
An Analytical Review of the Structural Features of Pentatricopeptide Repeats: Strategic Amino Acids, Repeat Arrangements and Superhelical Architecture


Abstract
:
1. Introduction
2. Results
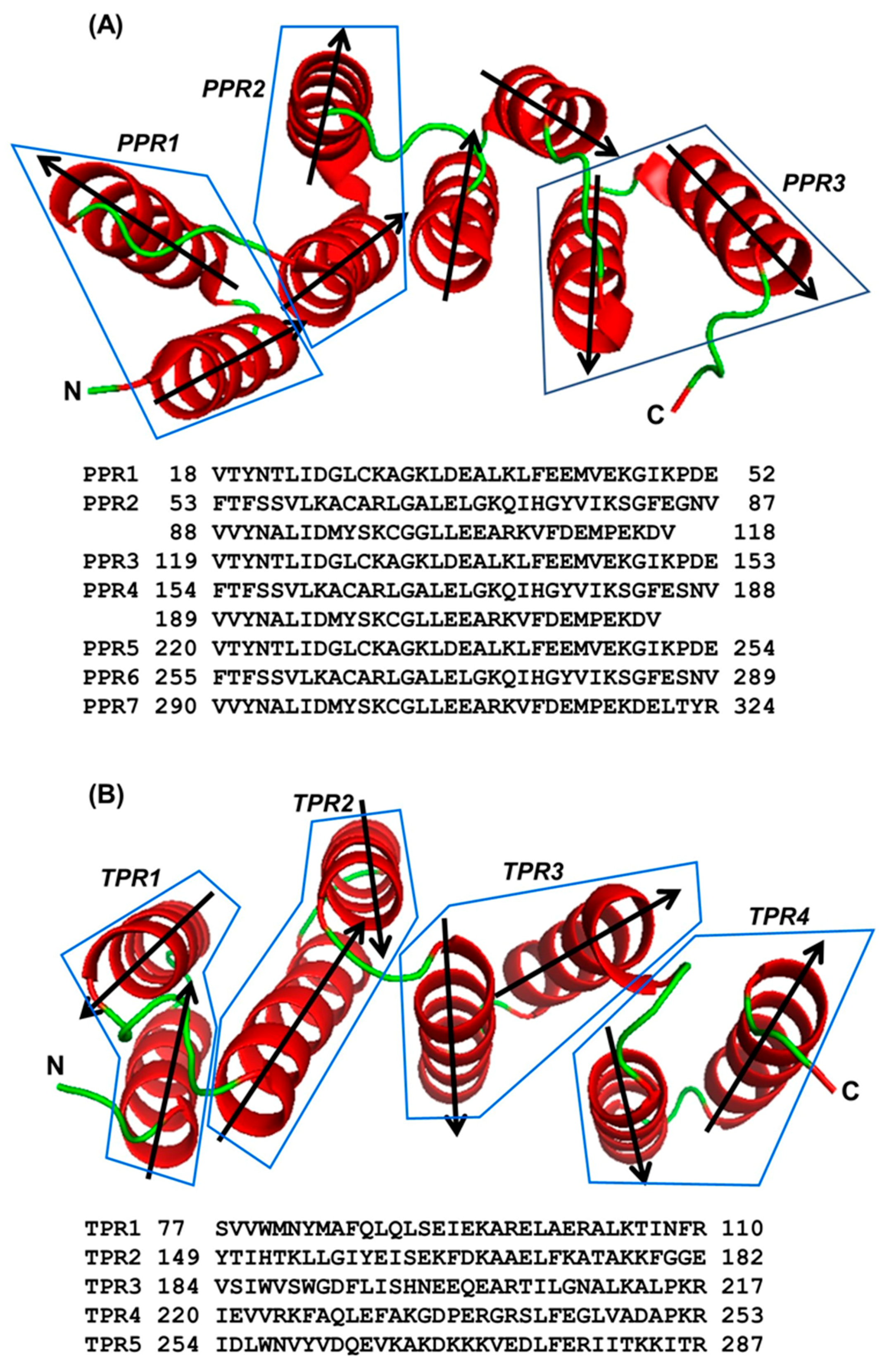
2.1. Location of Key Amino Acids in TPR and PPR; a Comparative Analysis
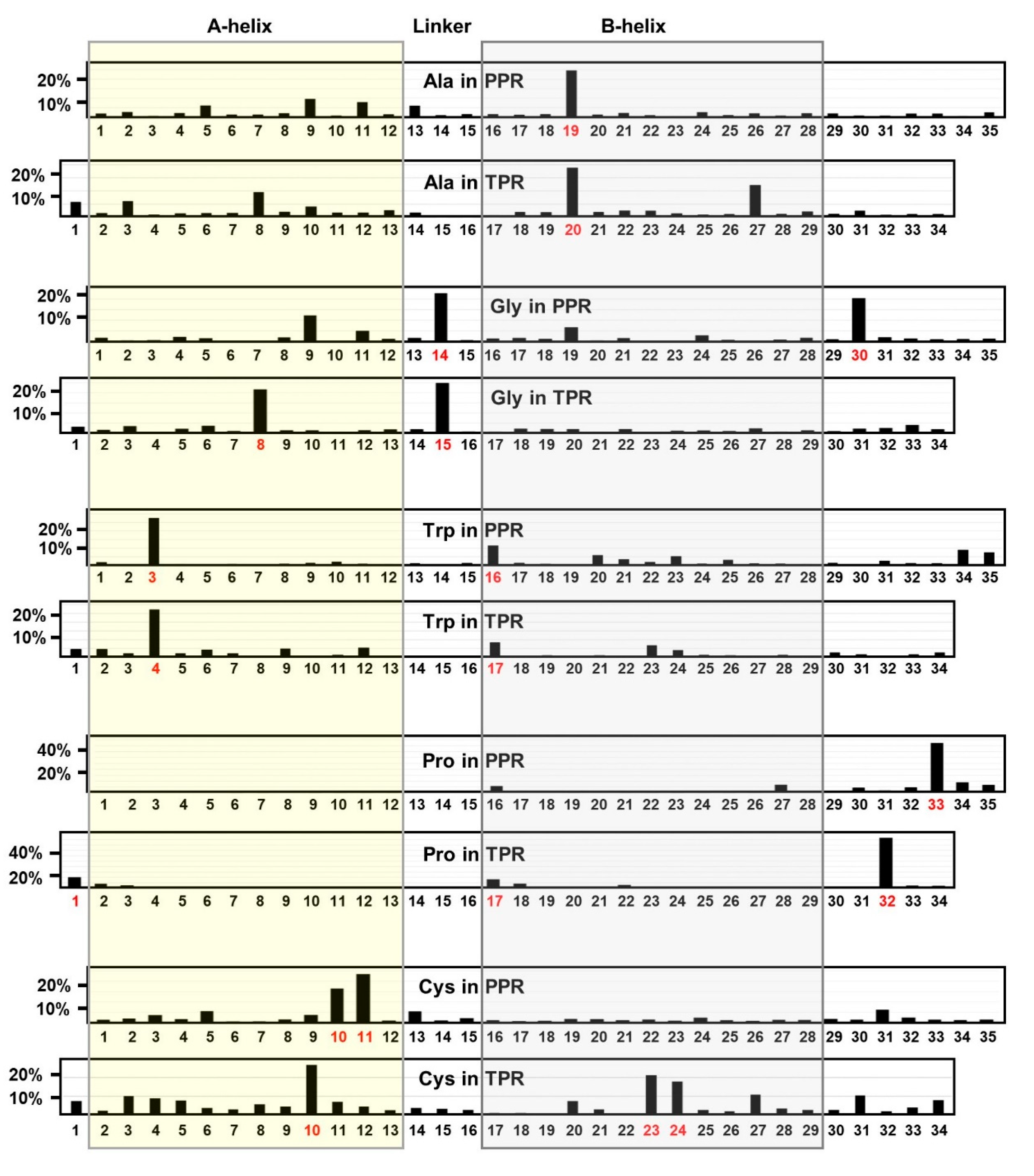
2.1.1. Alanine Locations in TPR and PPR
2.1.2. Glycine
2.1.3. Tryptophan Locations in TPR and PPR
2.1.4. Proline Locations in TPR and PPR
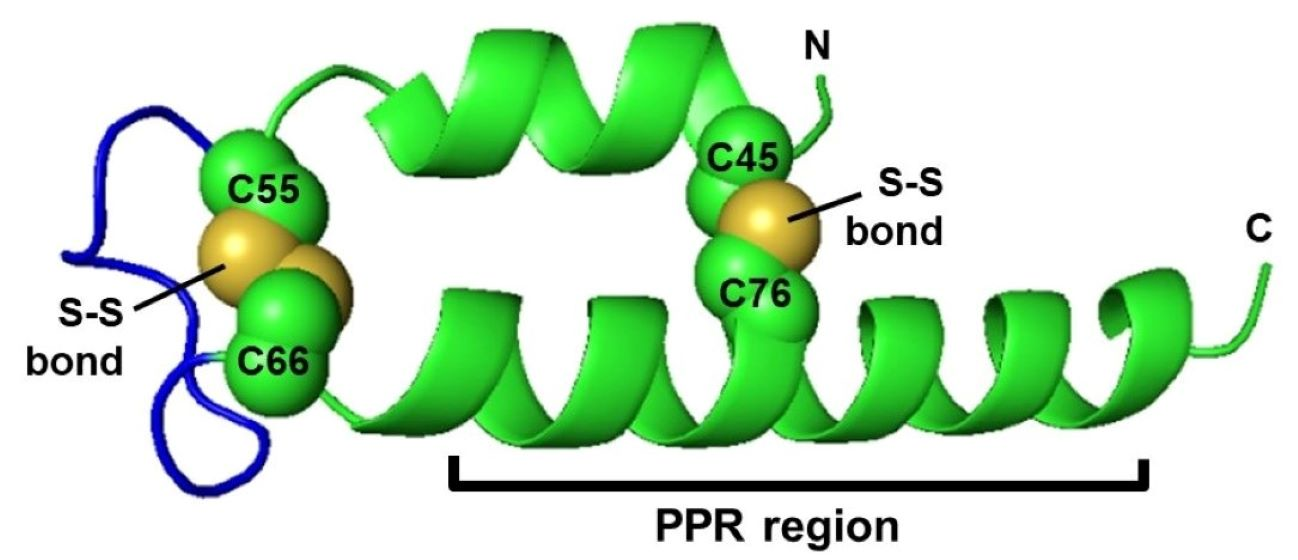
2.1.5. Cysteine
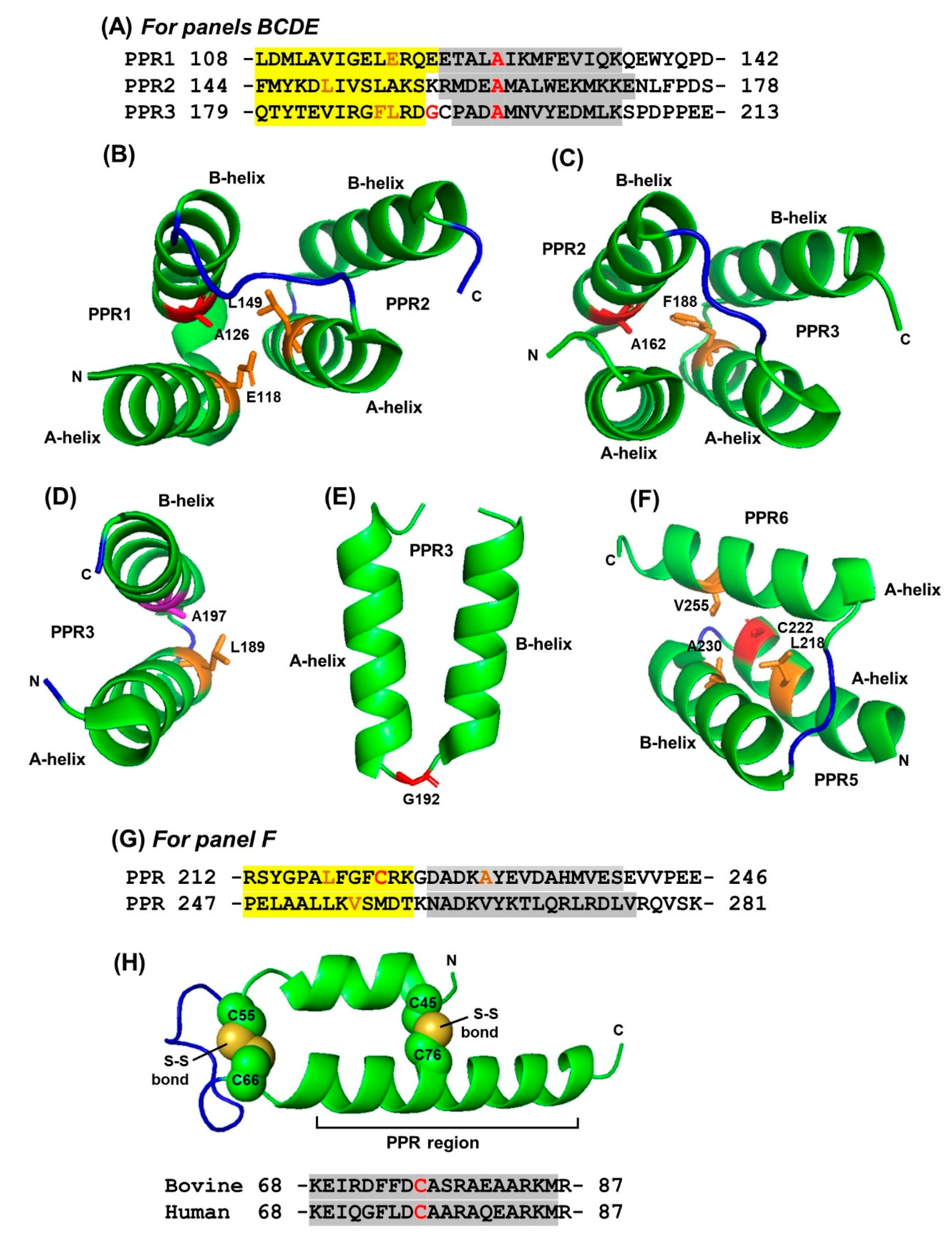
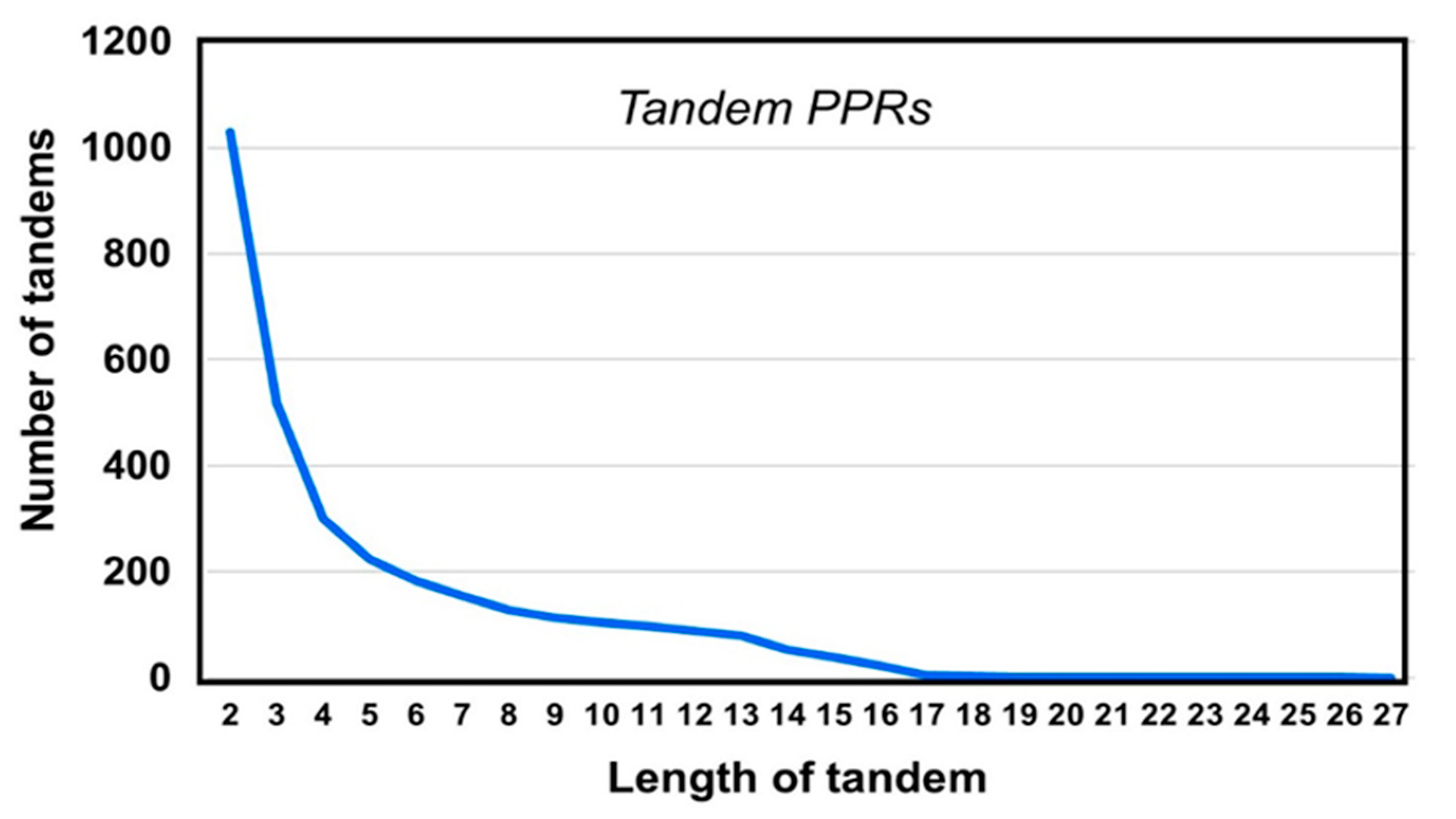
2.2. PPR Length and Clustering
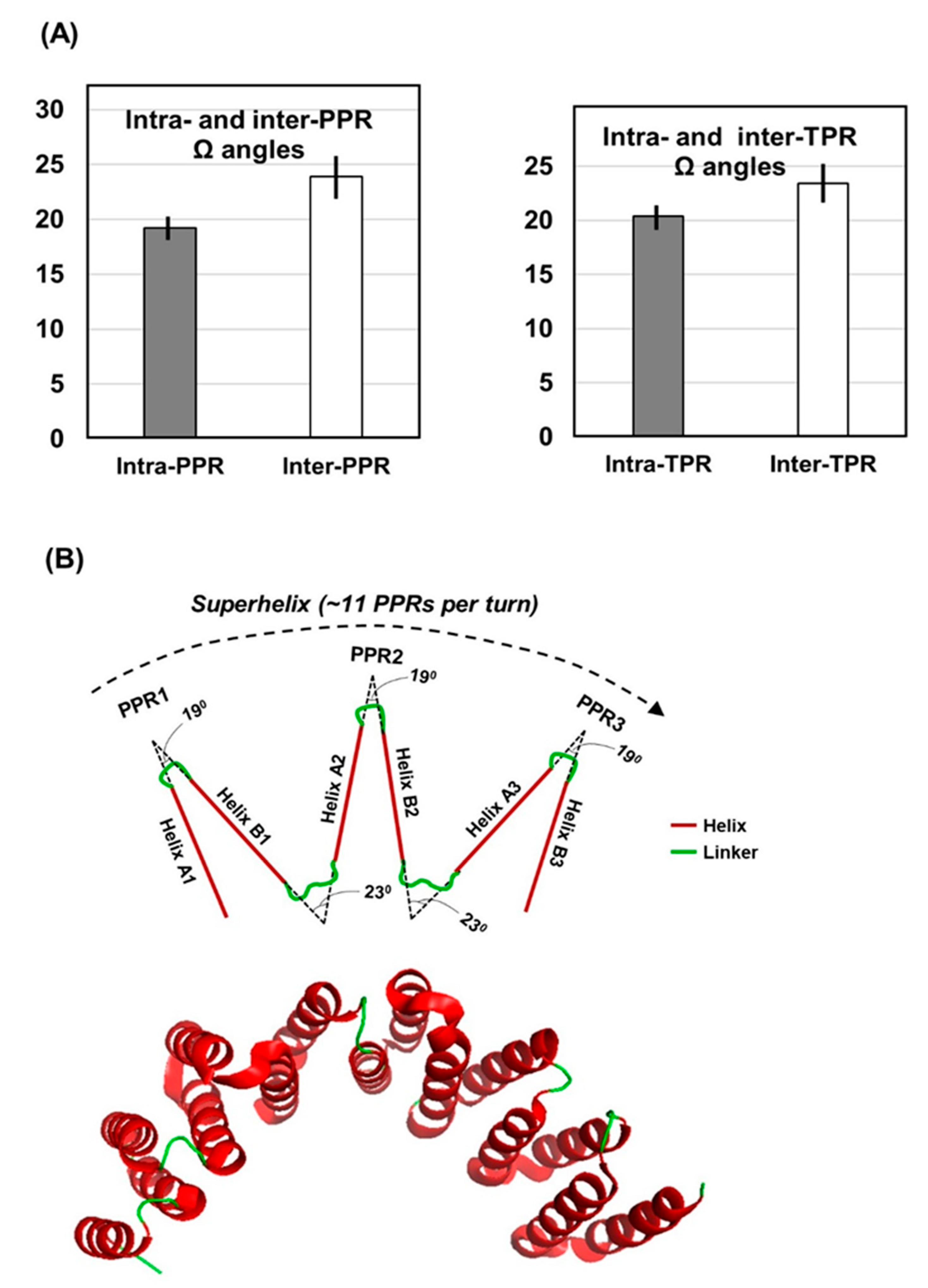
2.3. Interhelical Angles in PPR Domains and Superhelices
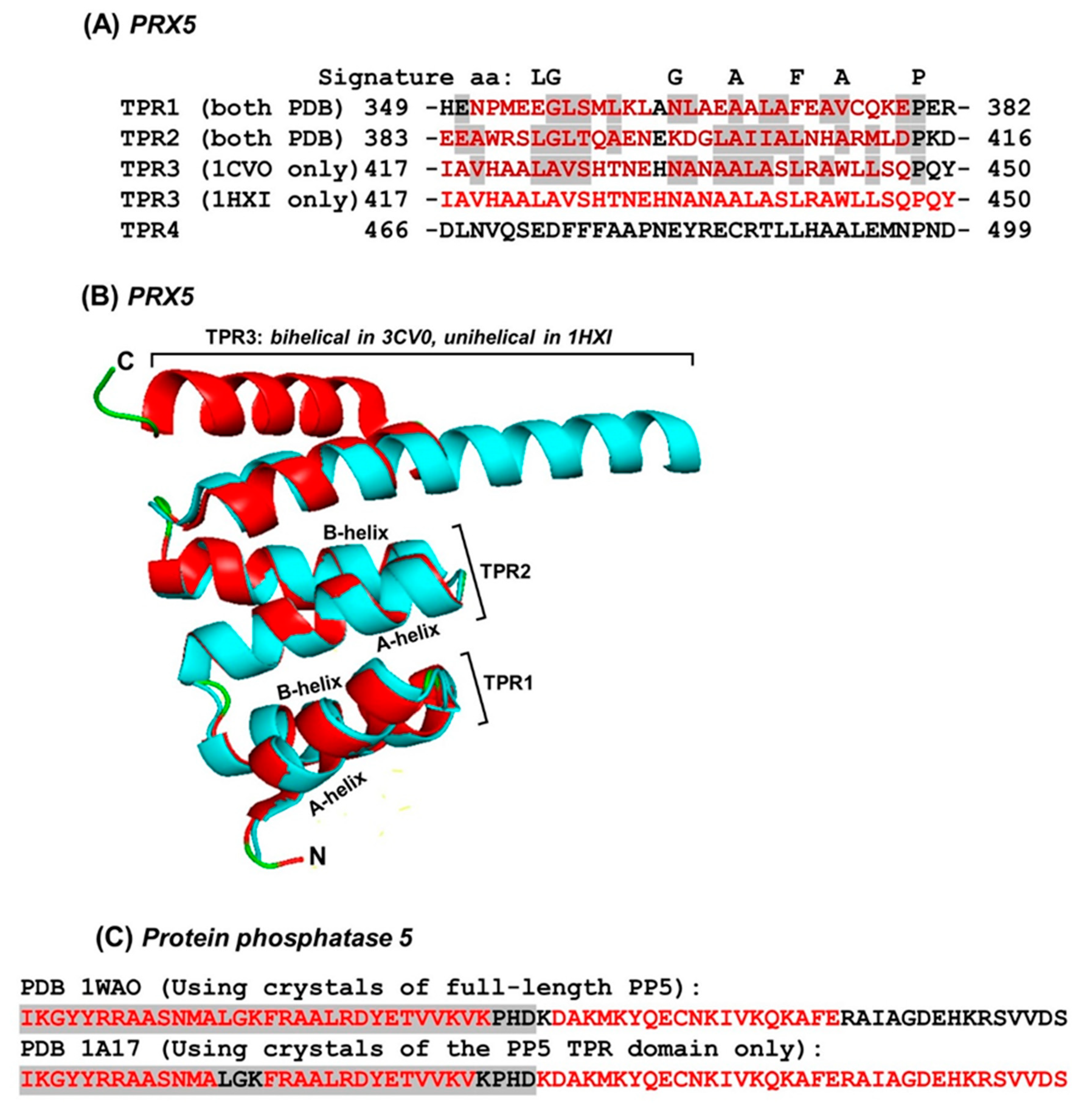
2.4. Regulation of Repeat Folding by Interaction between Neighboring Helices
3. Conclusions, Discussion, and Future Directions
4. Materials and Methods
4.1. Retrieval of Sequences
4.2. Analysis of Sequence Patterns and Interhelical Angles
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kajava, A.V. Tandem repeats in proteins: From sequence to structure. J. Struct. Biol. 2012, 179, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, N.; Chen, J.; Regan, L. All repeats are not equal: A module-based approach to guide repeat protein design. J. Mol. Biol. 2013, 425, 1826–1838. [Google Scholar] [CrossRef] [Green Version]
- Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 2015, 113, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, I.D.; Peeters, N. The PPR motif—A TPR-related motif prevalent in plant organellar proteins. Trends Biochem. Sci. 2000, 25, 46–47. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kawabata, M.; Hisano, K.; Kazama, T.; Matsuoka, K.; Sugita, M.; Nakamura, T. Identification and characterization of the RNA binding surface of the pentatricopeptide repeat protein. Nucleic Acids Res. 2012, 40, 2712–2723. [Google Scholar] [CrossRef] [Green Version]
- Yagi, Y.; Hayashi, S.; Kobayashi, K.; Hirayama, T.; Nakamura, T. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS ONE 2013, 8, e57286. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef]
- Perez-Riba, A.; Itzhaki, L.S. The tetratricopeptide-repeat motif is a versatile platform that enables diverse modes of molecular recognition. Curr. Opin. Struct. Biol. 2019, 54, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. Protein tetratricopeptide repeat and the companion non-tetratricopeptide repeat helices: Bioinformatic analysis of interhelical interactions. Bioinform. Biol. Insights 2019, 13, 1177932219863363. [Google Scholar] [CrossRef]
- Barik, S. The nature and arrangement of pentatricopeptide domains and the linker sequences between them. Bioinform. Biol. Insights 2020, 14, 1177932220906434. [Google Scholar] [CrossRef] [Green Version]
- Main, E.R.G.; Xiong, Y.; Cocco, M.J.; D’Andrea, L.; Regan, L. Design of stable alpha-helical arrays from an idealized TPR motif. Structure 2003, 11, 497–508. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, L.D.; Regan, L. TPR proteins: The versatile helix. Trends Biochem. Sci. 2003, 28, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, H.R.