
Anticancer Activity of Two Novel Hydroxylated Biphenyl Compounds toward Malignant Melanoma Cells

 , ,
, , 
Abstract
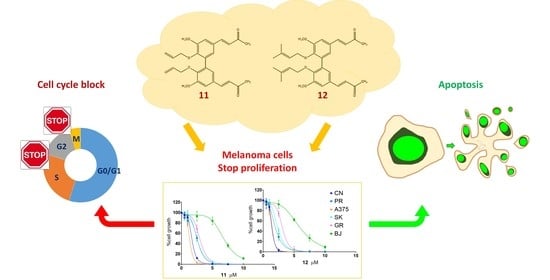
:
1. Introduction
2. Results
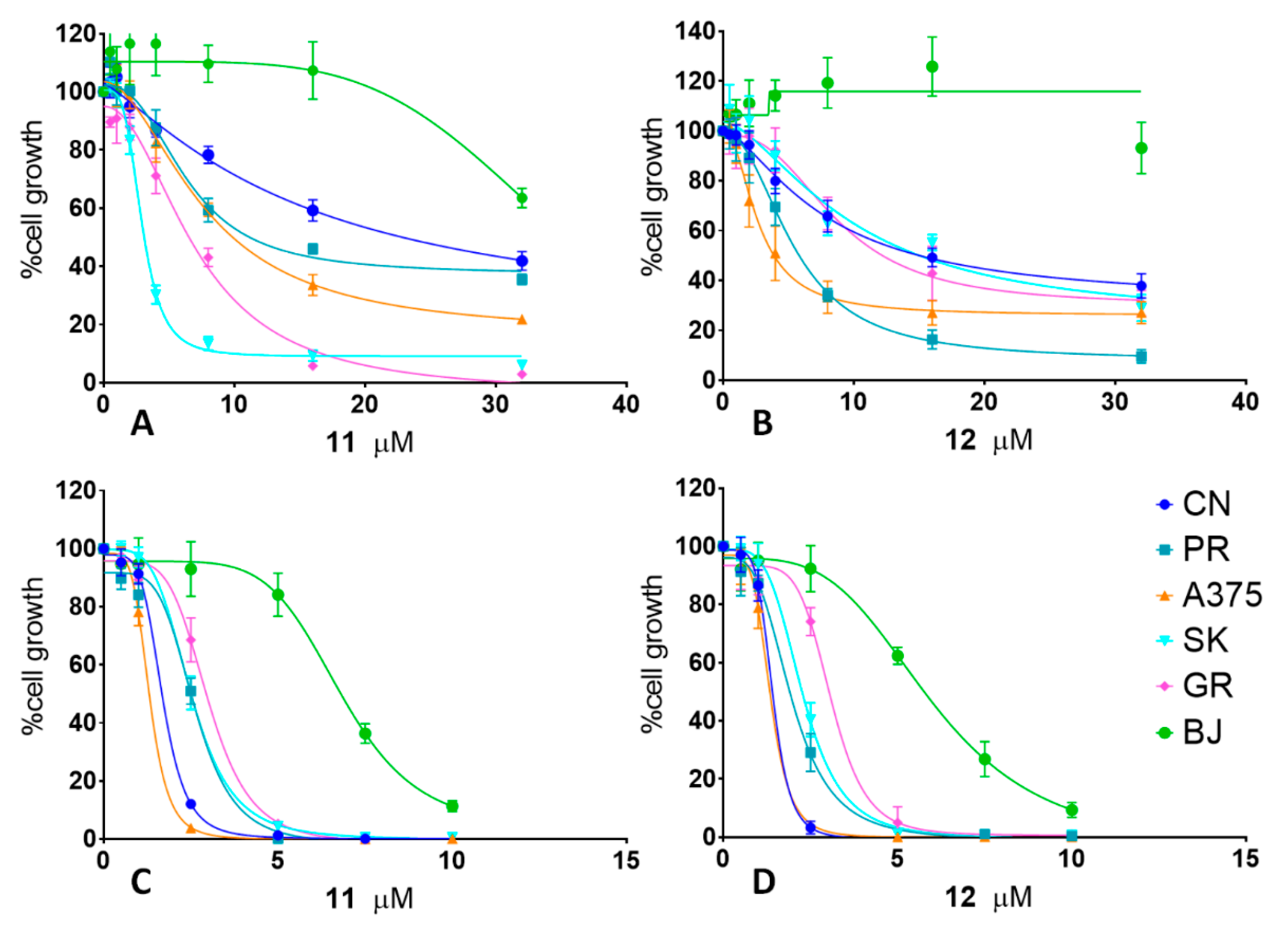
2.1. Antiproliferative Activity
2.2. Apoptosis Induction
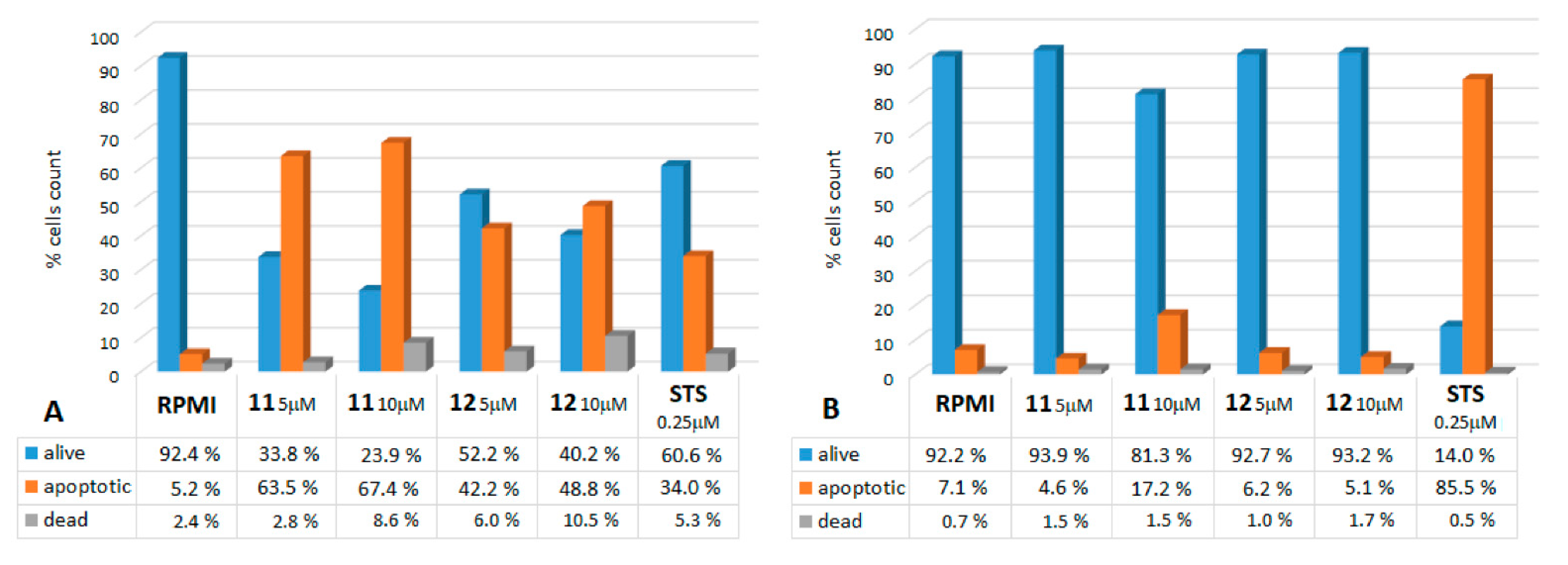
2.2.1. Annexin V Assays
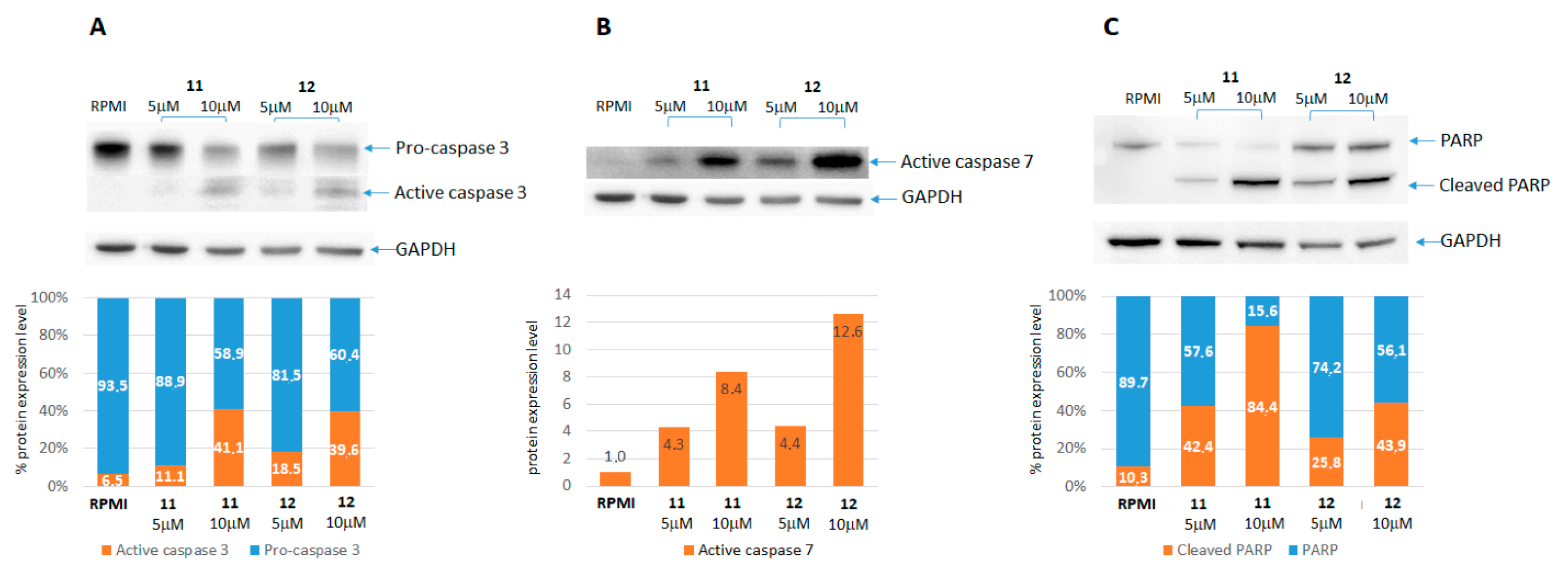
2.2.2. Caspases and PARP Activation
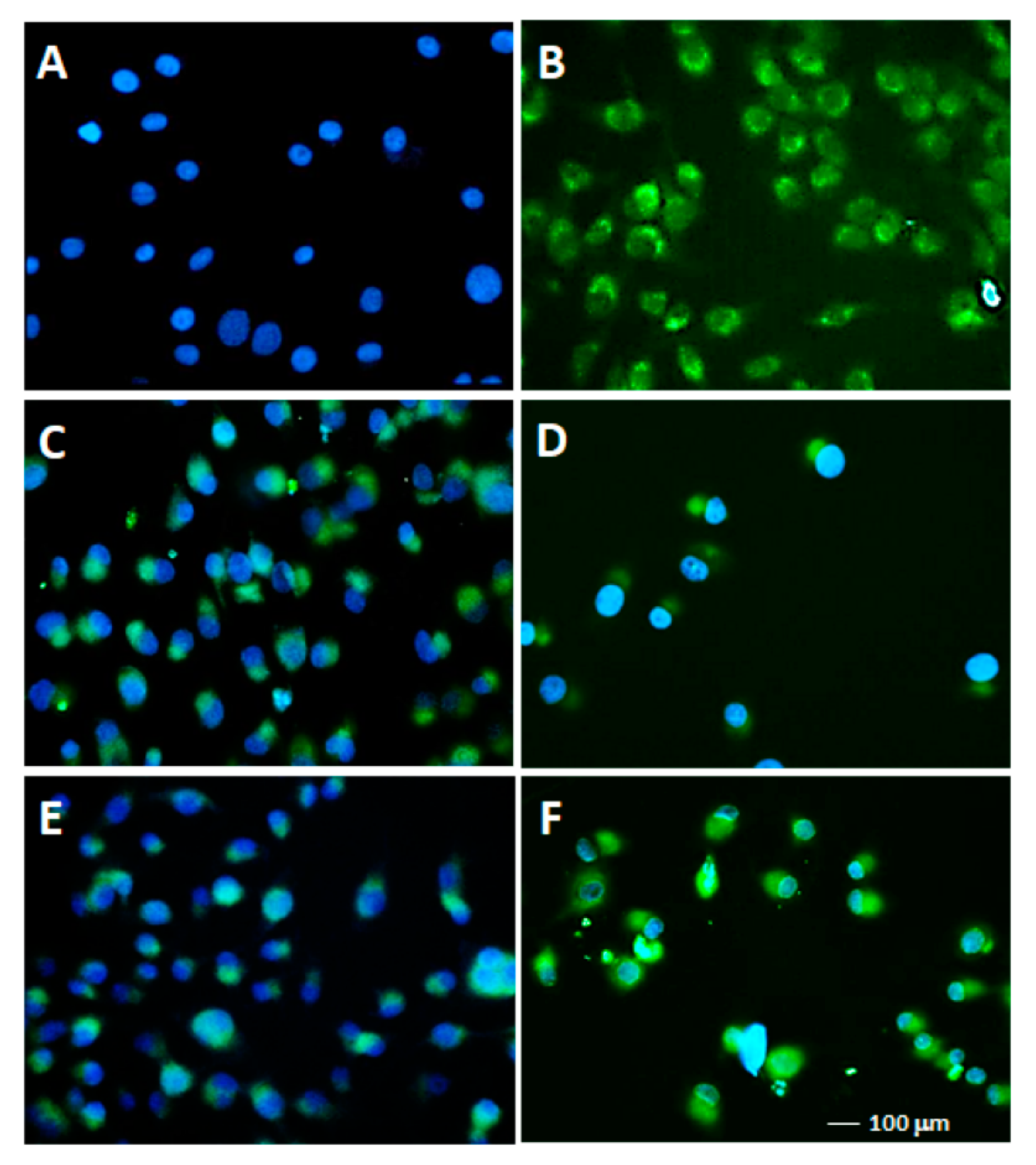
2.2.3. TUNEL Assays
