
Structure and Dynamics of Meprin β in Complex with a Hydroxamate-Based Inhibitor

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
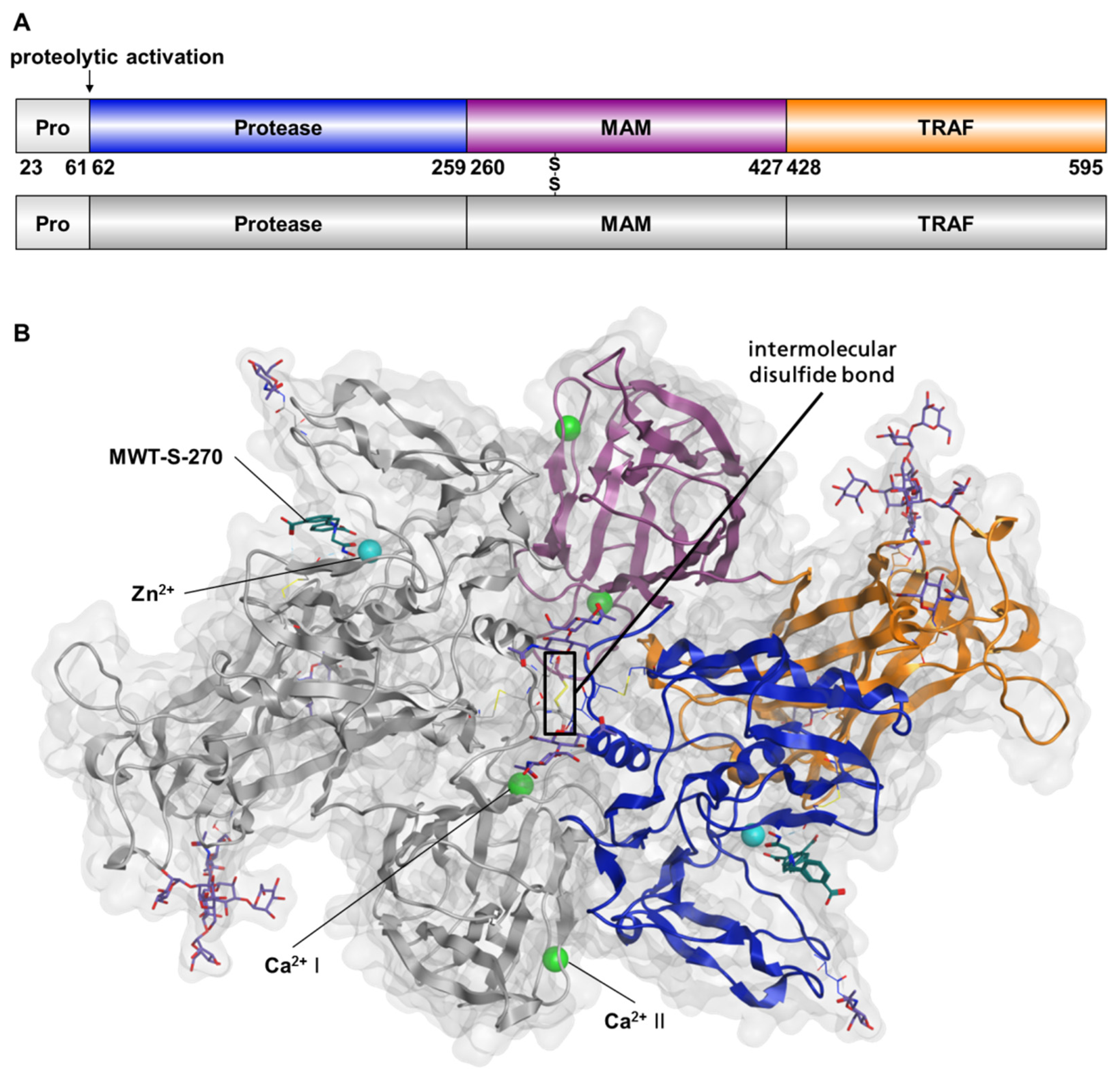
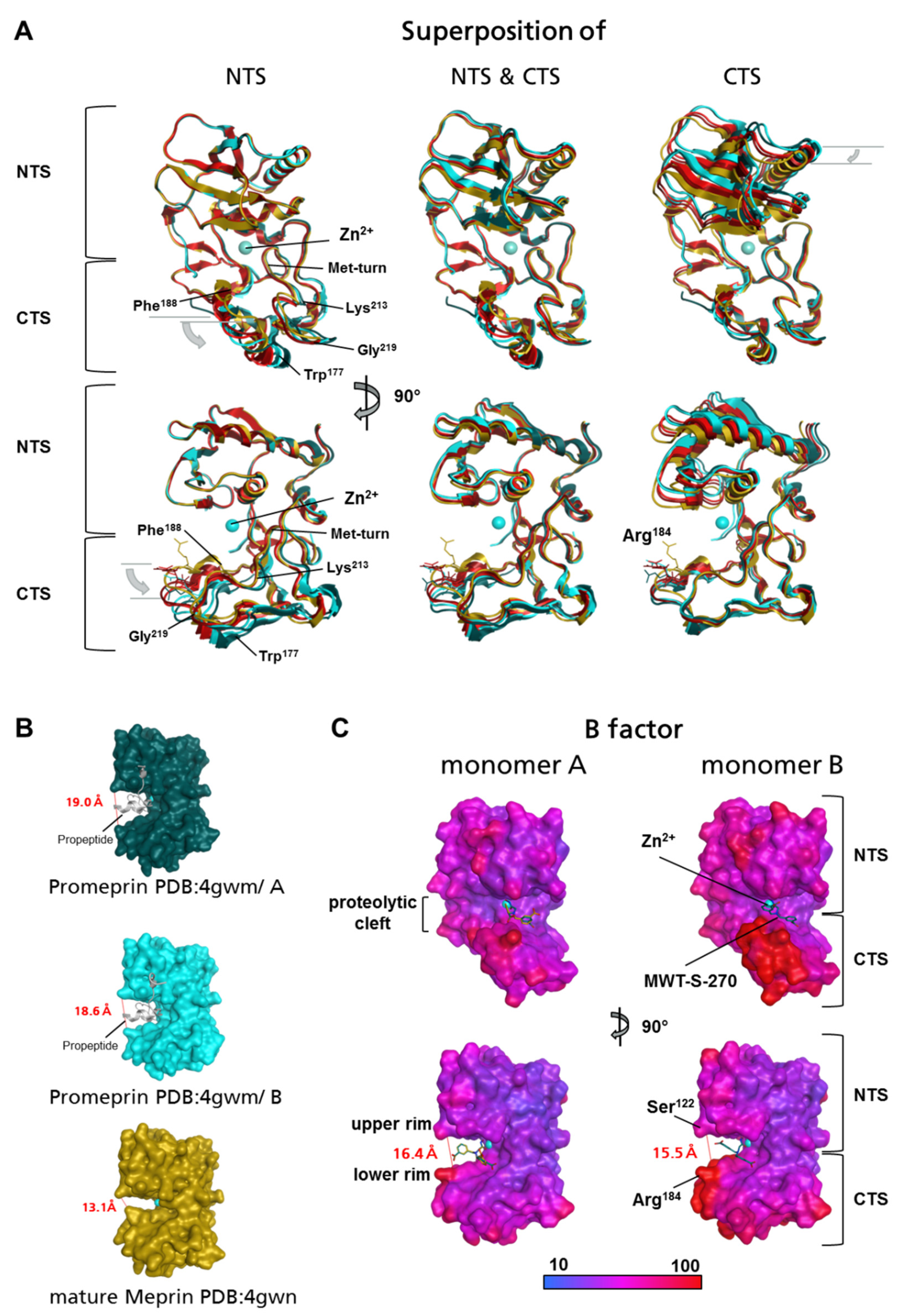
2.1. Preparation of Meprin β62–595 (MβΔC) for Crystallization
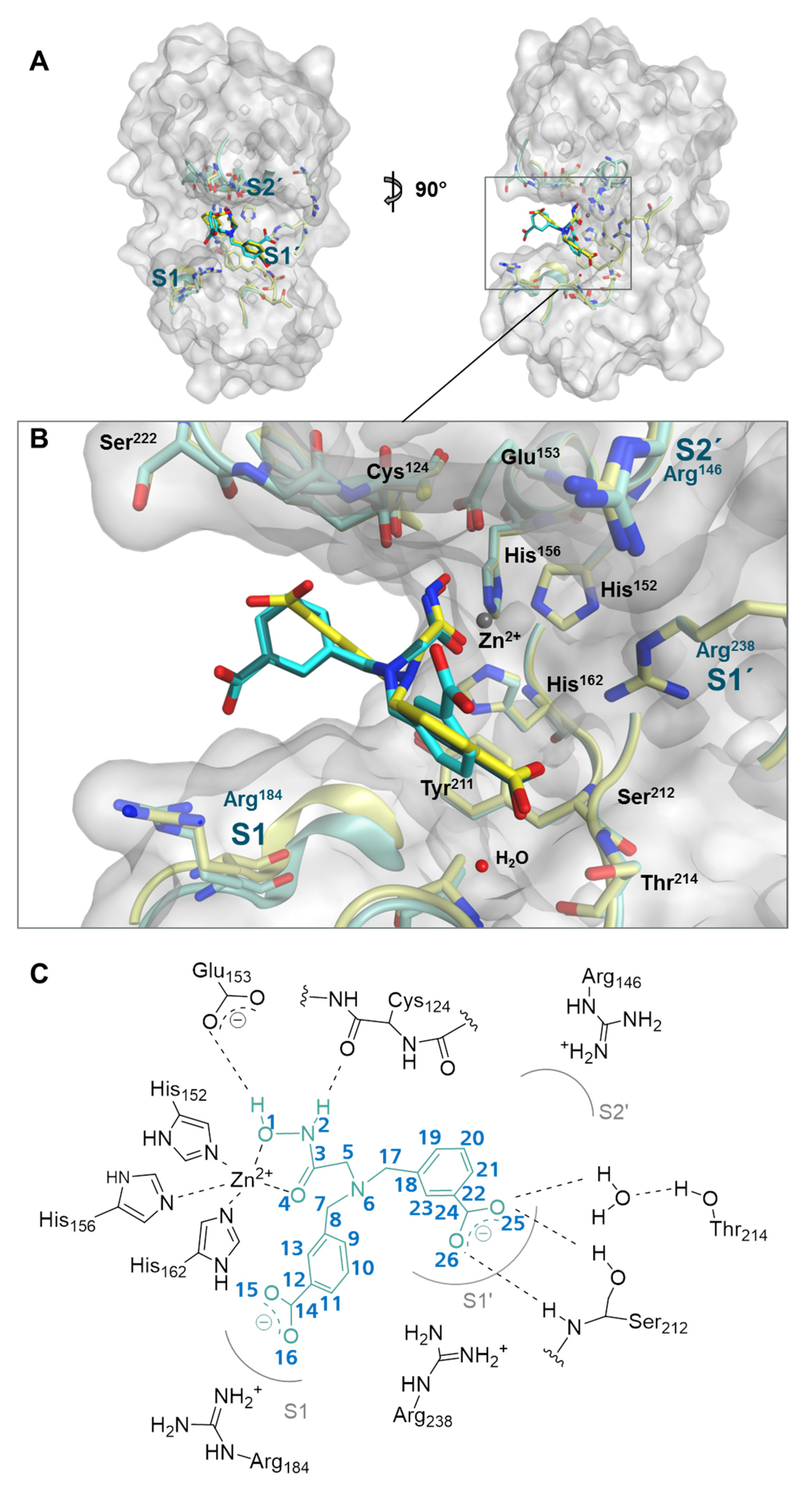
2.2. Structure of Meprin β62–595 (MβΔC) in Complex with MWT-S-270
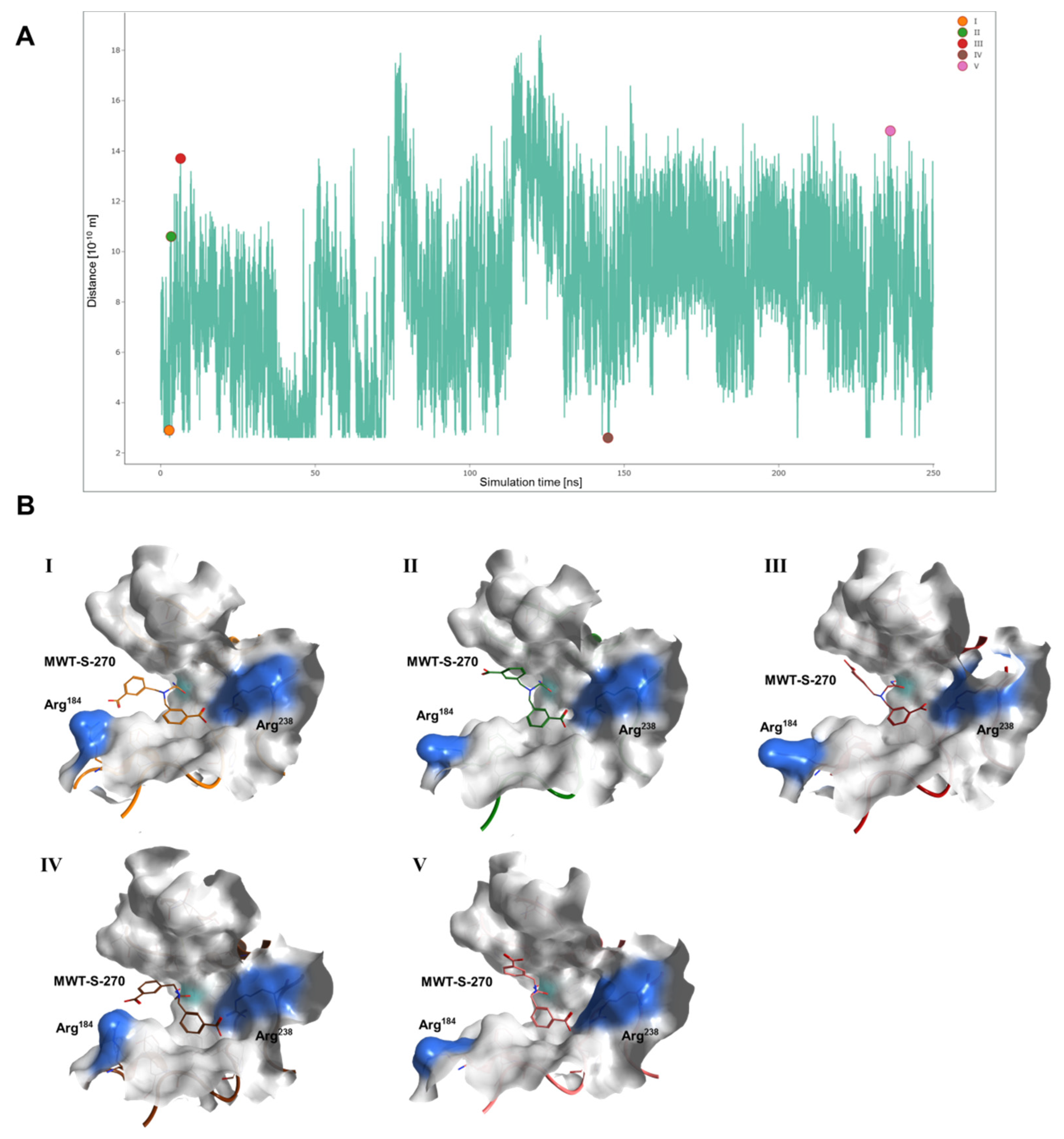
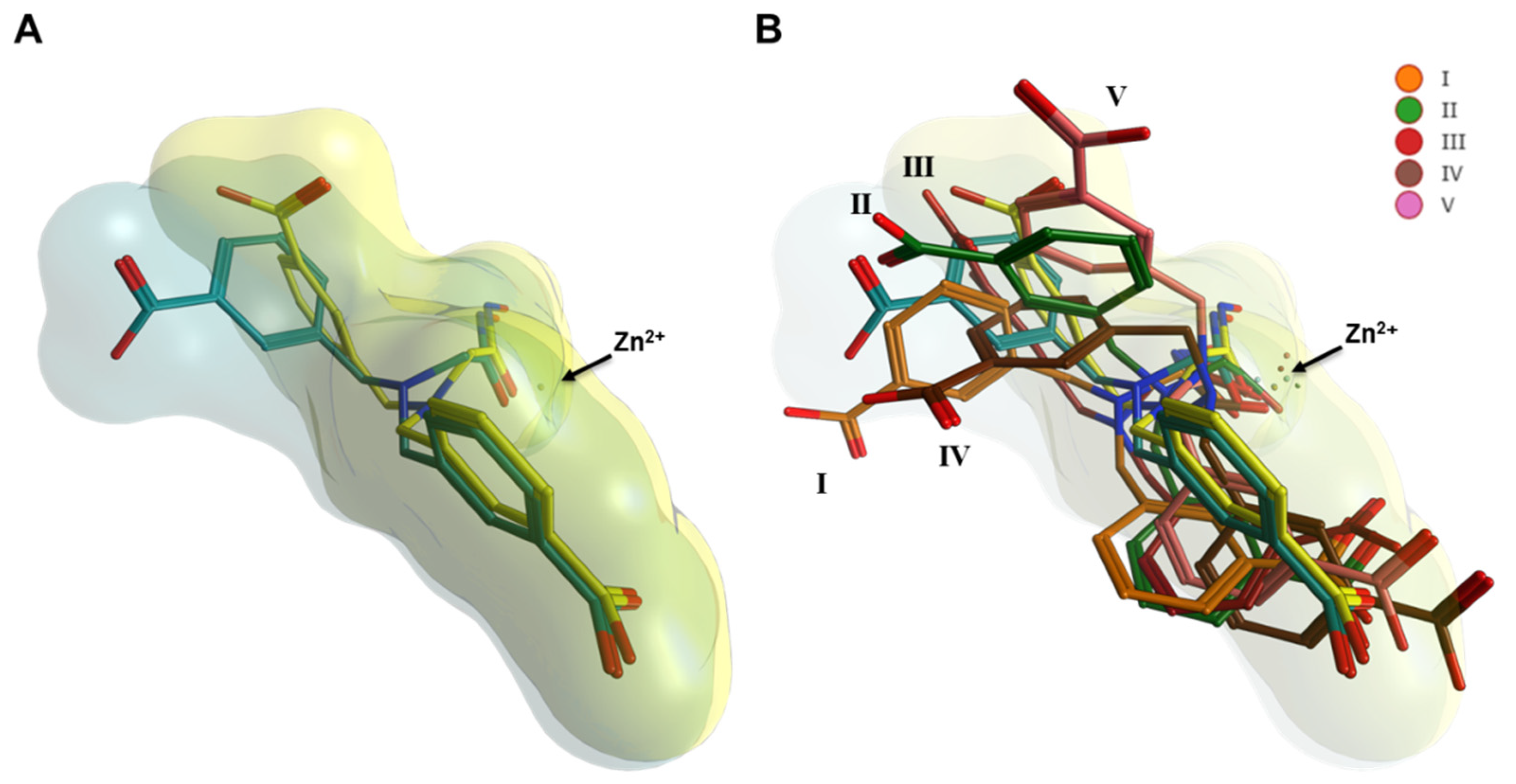
2.3. Interaction Analysis of Meprin β62–595 (MβΔC) and MWT-S-270 by Calorimetry and Molecular Dynamics (MD)–Simulations
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Mature Meprin β62–595 (MβΔC)
4.2. Deglycosylation of Meprin β62–595 (MβΔC)
4.3. Determination of Meprin β Activity
4.4. Crystallization of Meprin β62–595 (MβΔC), Data Collection and Structure Elucidation
4.5. Isothermal Titration Calorimetry (ITC)
4.6. Molecular Dynamic (MD) Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sterchi, E.E.; Green, J.R.; Lentze, M.J. Non-pancreatic hydrolysis of N-benzoyl-l-tyrosyl-p-aminobenzoic acid (PABA-peptide) in the human small intestine. Clin. Sci. 1982, 62, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Beynon, R.J.; Shannon, J.D.; Bond, J.S. Purification and characterization of a metallo-endoproteinase from mouse kidney. Biochem. J. 1981, 199, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Sterchi, E.E.; Stöcker, W.; Bond, J.S. Meprins, membrane-bound and secreted astacin metalloproteinases. Mol. Asp. Med. 2008, 29, 309–328. [Google Scholar] [CrossRef] [Green Version]
- Broder, C.; Arnold, P.; Vadon-Le Goff, S.; Konerding, M.A.; Bahr, K.; Müller, S.; Overall, C.M.; Bond, J.S.; Koudelka, T.; Tholey, A.; et al. Metalloproteases meprin α and meprin β are C- and N-procollagen proteinases important for collagen assembly and tensile strength. Proc. Natl. Acad. Sci. USA 2013, 110, 14219–14224. [Google Scholar] [CrossRef] [Green Version]