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Sasisekharan, V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968, 23, 283–438. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Ramachandran, G.N. Conformation of polypeptide chains. Annu. Rev. Biochem. 1969, 38, 45–82. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S.; Richardson, D.C. Amino acid preferences for specific locations at the ends of alpha helices. Science 1988, 240, 1648–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashton, M.; Chothia, C. The geometry of domain combination in proteins. J. Mol. Biol. 2002, 315, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Chothia, C. Principles that determine the structure of proteins. Annu. Rev. Biochem. 1984, 53, 537–572. [Google Scholar] [CrossRef]
- Chothia, C.; Finkelstein, A.V. The classification and origins of protein folding patterns. Annu. Rev. Biochem. 1990, 59, 1007–1039. [Google Scholar] [CrossRef]
- Taylor, P.; Dornan, J.; Carrello, A.; Minchin, R.F.; Ratajczak, T.; Walkinshaw, M.D. Two structures of cyclophilin 40: Folding and fidelity in the TPR domains. Structure 2001, 9, 431–438. [Google Scholar] [CrossRef]
- Wilson, C.G.; Kajander, T.; Regan, L. The crystal structure of NlpI. A prokaryotic tetratricopeptide repeat protein with a globular fold. FEBS J. 2005, 272, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Katibah, G.E.; Lee, H.J.; Huizar, J.P.; Vogan, J.M.; Alber, T.; Collins., K. tRNA binding, structure, and