
Clinical Characteristics of POC1B-Associated Retinopathy and Assignment of Pathogenicity to Novel Deep Intronic and Non-Canonical Splice Site Variants

 , , and
, , and
Abstract
:1. Introduction
2. Results
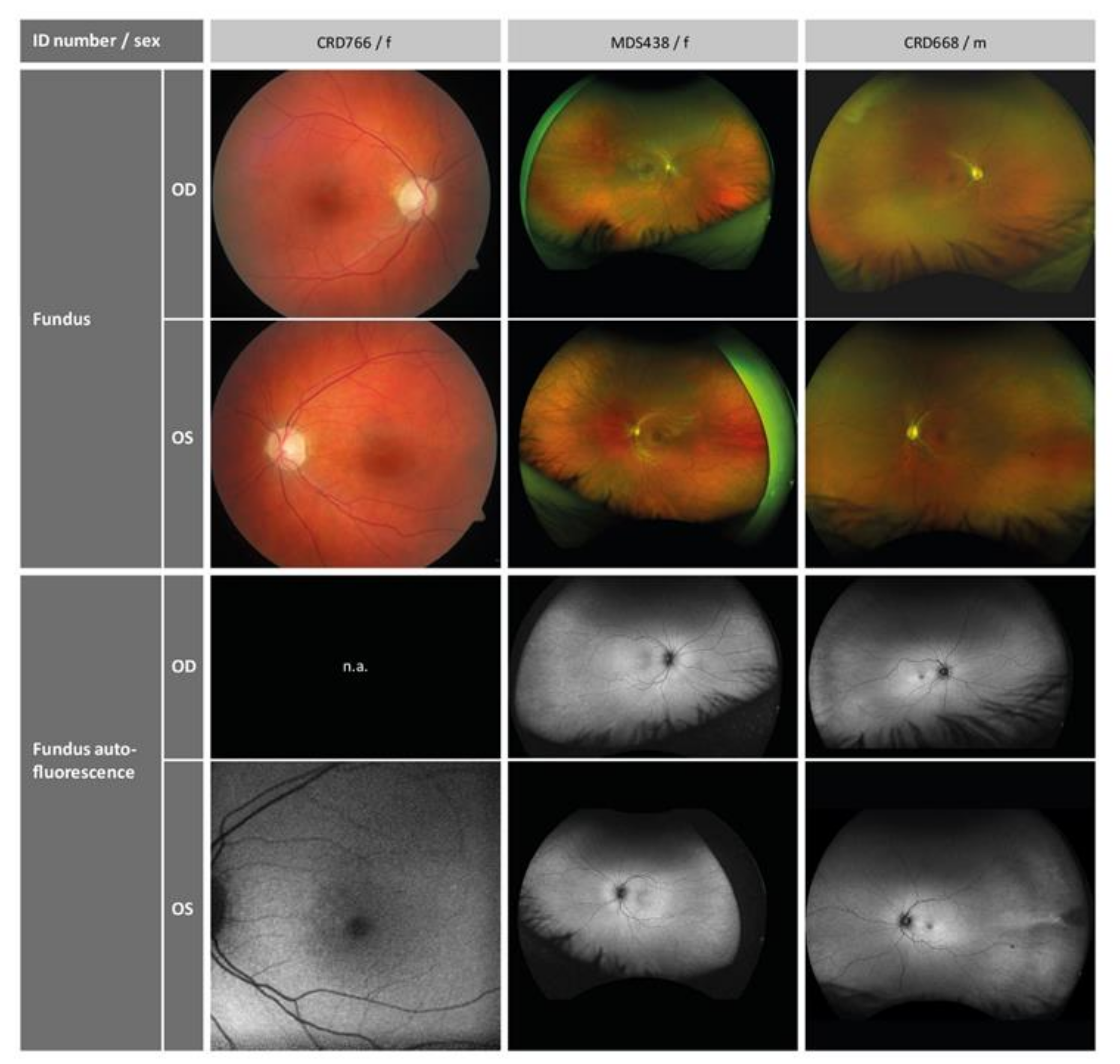
2.1. Clinical Phenotype
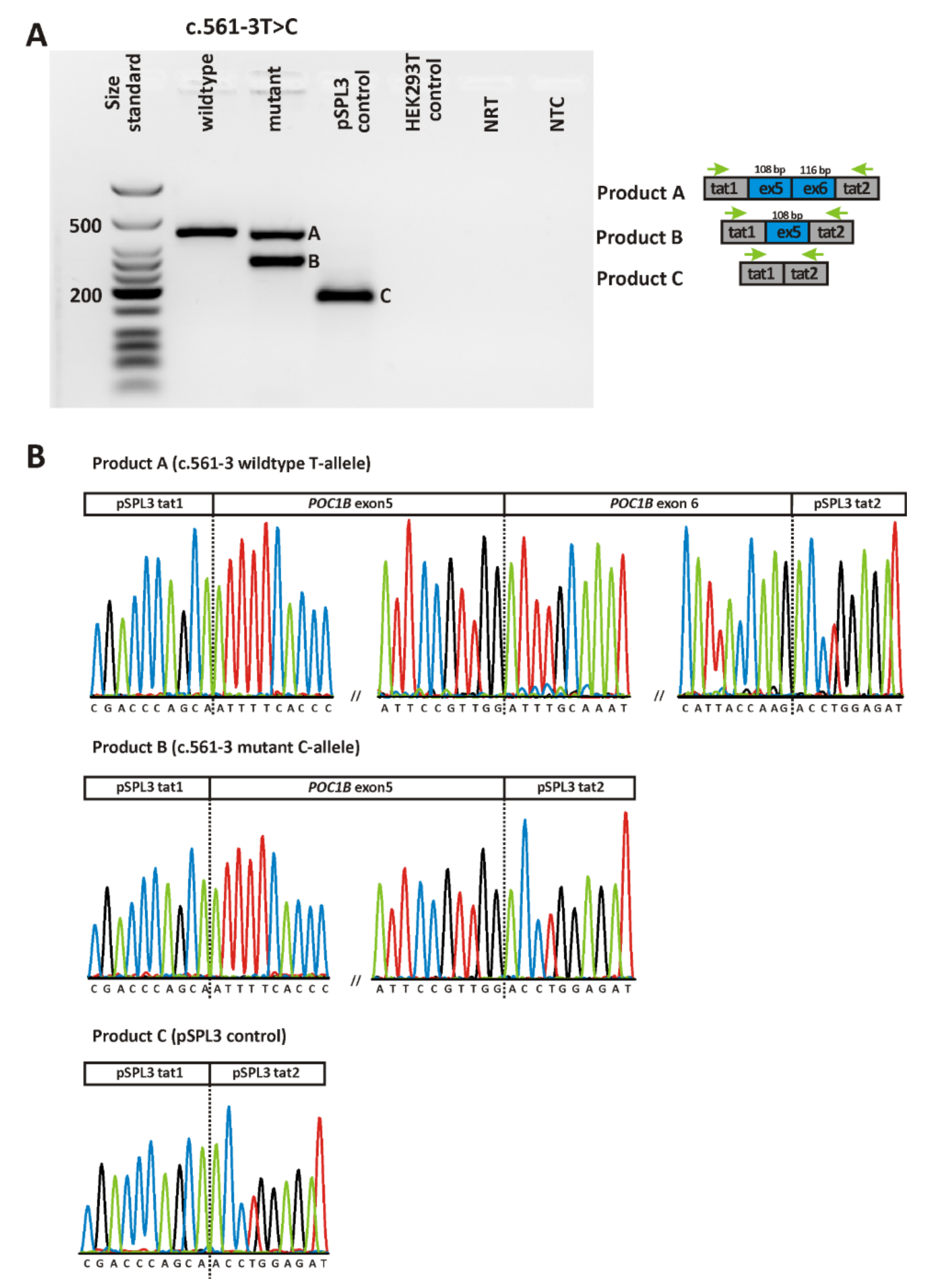
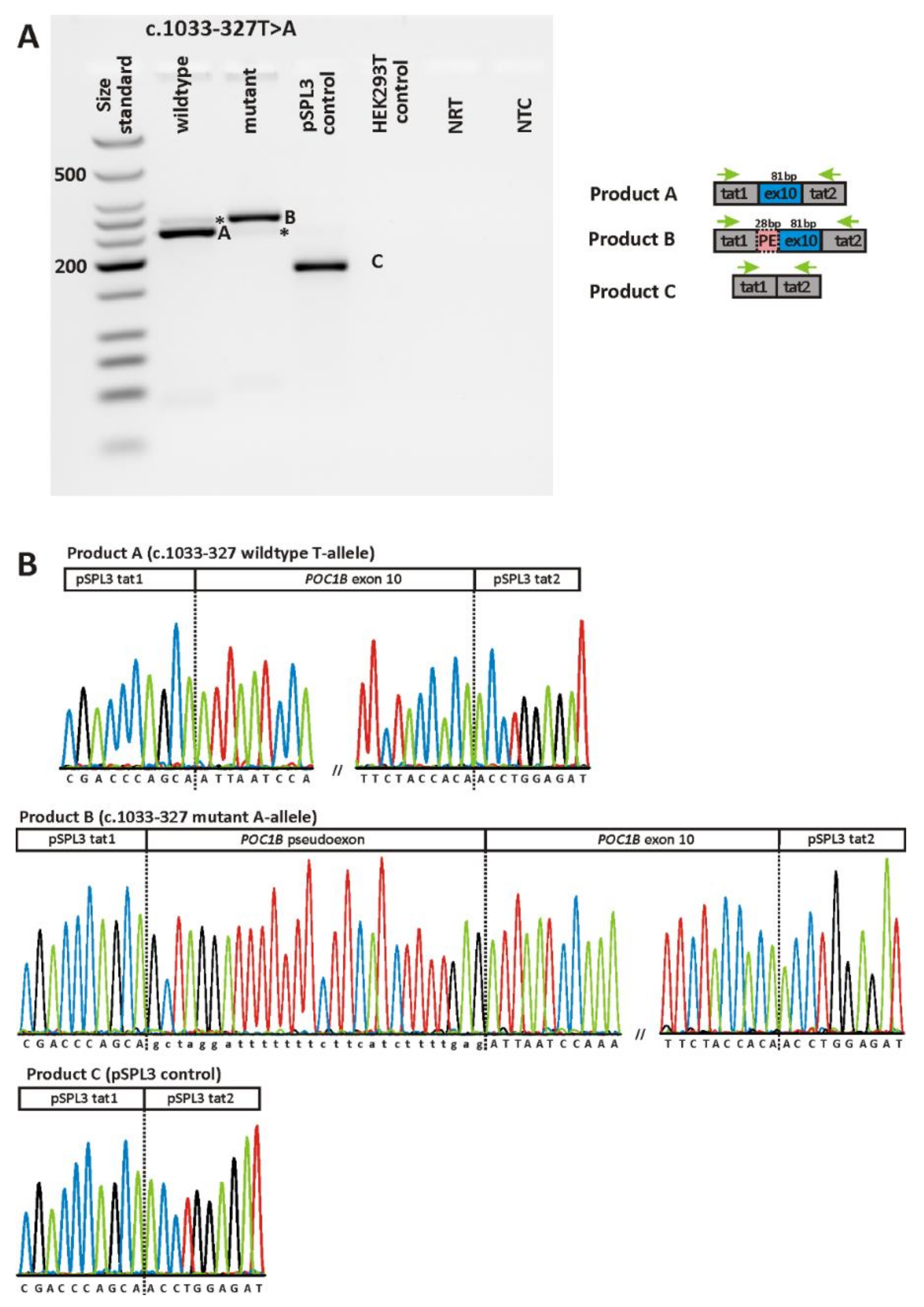
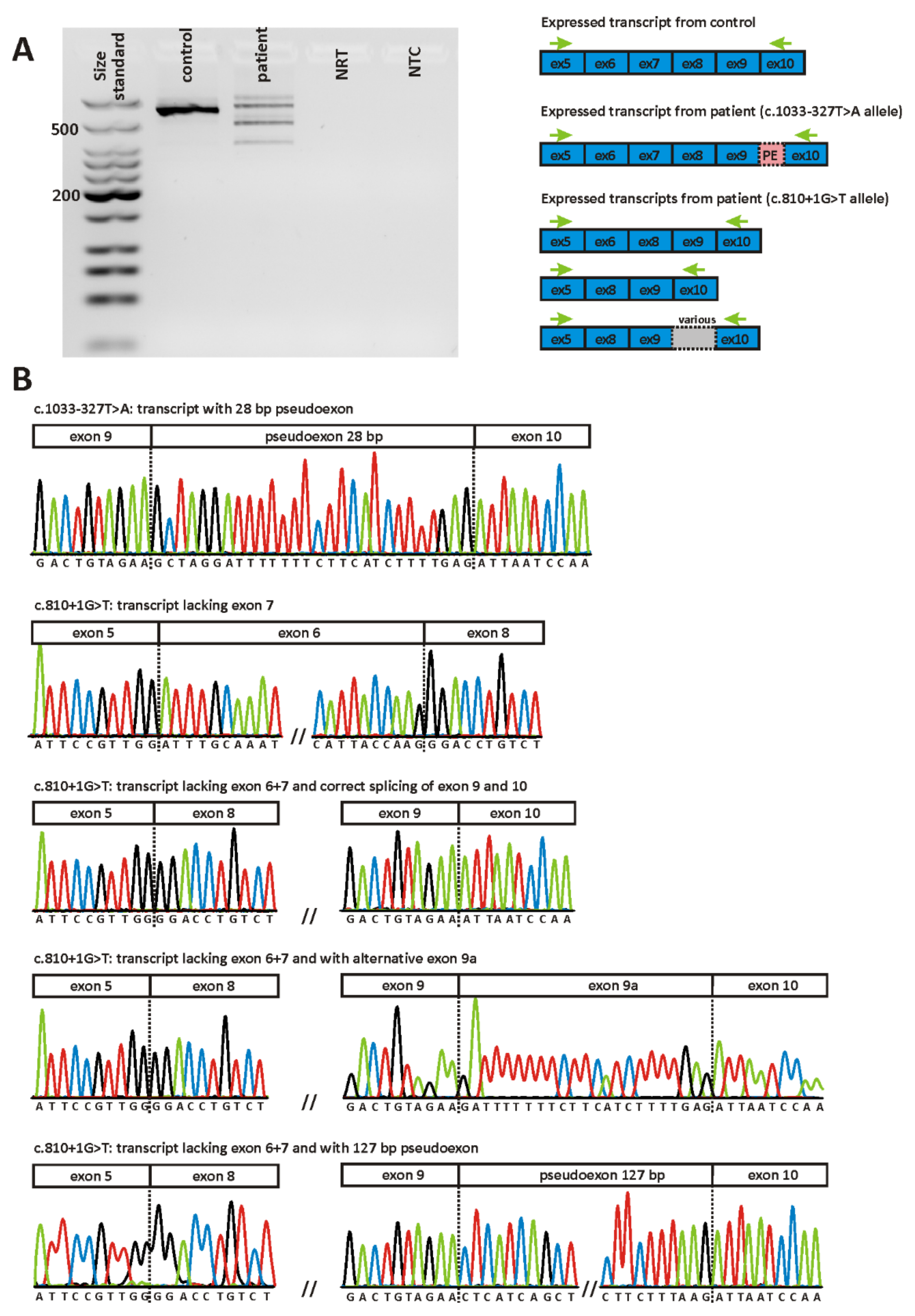
2.2. Validation of Variants
3. Discussion
4. Materials and Methods
4.1. Patient Enrollment and Retrieval of Blood Samples
4.2. Clinical Evaluation
4.3. Genetic Diagnostic Testing
4.4. In Silico Analysis
4.5. In Vitro Splice Assays
4.6. Direct mRNA Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keller, L.C.; Geimer, S.; Romijn, E.; Yates, J., 3rd; Zamora, I.; Marshall, W.F. Molecular architecture of the centriole proteome: The conserved WD40 domain protein POC1 is required for centriole duplication and length control. Mol. Biol. Cell. 2009, 20, 1150–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venoux, M.; Tait, X.; Hames, R.S.; Straatman, K.R.; Woodland, H.R.; Fry, A.M. Poc1A and Poc1B act together in human cells to ensure