- Prox, J.; Arnold, P.; Becker-Pauly, C. Meprin α and meprin β: Procollagen proteinases in health and disease. Matrix Biol. 2015, 44–46, 7–13. [Google Scholar] [CrossRef]
- Arnold, P.; Otte, A.; Becker-Pauly, C. Meprin metalloproteases: Molecular regulation and function in inflam-mation and fibrosis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Broder, C.; Becker-Pauly, C. The metalloproteases meprin α and meprin β: Unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem. J. 2013, 450, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, P.E.; Bond, J.S. A latent proteinase in mouse kidney membranes. Characterization and relationship to meprin. J. Biol. Chem. 1988, 263, 13419–13426. [Google Scholar] [CrossRef]
- Gorbea, C.M.; Flannery, A.V.; Bond, J.S. Homo- and heterotetrameric forms of the membrane-bound metal-loendopeptidases meprin A and B. Arch. Biochem. Biophys. 1991, 290, 549–553. [Google Scholar] [CrossRef]
- Bertenshaw, G.P.; Norcum, M.T.; Bond, J.S. Structure of homo- and hetero-oligomeric meprin metalloproteases. Dimers, tetramers, and high molecular mass multimers. J. Biol. Chem. 2003, 278, 2522–2532. [Google Scholar] [CrossRef] [Green Version]
- Peters, F.; Scharfenberg, F.; Colmorgen, C.; Armbrust, F.; Wichert, R.; Arnold, P.; Potempa, B.; Potempa, J.; Pie-trzik, C.U.; Häsler, R.; et al. Tethering soluble meprin α in an enzyme complex to the cell surface affects IBD-associated genes. FASEB J. 2019, 33, 7490–7504. [Google Scholar] [CrossRef]
- Jiang, W.; Gorbea, C.M.; Flannery, A.V.; Beynon, R.J.; Grant, G.A.; Bond, J.S. The alpha subunit of meprin A. Molecular cloning and sequencing, differential expression in inbred mouse strains, and evidence for divergent evolution of the alpha and beta subunits. J. Biol. Chem. 1992, 267, 9185–9193. [Google Scholar] [CrossRef]
- Beckmann, G.; Bork, P. An adhesive domain detected in functionally diverse receptors. Trends Biochem. Sci. 1993, 18, 40–41. [Google Scholar] [CrossRef]