localization of the human interferon-induced protein IFIT5. Mol. Cell 2013, 49, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Khoshnevis, S.; Askenasy, I.; Johnson, M.C.; Dattolo, M.D.; Young-Erdos, C.L.; Stroupe, M.E.; Karbstein, K. The DEAD-box protein Rok1 orchestrates 40S and 60S ribosome assembly by promoting the release of Rrp5 from pre-40S ribosomes to allow for 60S maturation. PLoS Biol. 2016, 14, e1002480. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhu, X.; Qi, J.; An, W.; Lan., P.; Tan, D.; Chen, R.; Wang, B.; Zheng, S.; Zhang, C.; et al. Molecular architecture of the 90S small subunit pre-ribosome. eLife 2017, 6, e22086. [Google Scholar] [CrossRef]
- Feng, F.; Yuan, L.; Wang, Y.E.; Crowley, C.; Lv, Z.; Li, J.; Liu, Y.; Cheng, G.; Zeng, S.; Liang, H. Crystal structure and nucleotide selectivity of human IFIT5/ISG58. Cell Res. 2013, 23, 1055–1058. [Google Scholar] [CrossRef]
- Fensterl, V.; Sen, G.C. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 2011, 31, 71–78. [Google Scholar] [CrossRef]
- Fujii, S.; Small, I. The evolution of RNA editing and pentatricopeptide repeat genes. New Phytol. 2011, 191, 37–47. [Google Scholar] [CrossRef]
- Gutmann, B.; Royan, S.; Schallenberg-Rüdinger, M.; Lenz, H.; Castleden, I.R.; McDowell, R.; Vacher, M.A.; Tonti-Filippini, J.; Bond, C.S.; Knoop, V.; et al. The expansion and diversification of pentatricopeptide repeat RNA-editing factors in plants. Mol. Plant 2020, 13, 215–230. [Google Scholar] [CrossRef]