centriole integrity. J. Cell Sci. 2013, 126, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosing, S.; Lamers, I.J.; de Vrieze, E.; van den Born, L.I.; Lambertus, S.; Arts, H.H.; POC1B Study Group; Peters, T.A.; Hoyng, C.B.; Kremer, H.; et al. Disruption of the basal body protein POC1B results in autosomal-recessive cone-rod dystrophy. Am. J. Hum. Genet. 2014, 95, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durlu, Y.K.; Köroğlu, Ç.; Tolun, A. Novel recessive cone-rod dystrophy caused by POC1B mutation. JAMA Ophthalmol. 2014, 132, 1185–1191. [Google Scholar] [CrossRef] [Green Version]
- Beck, B.B.; Phillips, J.B.; Bartram, M.P.; Wegner, J.; Thoenes, M.; Pannes, A.; Sampson, J.; Heller, R.; Göbel, H.; Koerber, F.; et al. Mutation of POC1B in a severe syndromic retinal ciliopathy. Hum. Mutat. 2014, 35, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Chen, L.; Wang, D.; Zhang, Y.; Chen, Z.; Huang, H. Novel compound heterozygous mutation in the POC1B gene underlie peripheral cone dystrophy in a Chinese family. Ophthalmic Genet. 2018, 39, 300–306. [Google Scholar] [CrossRef]