- Aricescu, A.R.; Hon, W.-C.; Siebold, C.; Lu, W.; van der Merwe, P.A.; Jones, E.Y. Molecular analysis of receptor protein tyrosine phosphatase mu-mediated cell adhesion. EMBO J. 2006, 25, 701–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uren, A.G.; Vaux, D.L. TRAF protiens and meprins share a conserved domain. Trends Biochem. Sci. 1996, 21, 244–245. [Google Scholar] [CrossRef]
- Laitaoja, M.; Valjakka, J.; Jänis, J. Zinc coordination spheres in protein structures. Inorg. Chem. 2013, 52, 10983–10991. [Google Scholar] [CrossRef] [PubMed]
- Marchand, P.; Tang, J.; Johnson, G.D.; Bond, J.S. COOH-terminal proteolytic processing of secreted and mem-brane forms of the alpha subunit of the metalloprotease meprin A. Requirement of the I domain for processing in the endoplasmic reticulum. J. Biol. Chem. 1995, 270, 5449–5456. [Google Scholar] [CrossRef] [Green Version]
- Hahn, D.; Lottaz, D.; Sterchi, E.E. C-cytosolic and transmembrane domains of the N-benzoyl-L-tyrosyl-p-aminobenzoic acid hydrolase alpha subunit (human meprin alpha) are essential for its retention in the endoplasmic reticulum and C-terminal processing. Eur. J. Biochem. 1997, 247, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Gomis-Rüth, F.X.; Huber, R.; Zwilling, R.; Stöcker, W. Structure of astacin and implications for acti-vation of astacins and zinc-ligation of collagenases. Nature 1992, 358, 164–167. [Google Scholar] [CrossRef]
- Gomis-Rüth, F.X.; Stöcker, W.; Huber, R.; Zwilling, R.; Bode, W. Refined 1.8 A X-ray crystal structure of astacin, a zinc-endopeptidase from the crayfish Astacus astacus L. Structure determination, refinement, molecular structure and comparison with thermolysin. J. Mol. Biol. 1993, 229, 945–968. [Google Scholar] [CrossRef]
- Arolas, J.L.; Broder, C.; Jefferson, T.; Guevara, T.; Sterchi, E.E.; Bode, W.; Stocker, W.; Becker-Pauly, C.; Go-mis-Ruth, F.X. Structural basis for the sheddase function of human meprin metalloproteinase at the plasma membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 16131–16136. [Google Scholar] [CrossRef] [Green Version]