- Lurin, C.; Andrés, C.; Aubourg, S.; Bellaoui, M.; Bitton, F.; Bruyère, C.; Caboche, M.; Debast, C.; Gualberto, J.; Hoffmann, B.; et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 2004, 16, 2089–2103. [Google Scholar] [CrossRef] [Green Version]
- Das, A.K.; Cohen, P.W.; Barford, D. The structure of the tetratricopeptide repeats of protein phosphatase 5: Implications for TPR-mediated protein-protein interactions. EMBO J. 1998, 17, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Barik, S. The uniqueness of tryptophan in biology: Properties, metabolism, interactions and localization in proteins. Int. J. Mol. Sci. 2020, 21, 8776. [Google Scholar] [CrossRef]
- MacArthur, M.W.; Thornton, J.M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991, 218, 397–412. [Google Scholar] [CrossRef]
- Woolfson, D.N.; Williams, D.H. The influence of proline residues on alpha-helical structure. FEBS Lett. 1990, 277, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Roach, C.; Hirsh, I.S.; Turley, S.; Walque, S.D.; Michels, P.A.; Hol, W.G. An unexpected extended conformation for the third TPR motif of the peroxin PEX5 from Trypanosoma brucei. J. Mol. Biol. 2001, 307, 271–282. [Google Scholar] [CrossRef]
- Koripella, R.K.; Sharma, M.R.; Haque, M.E.; Risteff, P.; Spremulli, L.L.; Agrawal, R.K. Structure of human mitochondrial translation initiation factor 3 bound to the small ribosomal subunit. iScience 2019, 12, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Khawaja, A.; Itoh, Y.; Remes, C.; Spåhr, H.; Yukhnovets, O.; Höfig, H.; Amunts, A.; Rorbach, J. Distinct pre-initiation steps in human mitochondrial translation. Nat. Commun. 