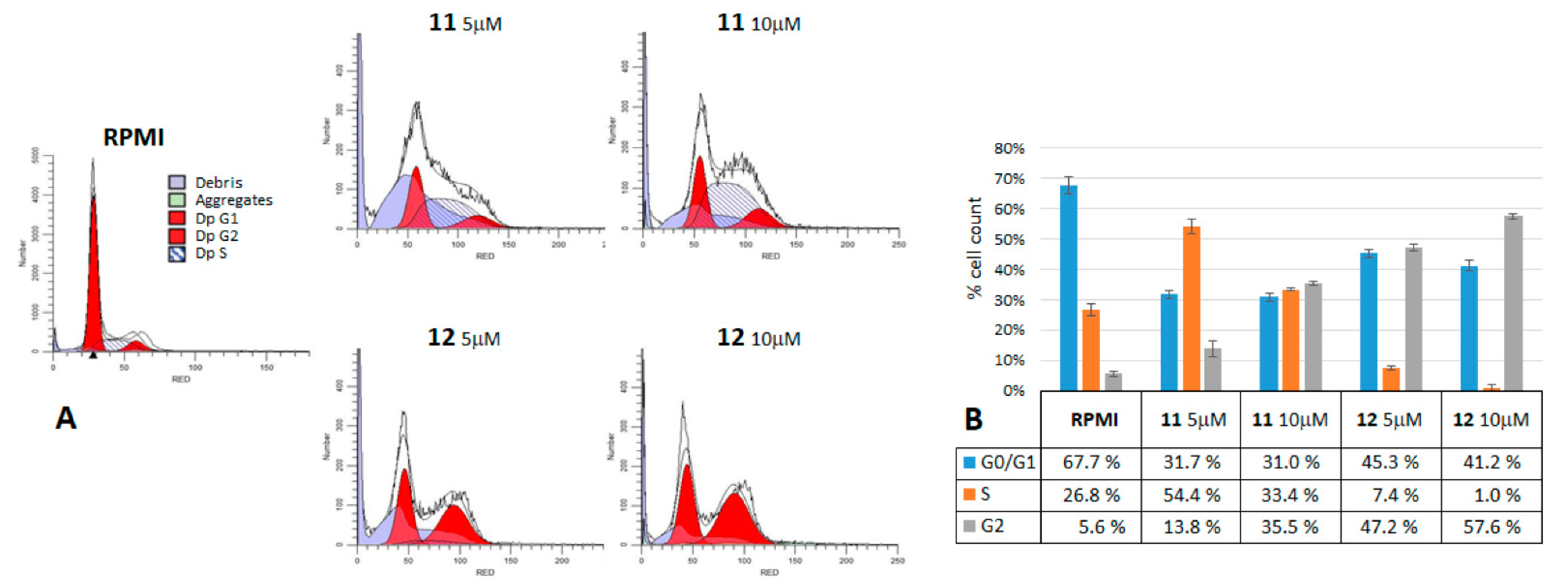
2.3. Cell Cycle Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Lines
- CN-mel (CN), derived from melanoma lymph node metastasis, carrying a NRASQ61R gene mutation;
- PR-mel (PR), derived from melanoma cutaneous metastasis, carrying a BRAFV600R gene mutation;
- A375, derived from melanoma cutaneous metastasis, carrying CDKN2AE61*-E69* gene mutations and a BRAFV600E gene mutation;
- SK-mel (SK), derived from melanoma lymph node metastasis, carrying a PTENT167A gene mutation;