- Bode, W.; Gomis-Rüth, F.-X.; Stöckler, W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. 1993, 331, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Gomis-Rüth, F.X.; Trillo-Muyo, S.; Stöcker, W. Functional and structural insights into astacin metallopeptidases. Biol. Chem. 2012, 393, 1027–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Bond, J.S. Families of metalloendopeptidases and their relationships. FEBS Lett. 1992, 312, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Doll, B.A.; Villa, J.P.; Ishmael, F.T.; Bond, J.S. Zinc ligands in an astacin family metalloprotease meprin A. Biol. Chem. 2002, 383, 1167–1173. [Google Scholar] [CrossRef]
- Stöcker, W.; Ng, M.; Auld, D.S. Fluorescent oligopeptide substrates for kinetic characterization of the specifici-ty of Astacus protease. Biochemistry 1990, 29, 10418–10425. [Google Scholar] [CrossRef]
- Grams, F.; Dive, V.; Yiotakis, A.; Yiallouros, I.; Vassiliou, S.; Zwilling, R.; Bode, W.; Stöcker, W. Structure of astacin with a transition-state analogue inhibitor. Nat. Struct. Biol. 1996, 3, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bertenshaw, G.P.; Turk, B.E.; Hubbard, S.J.; Matters, G.L.; Bylander, J.E.; Crisman, J.M.; Cantley, L.C.; Bond, J.S. Marked differences between metalloproteases meprin A and B in substrate and peptide bond specificity. J. Biol. Chem. 2001, 276, 13248–13255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, J.P.; Bertenshaw, G.P.; Bond, J.S. Critical amino acids in the active site of meprin metalloproteinases for substrate and peptide bond specificity. J. Biol. Chem. 2003, 278, 42545–42550. [Google Scholar] [CrossRef] [Green Version]
- Kruse, M.-N.; Becker, C.; Lottaz, D.; Köhler, D.; Yiallouros, I.; Krell, H.-W.; Sterchi, E.E.; Stöcker, W. Human meprin alpha and beta homo-oligomers: Cleavage of basement membrane proteins and sensitivity to metallo-protease inhibitors. Biochem. J. 2004, 378, 383–389. [Google Scholar] [CrossRef] [Green Version]