2020, 11, 2932. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; An, Y.; Xu, P.; Xiao, J. Functioning of PPR Proteins in organelle RNA metabolism and chloroplast biogenesis. Front. Plant Sci. 2021, 12, 627501. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Rojas, M.; Fujii, S.; Yap, A.; Chong, Y.S.; Bond, C.S.; Small, I. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 2012, 8, e1002910. [Google Scholar] [CrossRef] [PubMed]
- Magliery, T.J.; Regan, L. Beyond consensus: Statistical free energies reveal hidden interactions in the design of a TPR motif. J. Mol. Biol. 2004, 343, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Q.; Guan, Z.; Wang, Q.; Li, L.; Ruan, F.; Lin, R.; Zou, T.; Yin, P. MORF9 increases the RNA-binding activity of PLS-type pentatricopeptide repeat protein in plastid RNA editing. Nat. Plants 2017, 3, 17037. [Google Scholar] [CrossRef] [PubMed]
- Main, E.R.G.; Stott, K.; Jackson, S.E.; Regan, L. Local and long-range stability in tandemly arrayed tetratricopeptide repeats. Proc. Natl. Acad. Sci. USA 2005, 102, 5721–5726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gully, B.S.; Shah, K.R.; Lee, M.; Shearston, K.; Smith, N.M.; Sadowska, A.; Blythe, A.J.; Bernath-Levin, K.; Stanley, W.A.; Small, I.D.; et al. The design and structural characterization of a synthetic pentatricopeptide repeat protein. Acta. Crystallogr. D Biol. Crystallogr. 