- GR-mel (GR), derived from a primary melanoma, carrying a homozygous deletion in the CDKN2B gene.
4.2. Reagents
4.3. Cell Proliferation Assays
4.3.1. Dose-Response Assays
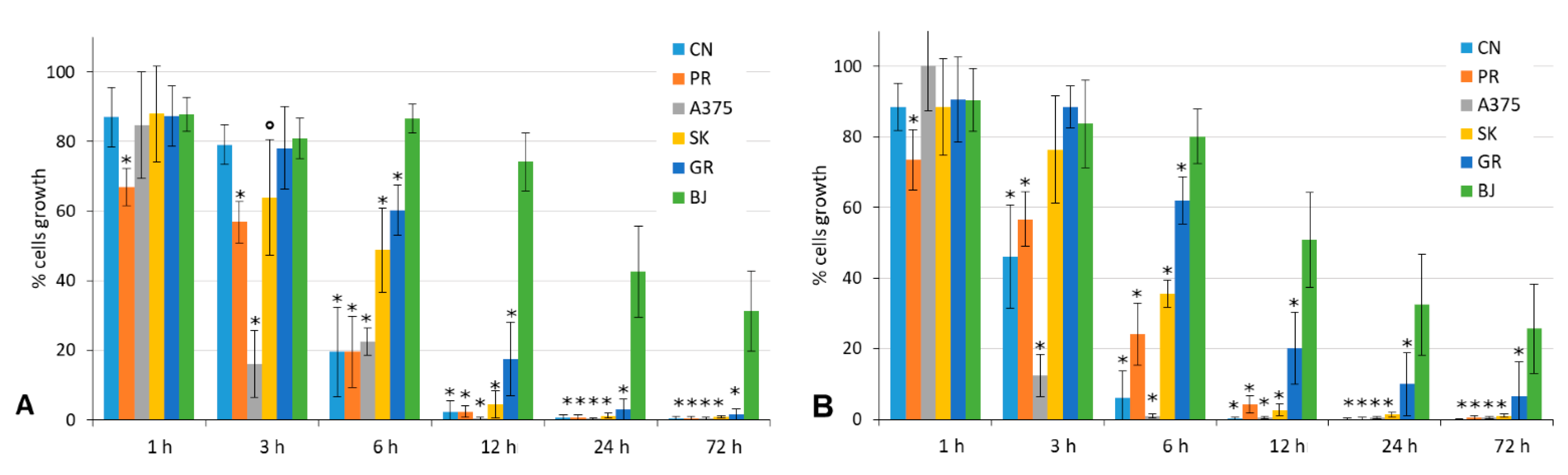
4.3.2. Wash-Out Assays
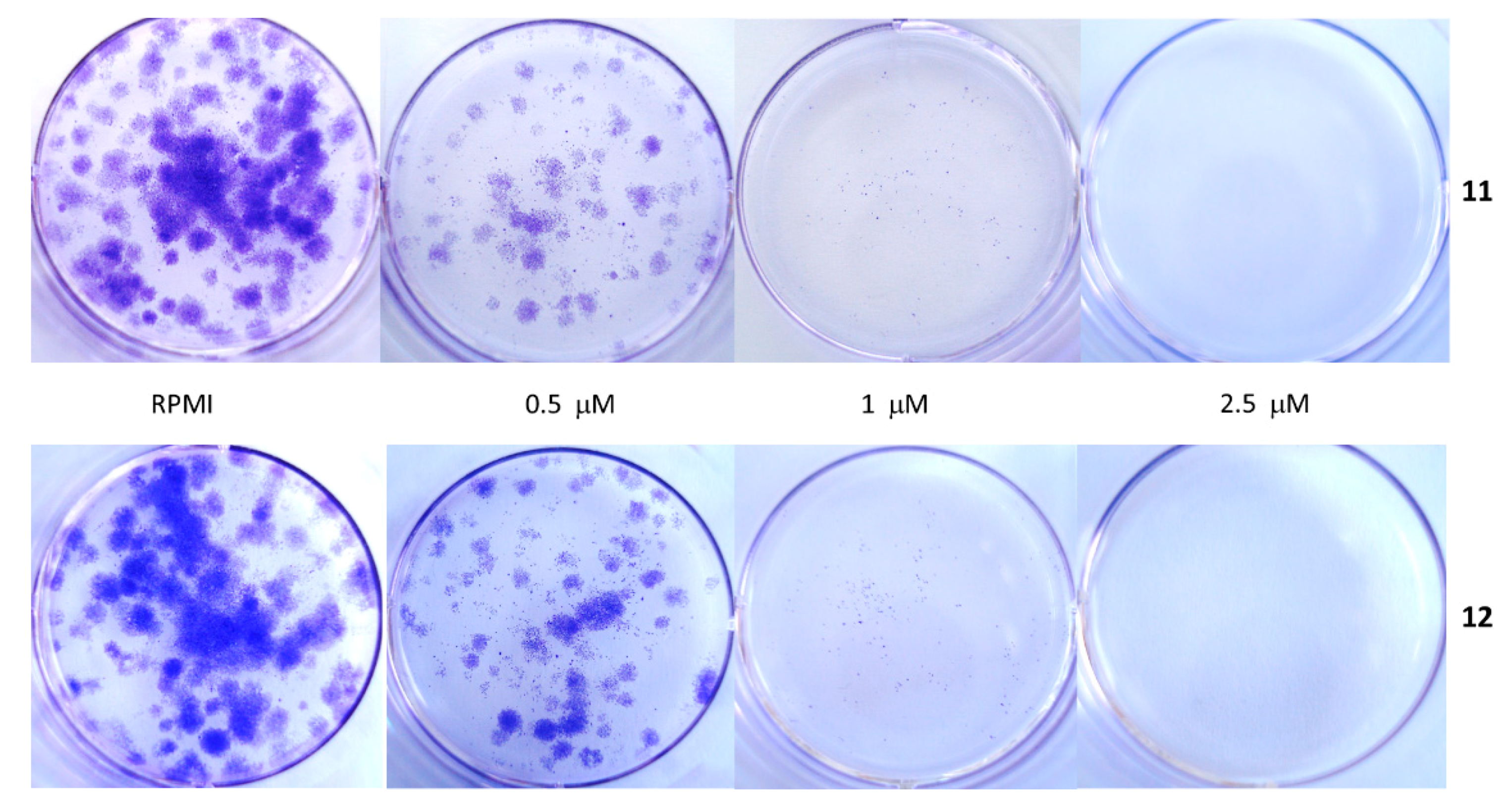
4.3.3. Clonogenic Cell Survival Assays
4.4. Apoptosis Assays
4.4.1. Annexin V
4.4.2. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) Assays
4.4.3. Western Blot
4.5. Cell Cycle Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berwick, M.; Buller, D.B.; Cust, A.; Gallagher, R.; Lee, T.K.; Meyskens, F.; Pandey, S.; Thomas, N.E.; Veierød, M.B.; Ward, S. Melanoma Epidemiology and Prevention. In Melanoma. Cancer Treatment and Research; Kaufman, H.L., Mehnert, J.M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 167, pp. 17–50. [Google Scholar] [CrossRef] [Green Version]
- Sacchetto, L.; Zanetti, R.; Comber, H.; Bouchardy, C.; Brewster, D.H.; Broganelli, P.; Chirlaque, M.D.; Coza, D.; Galceran, J.; Gavin, A.; et al. Trends in incidence of thick, thin and in situ melanoma in Europe. Eur. J. Cancer 2018, 92, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- AIOM, AIRTUM, Fondazione AIOM, PASSI Working Group. I numeri del cancro in Italia 2020. Intermedia Edizioni. 2019. Available online: https://www.aiom.it/wp-content/uploads/2020/10/2020_Numeri_Cancro-operatori_web.pdf (accessed on 8 April 2021).