- Ramsbeck, D.; Hamann, A.; Richter, G.; Schlenzig, D.; Geissler, S.; Nykiel, V.; Cynis, H.; Schilling, S.; Buchholz, M. Structure-guided design, synthesis, and characterization of next-generation meprin β inhibitors. J. Med. Chem. 2018, 61, 4578–4592. [Google Scholar] [CrossRef] [PubMed]
- Ramsbeck, D.; Hamann, A.; Schlenzig, D.; Schilling, S.; Buchholz, M. First insight into structure-activity rela-tionships of selective meprin β inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2428–2431. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Jäger, C.; Schlenzig, D.; Schilling, S.; Buchholz, M.; Ramsbeck, D. Tertiary-amine-based inhibitors of the astacin protease meprin α. ChemMedChem 2018, 13, 1619–1624. [Google Scholar] [CrossRef]
- Schlenzig, D.; Wermann, M.; Ramsbeck, D.; Moenke-Wedler, T.; Schilling, S. Expression, purification and initial characterization of human meprin β from Pichia pastoris. Protein Expr. Purif. 2015, 116, 75–81. [Google Scholar] [CrossRef]
- Yiallouros, I.; Kappelhoff, R.; Schilling, O.; Wegmann, F.; Helms, M.W.; Auge, A.; Brachtendorf, G.; Berkhoff, E.G.; Beermann, B.; Hinz, H.-J.; et al. Activation mechanism of pro-astacin: Role of the pro-peptide, tryptic and autoproteolytic cleavage and importance of precise amino-terminal processing. J. Mol. Biol. 2002, 324, 237–246. [Google Scholar] [CrossRef]
- Diederichs, K.; Karplus, P.A. Improved R-factors for diffraction data analysis in macromolecular crystallog-raphy. Nat. Struct. Biol. 1997, 4, 269–275. [Google Scholar] [CrossRef]
- Harding, M.M. Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Rüth, F.X.; Botelho, T.O.; Bode, W. A standard orientation for metallopeptidases. Biochim. Biophys. Acta 2012, 1824, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. ChemBioChem 2002, 3, 604. [Google Scholar] [CrossRef]
- Becker-Pauly, C.; Barre, O.; Schilling, O.; auf dem Keller, U.; Ohler, A.; Broder, C.; Schutte, A.; Kappelhoff, R.; Stöcker, W.; Overall, C.M. Proteomic analyses reveal an acidic prime side specificity for the astacin metal-loprotease family reflected by physiological substrates. Mol. Cell. Proteom. 2011, 10, M111.009233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, G.E.; Schirmer, R.H. Principles of Protein Structure; Springer: New York, NY, USA, 1979; ISBN 9780387903347. [Google Scholar]