- Kameya, S.; Fujinami, K.; Ueno, S.; Hayashi, T.; Kuniyoshi, K.; Ideta, R.; Kikuchi, S.; Kubota, D.; Yoshitake, K.; Katagiri, S.; et al. Japan Eye Genetics Consortium. Phenotypical Characteristics of POC1B-Associated Retinopathy in Japanese Cohort: Cone Dystrophy with Normal Funduscopic Appearance. Invest. Ophthalmol. Vis. Sci. 2019, 60, 3432–3446. [Google Scholar] [CrossRef] [Green Version]
- Peturson, A.C.; Noel, N.C.L.; MacDonald, I.M. A homozygous POC1B variant causes recessive cone-rod dystrophy. Ophthalmic Genet. 2021, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Kominami, A.; Ueno, S.; Kominami, T.; Nakanishi, A.; Ito, Y.; Fujinami, K.; Tsunoda, K.; Hayashi, T.; Kikuchi, S.; Kameya, S.; et al. Case of cone dystrophy with normal fundus appearance associated with biallelic POC1B variants. Ophthalmic Genet. 2018, 39, 255–262. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Toulis, V.; Cortés-González, V.; Castro-Miró, M.; Sallum, J.F.; Català-Mora, J.; Villanueva-Mendoza, C.; Ciccioli, M.; Gonzàlez-Duarte, R.; Valero, R.; Marfany, G. Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants. Genes 2020, 11, 378. [Google Scholar] [CrossRef] [Green Version]
