. [Google Scholar] [CrossRef]
- Matusova, R.; Rani, K.; Verstappen, F.W.A.; Franssen, M.C.R.; Beale, M.H.; Bouwmeester, H.J. The strigolactone germination stimulants of the plant-parasitic Striga and Orobanche spp. are derived from the carotenoid pathway. Plant Physiol. 2005, 139, 920–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangappa, S.N.; Botto, J.F. The Multifaceted Roles of HY5 in Plant Growth and Development. Mol. Plant 2016, 9, 1353–1365. [Google Scholar] [CrossRef] [Green Version]
- Petersen, H.V.; Jensen, J.N.; Stein, R.; Serup, P. Glucose induced MAPK signalling influences NeuroD1-mediated activation and nuclear localization. FEBS Lett. 2002, 528, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Bi, X.; Tao, C.; Fei, Y.; Gao, S.; Liang, J.; Bai, W. Comparative transcriptomics analysis of Zygosaccharomyces mellis under high-glucose stress. Food Sci. Hum. Wellness 2020, 10, 54–62. [Google Scholar] [CrossRef]
- Lee, J.; He, K.; Stolc, V.; Lee, H.; Figueroa, P.; Gao, Y.; Tongprasit, W.; Zhao, H.Y.; Lee, I.; Deng, X. Analysis of transcription factor HY5 genomic binding sites revealed its hierarchical role in light regulation of development. Plant Cell 2007, 19, 731–749. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Z.; Liu, H.; Liu, B.; Liu, X.; Lin, C. Blue Light-Dependent Interaction of CRY2 with SPA1 Regulates COP1 activity and Floral Initiation in Arabidopsis. Curr. Biol. 2011, 21, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Ouyang, X.H.; Deng, X.W. Beyond repression of photomorphogenesis: Role switching of COP/DET/FUS in light signaling. Curr. Opin. Plant Biol. 2014, 21, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Xu, P.; He, S.; Wu, J.; Pan, J.; Wang, W.; Xu, F.; Wang, S.; Pan, J.; Huang, J.; et al. Photoexcited CRYPTOCHROME 1 Interacts Directly with G-Protein β Subunit AGB1 to Regulate the DNA-Binding Activity of HY5 and Photomorphogenesis in Arabidopsis. Mol. Plant 2018, 11, 1248–1263. [Google Scholar] [CrossRef] [Green Version]
- Osterlund, M.T.; Hardtke, C.S.; Wei, N.; Deng, X.W. Targeted destabilization of HY5 during light-regulated development of Arabidopsis. Nature 2000, 405, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Castells, E.; Molinier, J.; Drevensek, S.; Genschik, P.; Barneche, F.; Bowler, C. det1-1-induced UV-C hyposensitivity through UVR3 and PHR1 photolyase gene over-expression. Plant J. 2010, 63, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 2003, 103, 2203–2237. [Google Scholar] [CrossRef]
- Biever, J.J.; Gardner, G. The relationship between multiple UV-B perception mechanisms and DNA repair pathways in plants. Environ. Exp. Bot. 2016, 124, 89–99. [Google Scholar] [CrossRef]
- Li, H.; Ding, Y.; Shi, Y.; Zhang, X.; Zhang, S.; Gong, Z.; Yang, S. MPK3- and MPK6-Mediated ICE1 Phosphorylation Negatively Regulates ICE1 Stability and Freezing Tolerance in Arabidopsis. Dev. Cell 2017, 43, 630–642.e4. [Google Scholar] [CrossRef]
- Shao, Y.; Yu, X.; Xu, X.; Li, Y.; Yuan, W.; Xu, Y.; Mao, C.; Zhang, S.; Xu, J. The YDA-MKK4/MKK5-MPK3/MPK6 Cascade Functions Downstream of the RGF1-RGI Ligand–Receptor Pair in Regulating Mitotic Activity in Root Apical Meristem. Mol. Plant 2020, 13, 1608–1623. [Google Scholar] [CrossRef]
- Yu, F.F.; Wu, Y.R.; Xie, Q. Ubiquitin-Proteasome System in ABA Signaling: From Perception to Action. Mol. Plant 2016, 9, 21–33. [Google Scholar] [CrossRef] [Green Version]
- De Zelicourt, A.; Colcombet, J.; Hirt, H. The Role of MAPK Modules and ABA during Abiotic Stress Signaling. Trends Plant Sci. 2016, 21, 677–685. [Google Scholar] [CrossRef]
- Jamshed, M.; Sankaranarayanan, S.; Abhinandan, K.; Samuel, M.A. Stigma Receptivity Is Controlled by Functionally Redundant MAPK Pathway Components in Arabidopsis. Mol. Plant 2020, 13, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- Tena, G.; Boudsocq, M.; Sheen, J. Protein kinase signaling networks in plant innate immunity. Curr. Opin. Plant Biol. 2011, 14, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. Plant protein-serine/threonine kinases: Classification into subfamilies and overview of function. In Advances in Botanical Research; Academic Press: Cambridge, MA, USA, 2000; Volume 32, pp. 1–44. [Google Scholar]
- Pokora, W.; Aksmann, A.; Baścik-Remisiewicz, A.; Dettlaff-Pokora, A.; Rykaczewski, M.; Gappa, M.; Tukaj, Z. Changes in nitric oxide/hydrogen peroxide content and cell cycle progression: Study with synchronized cultures of green alga Chlamydomonas reinhardtii. J. Plant Physiol. 2017, 208, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Schluttenhofer, C. Origin and evolution of jasmonate signaling. Plant Sci. 2020, 298, 110542. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, B.; Jiang, Z.; Wang, S. Calcium-mediation of jasmonate biosynthesis and signaling in plants. Plant Sci. 2019, 287, 110192. [Google Scholar] [CrossRef]
- Lee, J.S.; Kuroha, T.; Hnilova, M.; Khatayevich, D.; Kanaoka, M.M.; McAbee, J.M.; Sarikaya, M.; Tamerler, C.; Torii, K.U. Direct interaction of ligand-receptor pairs specifying stomatal patterning. Genes Dev. 2012, 26, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Subramanian, B.; Gao, S.H.; Lercher, M.J.; Hu, S.N.; Chen, W.H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Zhang, X. Transcriptome and Metabolomic Analyses Reveal Regulatory Networks Controlling Maize Stomatal Development in Response to Blue Light. Int. J. Mol. Sci. 2021, 22, 5393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105393
Liu T, Zhang X. Transcriptome and Metabolomic Analyses Reveal Regulatory Networks Controlling Maize Stomatal Development in Response to Blue Light. International Journal of Molecular Sciences. 2021; 22(10):5393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105393
Chicago/Turabian StyleLiu, Tiedong, and Xiwen Zhang. 2021. "Transcriptome and Metabolomic Analyses Reveal Regulatory Networks Controlling Maize Stomatal Development in Response to Blue Light" International Journal of Molecular Sciences 22, no. 10: 5393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105393