2015, 71, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Sampathkumar, P.; Roach, C.; Michels, P.A.M.; Hol, W.G.J. Structural insights into the recognition of peroxisomal targeting signal 1 by Trypanosoma brucei peroxin 5. J. Mol. Biol. 2008, 381, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roe, S.M.; Cliff, M.J.; Williams, M.A.; Ladbury, J.E.; Cohen, P.T.; Barford, D. Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. EMBO J. 2005, 24, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Sayner, S.L.; Gross, K.L.; Russell, L.C.; Chinkers, M. Identification of amino acids in the tetratricopeptide repeat and C-terminal domains of protein phosphatase 5 involved in autoinhibition and lipid activation. Biochemistry 2001, 40, 10485–10490. [Google Scholar] [CrossRef] [PubMed]
- Kohl, A.; Binz, H.K.; Forrer, P.; Stumpp, M.T.; Plückthun, A.; Grütter, M.G. Designed to be stable: Crystal structure of a consensus ankyrin repeat protein. Proc. Natl. Acad. Sci. USA 2003, 100, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Grove, T.Z.; Cortajarena, A.L.; Regan, L. Ligand binding by repeat proteins: Natural and designed. Curr. Opin. Struct. Biol. 2008, 18, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Christenhusz, M.J.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, I.; Dujeancourt, L.; Gaisne, M.; Herbert, C.J.; Bonnefoy, N. A genome wide study in fission yeast reveals nine PPR proteins that regulate mitochondrial gene expression. Nucleic Acids Res. 2011, 39, 8029–8041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lano, W.L.D. PyMOL User’s Manual; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPR3 B-Helix aa number | Interacting aa in TPR4 | Interaction Energy (kJ/mol) |