- Koshenkov, V.P.; Broucek, J.; Kaufman, H.L. Surgical Management of Melanoma. In Melanoma. Cancer Treatment and Research; Kaufman, H.L., Mehnert, J.M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 167, pp. 149–180. [Google Scholar] [CrossRef]
- Wilson, M.A.; Schuchter, L.M. Chemotherapy for Melanoma. In Melanoma. Cancer Treatment and Research; Kaufman, H.L., Mehnert, J.M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 167, pp. 209–229. [Google Scholar] [CrossRef]
- Lombardo, N.; Della Corte, M.; Pelaia, C.; Piazzetta, G.; Lobello, N.; Del Duca, E.; Bennardo, L.; Nisticò, S.P. Primary Mucosal Melanoma Presenting with a Unilateral Nasal Obstruction of the Left Inferior Turbinate. Medicina 2021, 57, 359. [Google Scholar] [CrossRef]
- Coricovac, D.; Dehelean, C.; Moaca, E.A.; Pinzaru, I.; Bratu, T.; Navolan, D.; Boruga, O. Cutaneous Melanoma—A Long Road from Experimental Models to Clinical Outcome: A Review. Int. J. Mol. Sci. 2018, 19, 1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: Current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef]
- Palmieri, G.; Ombra, M.N.; Colombino, M.; Casula, M.; Sini, M.C.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple molecular pathways in melanomagenesis: Characterization of therapeutic targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Agarwala, S.S.; Botti, G.; Budillon, A.; Davies, M.A.; Dummer, R.; Ernstoff, M.; Ferrone, S.; Formenti, S.; Gajewski, T.F.; et al. Perspectives in melanoma: Meeting report from the Melanoma Bridge (November 29th–1 December 1st, 2018, Naples, Italy). J. Transl. Med. 2019, 17, 234. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Flaherty, K.; Goff, S. Emerging Strategies in Systemic Therapy for the Treatment of Melanoma. Am. Soc. Clin. Oncol. Educ. Book 2018, 23, 751–758. [Google Scholar] [CrossRef]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving impact of long-term survival results on metastatic melanoma treatment. J. Immunother. Cancer 2020, 8, e000948. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Gibney, G.T.; Sondak, V.K.; Smalley, K.S. Beyond BRAF: Where next for melanoma therapy? Br. J. Cancer 2015, 112, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.; Heinzerling, L.; Bhardwaj, N.; Friedlander, P. Current Melanoma Treatments: Where Do We Stand? Cancers 2021, 13, 221. [Google Scholar] [CrossRef]
- Joshi, S.; Durden, D.L. Combinatorial Approach to Improve Cancer Immunotherapy: Rational Drug Design Strategy to Simultaneously Hit Multiple Targets to Kill Tumor Cells and to Activate the Immune System. J. Oncol. 2019, 2019, 5245034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaria, B.; Sood, S.; Raad, C.; Khanafer, J.; Jayachandiran, R.; Pupulin, A.; Grewal, S.; Okoko, M.; Arora, M.; Miles, L.; et al. Natural Health Products (NHP’s) and Natural Compounds as Therapeutic Agents for the Treatment of Cancer; Mechanisms of Anti-Cancer Activity of Natural Compounds and Overall Trends. Int. J. Mol. Sci. 2020, 21, 8480. [Google Scholar] [CrossRef] [PubMed]
- Ombra, M.N.; Paliogiannis, P.; Stucci, L.S.; Colombino, M.; Casula, M.; Sini, M.C.; Manca, A.; Palomba, G.; Stanganelli, I.; Mandalà, M.; et al. Dietary compounds and cutaneous malignant melanoma: Recent advances from a biological perspective. Nutr. Metab. 2019, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Chinembiri, T.N.; du Plessis, L.H.; Gerber, M.; Hamman, J.H.; du Plessis, J. Review of natural compounds for potential skin cancer treatment. Molecules 2014, 19, 11679–11721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Luo, Y.; Tian, M.; Zhang, S.Y.; Lu, R.; Wang, J.H.; Kasimu, R.; Li, X. Plant natural products: From traditional compounds to new emerging drugs in cancer therapy. Cell Prolif. 2014, 47, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Marchiani, A.; Rozzo, C.; Fadda, A.; Delogu, G.; Ruzza, P. Curcumin and curcumin-like molecules: From spice to drugs. Curr. Med. Chem. 2014, 21, 204–222. [Google Scholar] [CrossRef]