- Marcus, Y.; Hefter, G. Ion pairing. Chem. Rev. 2006, 106, 4585–4621. [Google Scholar] [CrossRef] [PubMed]
- Shiroishi, M.; Yokota, A.; Tsumoto, K.; Kondo, H.; Nishimiya, Y.; Horii, K.; Matsushima, M.; Ogasahara, K.; Yu-tani, K.; Kumagai, I. Structural evidence for entropic contribution of salt bridge formation to a protein anti-gen-antibody interaction: The case of hen lysozyme-HyHEL-10 Fv complex. J. Biol. Chem. 2001, 276, 23042–23050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, A.; Wermann, M.; Demuth, H.-U.; Yoshimoto, T.; Ramsbeck, D.; Schlenzig, D.; Schilling, S. Continuous assays for meprin alpha and beta using prolyl tripeptidyl aminopeptidase (PtP) from Porphyromonas gingivalis. Anal. Biochem. 2018, 559, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Nettleship, J. Structural biology of glycoproteins. In Glycosylation; Petrescu, S., Ed.; InTech: Rijeka, Croatia, 2012; ISBN 978-953-51-0771-2. [Google Scholar]
- Trimble, R.B.; Atkinson, P.H.; Tschopp, J.F.; Townsend, R.R.; Maley, F. Structure of oligosaccharides on Saccharomyces SUC2 invertase secreted by the methylotrophic yeast Pichia pastoris. J. Biol. Chem. 1991, 266, 22807–22817. [Google Scholar] [CrossRef]
- Zheng, H.; Chordia, M.D.; Cooper, D.R.; Chruszcz, M.; Müller, P.; Sheldrick, G.M.; Minor, W. Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 2014, 9, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Agirre, J.; Iglesias-Fernández, J.; Rovira, C.; Davies, G.J.; Wilson, K.S.; Cowtan, K.D. Privateer: Software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 2015, 22, 833–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 2015, 43, W314–W319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M. Rational design of particle mesh Ewald compatible Lennard-Jones parameters for +2 metal cations in explicit solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Merz, K.M. Taking into account the ion-induced dipole interaction in the nonbonded model of ions. J. Chem. Theory Comput. 