|---|---|---|
| Ala 433 | Ala 477 | −1.58 |
| – | Phe 475 | −1.09 |
| Asn 434 | Ala 478 | −3.3 |
| Leu 437 | Tyr 482 | −5.7 |
| – | Glu 481 | −5.33 |
| – | Cys 485 | −5.08 |
| – | Phe 475 | −3.88 |
| – | Ala 478 | −1.68 |
| Leu 440 | Cys 485 | −2.87 |
| – | Leu 489 | −2.63 |
| Arg441 | Glu 484 | −60.92 |
| – | Leu 488 | −3.47 |
| – | Cys 485 | −3.26 |
| – | Glu 481 | −2.47 |
| Trp 443 | Leu 489 | −3.2 |
| Leu 444 | Leu 489 | −4.06 |
| Leu 445 | Leu 488 | −5.83 |
| – | Glu 484 | −1.04 |
| Gln 447 | Asp 499 | −2.04 |
| Gln 449 | Asp499 | −1.23 |
| Tyr 450 | Asn 496 | −23.44 |
| – | Ala 492 | −4.18 |
| – | Met 495 | −2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barik, S. An Analytical Review of the Structural Features of Pentatricopeptide Repeats: Strategic Amino Acids, Repeat Arrangements and Superhelical Architecture. Int. J. Mol. Sci. 2021, 22, 5407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105407
Barik S. An Analytical Review of the Structural Features of Pentatricopeptide Repeats: Strategic Amino Acids, Repeat Arrangements and Superhelical Architecture. International Journal of Molecular Sciences. 2021; 22(10):5407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105407
Chicago/Turabian StyleBarik, Sailen. 2021. "An Analytical Review of the Structural Features of Pentatricopeptide Repeats: Strategic Amino Acids, Repeat Arrangements and Superhelical Architecture" International Journal of Molecular Sciences 22, no. 10: 5407. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105407