- Faião-Flores, F.; Quincoces Suarez, J.A.; Maria-Engler, S.S.; Soto-Cerrato, V.; Pérez-Tomás, R.; Maria, D.A. The curcumin analog DM-1 induces apoptotic cell death in melanoma. Tumor Biol. 2013, 34, 1119–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faião-Flores, F.; Suarez, J.A.Q.; Fruet, A.C.; Maria-Engler, S.S.; Pardi, P.C.; Maria, D.A. Curcumin analog DM-1 in monotherapy or combinatory treatment with Dacarbazine as a strategy to inhibit in vivo melanoma progression. PLoS ONE 2015, 10, e0118702. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Evangelopoulos, A.; Schizas, N.; Kazazis, C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer. Res. 2015, 35, 645–651. [Google Scholar]
- Pignanelli, C.; Ma, D.; Noel, M.; Ropat, J.; Mansour, F.; Curran, C.; Pupulin, S.; Larocque, K.; Wu, J.; Liang, G.; et al. Selective Targeting of Cancer Cells by Oxidative Vulnerabilities with Novel Curcumin Analogs. Sci. Rep. 2017, 7, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, F.C.; Kumar, N.V.A.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019, 177, 76–104. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohamamdinejad, R.; Yaribeygi, H.; Serban, M.C.; Orafai, H.M.; Sahebkar, A. Curcumin Therapeutic Modulation of the Wnt Signaling Pathway. Curr. Pharm. Biotechnol. 2020, 21, 1006–1015. [Google Scholar] [CrossRef]
- Nisticò, S.; Tamburi, F.; Bennardo, L.; Dastoli, S.; Schipani, G.; Caro, G.; Fortuna, M.C.; Rossi, A. Treatment of telogen effluvium using a dietary supplement containing Boswellia serrata, Curcuma longa, and Vitis vinifera: Results of an observational study. Dermatol. Ther. 2019, 32, e12842. [Google Scholar] [CrossRef] [PubMed]
- Pisano, M.; Pagnan, G.; Dettori, M.A.; Cossu, S.; Caffa, I.; Sassu, I.; Emionite, L.; Fabbri, D.; Cilli, M.; Pastorino, F.; et al. Enhanced anti-tumor activity of a new curcumin-related compound against melanoma and neuroblastoma cells. Mol. Cancer 2010, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozzo, C.; Fanciulli, M.; Fraumene, C.; Corrias, A.; Cubeddu, T.; Sassu, I.; Cossu, S.; Nieddu, V.; Galleri, G.; Azara, E.; et al. Molecular changes induced by the curcumin analogue D6 in human melanoma cells. Mol. Cancer 2013, 12, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisano, M.; Palomba, A.; Tanca, A.; Pagnozzi, D.; Uzzau, S.; Addis, M.F.; Dettori, M.A.; Fabbri, D.; Palmieri, G.; Rozzo, C. Protein expression changes induced in a malignant melanoma cell line by the curcumin analogue compound D6. BMC Cancer 2016, 16, 317. [Google Scholar] [CrossRef] [Green Version]
- Dettori, M.A.; Pisano, M.; Rozzo, C.; Delogu, G.; Fabbri, D. Synthesis of hydroxylated biphenyl derivatives bearing an α,β-unsaturated ketone as a lead structure for the development of drug candidates against malignant melanoma. ChemMedChem 2021, 16, 1022–1033. [Google Scholar] [CrossRef]
- Casula, M.; Muggiano, A.; Cossu, A.; Budroni, M.; Caracò, C.; Ascierto, P.A.; Pagani, E.; Stanganelli, I.; Canzanella, S.; Sini, M.; et al. Role of key-regulator genes in melanoma susceptibility and pathogenesis among patients from South Italy. BMC Cancer 2009, 9, 352. [Google Scholar] [CrossRef] [Green Version]
- GraphPad Prism, Version 7.00 for Windows. Scientific 2D Graphing and Statistics Software. GraphPad Software: La Jolla, CA, USA, 2016. Available online: www.graphpad.com (accessed on 25 May 2021).
- UVI-1D, Version 16.12.0.0 for Windows. Image Acquisition, Enhancement and Analysis Software. UVITec Ltd.: Cambridge, UK, 2015. Available online: www.uvitec.co.uk (accessed on 25 May 2021).
- Modfit LT, Version 4.1 for Windows. Modeling Software for Flow Cytometry Histograms. Verity Software House: Topsham, ME, USA, 2014. Available online: www.vsh.com (accessed on 25 May 2021).