2014, 10, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Panteva, M.T.; Giambaşu, G.M.; York, D.M. Force field for Mg(2+), Mn(2+), Zn(2+), and Cd(2+) ions that have balanced interactions with nucleic acids. J. Phys. Chem. B 2015, 119, 15460–15470. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stone, J. An Efficient Library for Parallel Ray Tracing and Animation. Master’s Thesis, University of Missouri-Rolla, Rolla, MO, USA, 1998. [Google Scholar]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2016. [Google Scholar]
- R Core Team. R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the com-parative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skjærven, L.; Yao, X.-Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Skjærven, L.; Jariwala, S.; Yao, X.-Q.; Grant, B.J. Online interactive analysis of protein structure ensembles with Bio3D-web. Bioinformatics 2016, 32, 3510–3512. [Google Scholar] [CrossRef] [Green Version]
- Plotly Technologies Inc. Collaborative Data Science; Plotly Technologies Inc.: Montréal, QC, Canada, 2015. [Google Scholar]
- Tan, K.; Jäger, C.; Körschgen, H.; Geissler, S.; Schlenzig, D.; Buchholz, M.; Stöcker, W.; Ramsbeck, D. Heteroaromatic inhibitors of the astacin proteinases meprin α, meprin β and ovastacin discovered by a scaffold-hopping approach. ChemMedChem 2020. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Meprin β/MWT-S-270 | |
|---|---|
| Data collection statistics | |
| Radiation source | Rotating anode |
| Wavelength (Å) | 1.5418 |
| Space group | C 1 2 1 |
| Unit cell length (Å) | 162.25, 72.44, 135.47 |
| Unit cell angles (°) | 90, 118.43, 90 |
| Resolution range (Å) Highest resolution shell (Å) | 50–2.41 2.48–2.41 |
| Rmeas | 13.1 (112.9) |
| I/σI | 11.48 (1.79) |
| Completeness (%) | 99.1 (95.5) |
| CC (1/2) Multiplicity | 99.7 (74.6) 5.6 (5.1) |
| Solvent content/Meprin β per ASU | 58%/2 |