- Weisschuh, N.; Mazzola, P.; Heinrich, T.; Haack, T.; Wissinger, B.; Tonagel, F.; Kelbsch, C. First submicroscopic inversion of the OPA1 gene identified in dominant optic atrophy—A case report. BMC Med. Genet. 2020, 21, 236. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Frischmeyer, P.A.; Dietz, H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999, 8, 1893–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Thermann, R.; Neu-Yilik, G.; Deters, A.; Frede, U.; Wehr, K.; Hagemeier, C.; Hentze, M.W.; Kulozik, A.E. Binary specification of nonsense codons by splicing and ctoplasmic translation. EMBO J. 1998, 17, 3484–3494. [Google Scholar] [CrossRef] [PubMed]
- Takahara, K.; Schwarze, U.; Imamura, Y.; Hoffman, G.G.; Toriello, H.; Smith, L.T.; Byers, P.H.; Greenspan, D.S. Order of intron removal influences multiple splice outcomes, including a two-exon skip, in a COL5A1 acceptor-site mutation that results in abnormal pro-alpha 1(V) N-propeptides and Ehlers-Danlos syndrome type I. Am. J. Hum. Genet. 2002, 71, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Hori, T.; Fukao, T.; Murase, K.; Sakaguchi, N.; Harding, C.O.; Kondo, N. Molecular basis of two-exon skipping (exons 12 and 13) by c.1248 + 5g > a in OXCT1 gene: Study on intermediates of OXCT1 transcripts in fibroblasts. Hum. Mutat. 2013, 34, 473–480. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Weisschuh, N.; Wissinger, B.; Gramer, E. A splice site mutation in the PAX6 gene which induces exon skipping causes autosomal dominant inherited aniridia. Mol. Vis. 2012, 18, 751–757. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | cDNA (NM_172240.3) | Protein (NP_758440.1) | Zygosity | Trans Config. Validated | gnomAD MAF | SpliceAI Prediction (0–1) | NNSplice Prediction (0–1) |
|---|---|---|---|---|---|---|---|
| CRD766 | c.1331_1332dup | p.(T445Rfs*10) | hom | no | - | N/A | N/A |
| MDS438 | c.810+1G>T | p.(V226Gfs*30) AND p.(F188Dfs*73) | het | yes | 2.715E-05 | Donor loss (0.99) | Wildtype donor not recognized |
| c.1033-327T>A | p.(I345Afs*9) | het | 4.789E-05 | Acceptor gain (0.6) | Acceptor gain (0.61) | ||
| CRD668 | c.561-3T>C | p.(F188Sfs*6) | het | no | - | Acceptor loss (0.21) | No effect |
| c.1340C>G | p.(S447*) | het | - | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weisschuh, N.; Mazzola, P.; Bertrand, M.; Haack, T.B.; Wissinger, B.; Kohl, S.; Stingl, K. Clinical Characteristics of POC1B-Associated Retinopathy and Assignment of Pathogenicity to Novel Deep Intronic and Non-Canonical Splice Site Variants. Int. J. Mol. Sci. 2021, 22, 5396. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105396
Weisschuh N, Mazzola P, Bertrand M, Haack TB, Wissinger B, Kohl S, Stingl K. Clinical Characteristics of POC1B-Associated Retinopathy and Assignment of Pathogenicity to Novel Deep Intronic and Non-Canonical Splice Site Variants. International Journal of Molecular Sciences. 2021; 22(10):5396. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105396
Chicago/Turabian StyleWeisschuh, Nicole, Pascale Mazzola, Miriam Bertrand, Tobias B. Haack, Bernd Wissinger, Susanne Kohl, and Katarina Stingl. 2021. "Clinical Characteristics of POC1B-Associated Retinopathy and Assignment of Pathogenicity to Novel Deep Intronic and Non-Canonical Splice Site Variants" International Journal of Molecular Sciences 22, no. 10: 5396. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105396