- Fujisawa, S.; Atsumi, T.; Murakami, Y.; Kadoma, Y. Dimerization, ROS formation, and biological activity of o-methoxyphenols. Arch. Immunol. Ther. Exp. 2005, 53, 28–38. [Google Scholar]
- Bringmann, G.; Gulder, T.; Gulder, T.A.; Breuning, M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 2011, 111, 563–639. [Google Scholar] [CrossRef] [PubMed]
- Alhassan, A.M.; Abdullahi, M.I.; Uba, A.; Umar, A. Prenylation of Aromatic Secondary Metabolites: A New Frontier for Development of Novel Drugs. Trop. J. Pharm. Res. 2014, 13, 307–314. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberle, J.; Fecker, L.F.; Hossini, A.M.; Kurbanov, B.M.; Fechner, H. Apoptosis pathways and oncolytic adenoviral vectors: Promising targets and tools to overcome therapy resistance of malignant melanoma. Exp. Dermatol. 2008, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, 6. [Google Scholar] [CrossRef]
- Rozzo, C.; Sanna, D.; Garribba, E.; Serra, M.; Cantara, A.; Palmieri, G.; Pisano, M. Antitumoral effect of vanadium compounds in malignant melanoma cell lines. J. Inorg. Biochem. 2017, 174, 14–24. [Google Scholar] [CrossRef]
- Keshelava, N.; Tsao-Wei, D.; Reynolds, C.P. Pyrazoloacridine is active in multidrug-resistant neuroblastoma cell lines with nonfunctional p53. Clin. Cancer Res. 2003, 9, 3492–3502. [Google Scholar]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | 11 | 12 | ||||||
|---|---|---|---|---|---|---|---|---|
| T24 | T72 | T24 | T72 | |||||
| IC50 | CI 95% | IC50 | CI 95% | IC50 | CI 95% | IC50 | CI 95% | |
| CN-mel | 15.5 μM | 11.14 to 28.58 | 1.7 μM | 1.63 to 1.75 | 14.5 μM | 6.46 to 10.30 | 1.5 μM | 1.33 to 1.49 |
| PR-mel | 7.1 μM | 6.55 to 7.79 | 2.5 μM | 2.54 to 2.73 | 7.7μM | 4.93 to 5.60 | 1.7 μM | 1.85 to 2.08 |
| A375 | 2.9 μM | 2.84 to 3.03 | 1.5 μM | 1.27 to 1.36 | 3.8 μM | 2.32 to 3.10 | 1.4 μM | 1.27 to 1.42 |
| SK-mel | 7.7 μM | 6.91 to 8.84 | 2.5 μM | 2.46 to 2.56 | 19 μM | 7.02 to 18.90 | 2.2 μM | 2.19 to 2.36 |
| GR-mel | 6.4 μM | 5.86 to 7.06 | 3.2 μM | 2.88 to 3.15 | 12.3 μM | 7.41 to 11.02 | 3.4 μM | 2.75 to 3.28 |
| BJ | >32 μM | 6.8 μM | 6.50 to 7.26 | >32 μM | 6.0 μM | 5.55 to 6.90 | ||
| A | B | ||||||
|---|---|---|---|---|---|---|---|
| MM Cell Line | % Clonal Growth | 11 | 12 | ||||
| Liquid Medium | Semi-Solid Medium | Liquid Medium | Semi-Solid Medium | ||||
| In Continuum | Wash Out | In Continuum | Wash Out | ||||
| A375 | 4.65 | -- | -- | 2.5 μM | -- | -- | 2.5 μM |
| PR-mel | 9.86 | -- | -- | 10 μM | -- | -- | 10 μM |
| CN-mel | 9.61 | 2.5 μM | 10 μM | 5 μM | 1 μM | 10 μM | 5 μM |
| SK-mel | 7.00 | 1 μM | 5 μM | -- | 1 μM | 5 μM | -- |
| GR-mel | not able to develop clonal growth from single cells, both in the liquid and semi-solid medium | ||||||
| % of Apoptotic Cells | ||||||
|---|---|---|---|---|---|---|
| Cell Line | RPMI | 11 | 12 | |||
| 5 μM | 10 μM | 5 μM | 10 μM | |||
| 1 | SK-mel | 4.2 ± 0.9 | 62.6 ± 1.8 * | 67.1 ± 1.1 * | 42.0 ± 1.2 * | 49.3 ± 0.5 * |
| 2 | PR-mel | 5.5 ± 0.7 | 17.7 ± 0.9 * | 68.6 ± 0.4 * | 21.7 ± 0.9 * | 33.7 ± 1.7 * |
| 3 | CN-mel | 2.2 ± 0.4 | 12.6 ± 0.9 * | 31.4 ± 1.1 * | 18.2 ± 1.2 * | 34.2 ± 0.5 * |
| 4 | GR-mel | 3.7 ± 1.0 | 18.1 ± 0.2 * | 34.6 ± 1.0 * | 6.7 ± 1.1 | 21.5 ± 2.2 * |
| 5 | A375 | 1.7 ± 0.7 | 10.3 ± 1.3 * | 19.2 ± 1.1 | 12.3 ± 2.3 | 18.7 ± 0.7 * |
| 6 | BJ | 6.1 ± 1.3 | 5.3 ± 0.8 | 16.3 ± 3.0 | 6.9 ± 2.4 | 4.7 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisano, M.; Dettori, M.A.; Fabbri, D.; Delogu, G.; Palmieri, G.; Rozzo, C. Anticancer Activity of Two Novel Hydroxylated Biphenyl Compounds toward Malignant Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 5636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115636
Pisano M, Dettori MA, Fabbri D, Delogu G, Palmieri G, Rozzo C. Anticancer Activity of Two Novel Hydroxylated Biphenyl Compounds toward Malignant Melanoma Cells. International Journal of Molecular Sciences. 2021; 22(11):5636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115636
Chicago/Turabian StylePisano, Marina, Maria Antonietta Dettori, Davide Fabbri, Giovanna Delogu, Giuseppe Palmieri, and Carla Rozzo. 2021. "Anticancer Activity of Two Novel Hydroxylated Biphenyl Compounds toward Malignant Melanoma Cells" International Journal of Molecular Sciences 22, no. 11: 5636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115636