| Wilson B factor | 41.01 |
| Refinement statistics | |
| Number of reflections (working/test set) | 103,499/ 5195 |
| Rwork/Rfree | 0.20/0.24 |
| No. atoms | |
| Protein | 8542 |
| Ligand | 431 |
| Water | 417 |
| Average B-factors (Å2) | 52.27 |
| Protein | 51.77 |
| Ligand | 68.94 |
| Water | 45.31 |
| Bond length r.m.s.d. (Å) | 0.003 |
| Bond Angles r.m.s.d. (°) | 0.63 |
| Ramachandran plot (%): favored/allowed/outliers | 97.8/1.98/0.19 |
| MolProbity clashscore | 0.75 |
 | ITC Buffer | IC50 [nM] | Ki [nM] | KD [nM] | ΔH [cal/mol] | −ΔS * T [cal/mol] | |
|---|---|---|---|---|---|---|---|
| R | |||||||
| 1 (270) |  | 40 mM Tris pH 8.0 | 45,5 | 28,6 | 16 | −3884 | −6939 |
| 1 (270) |  | 150 mM NaCl, 40 mM Tris pH 8.0 | 176 | 198 | 400 | −4554 | −4303 |
| 2 (416) |  | 40 mM Tris pH 8.0 | 285 | 192 | 237 | −3966 | −5212 |
| 3 (396) |  | 40 mM Tris pH 8.0 | 1283 | 1395 | 746 | −1138 | −7363 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linnert, M.; Fritz, C.; Jäger, C.; Schlenzig, D.; Ramsbeck, D.; Kleinschmidt, M.; Wermann, M.; Demuth, H.-U.; Parthier, C.; Schilling, S. Structure and Dynamics of Meprin β in Complex with a Hydroxamate-Based Inhibitor. Int. J. Mol. Sci. 2021, 22, 5651. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115651
Linnert M, Fritz C, Jäger C, Schlenzig D, Ramsbeck D, Kleinschmidt M, Wermann M, Demuth H-U, Parthier C, Schilling S. Structure and Dynamics of Meprin β in Complex with a Hydroxamate-Based Inhibitor. International Journal of Molecular Sciences. 2021; 22(11):5651. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115651
Chicago/Turabian StyleLinnert, Miriam, Claudia Fritz, Christian Jäger, Dagmar Schlenzig, Daniel Ramsbeck, Martin Kleinschmidt, Michael Wermann, Hans-Ulrich Demuth, Christoph Parthier, and Stephan Schilling. 2021. "Structure and Dynamics of Meprin β in Complex with a Hydroxamate-Based Inhibitor" International Journal of Molecular Sciences 22, no. 11: 5651. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115651