
Physical Exercise-Induced Myokines in Neurodegenerative Diseases
by

Banseok Lee
1,†, 
Myeongcheol Shin
1,†,
Youngjae Park
1,
So-Yoon Won
1,2,* and
Kyoung Sang Cho
1,2,* 1
Department of Biological Sciences, Konkuk University, Seoul 05029, Korea
2
Korea Hemp Institute, Konkuk University, Seoul 05029, Korea
*
Authors to whom correspondence should be addressed.
†
The authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(11), 5795; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115795
Submission received: 19 April 2021
/
Revised: 24 May 2021
/
Accepted: 25 May 2021
/
Published: 28 May 2021
(This article belongs to the Special Issue Adipokines, Myokines and Physical Exercise in Health and Disease)
Abstract
:Neurodegenerative diseases (NDs), such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), are disorders characterized by progressive degeneration of the nervous system. Currently, there is no disease-modifying treatments for most NDs. Meanwhile, numerous studies conducted on human and animal models over the past decades have showed that exercises had beneficial effects on NDs. Inter-tissue communication by myokine, a peptide produced and secreted by skeletal muscles during exercise, is thought to be an important underlying mechanism for the advantages. Here, we reviewed studies about the effects of myokines regulated by exercise on NDs and their mechanisms. Myokines could exert beneficial effects on NDs through a variety of regulatory mechanisms, including cell survival, neurogenesis, neuroinflammation, proteostasis, oxidative stress, and protein modification. Studies on exercise-induced myokines are expected to provide a novel strategy for treating NDs, for which there are no adequate treatments nowadays. To date, only a few myokines have been investigated for their effects on NDs and studies on mechanisms involved in them are in their infancy. Therefore, future studies are needed to discover more myokines and test their effects on NDs.
1. Introduction
Neurodegenerative diseases (NDs) such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) are characterized by progressive loss of neurons and accumulation of abnormal protein aggregates [1]. They are accompanied by cognitive impairments, memory loss, and locomotor deficits and share several fundamental processes, including cell survival, neurogenesis, neuroinflammation, proteostasis, oxidative stress, and protein modification [2,3,4,5,6,7]. The pathological hallmark of each ND contains an abnormal aggregation of different proteins: amyloid-β (Aβ) and tau in AD, α-synuclein in PD, huntingtin in HD, and several proteins including Tar DNA-binding protein of 43 kDa (TDP-43) and mutant superoxide dismutase 1 (SOD1) in ALS [8]. Pathological deposition of these proteins in neurons takes place long before psychological symptoms of each ND appear, and substances that can block their abnormal aggregation are expected to be used as preventive or therapeutic agents for NDs [9,10,11,12,13].
Results of numerous studies conducted on human and animal models over the past decades have shown that exercise has beneficial effects not only on physical health, but also on neuronal functions, resulting in improved learning and memory, inhibition of neurodegeneration, and reduction of depression [14]. Exercise can increase brain volume or connectivity by enhancing neurogenesis and synaptic plasticity and changing metabolism and vascular function [14,15]. Beneficial effects of exercise on brain health are thought to be mediated by several factors, including increased trophic factors such as brain-derived neurotrophic factor (BDNF), changed expression levels of many genes, decreased inflammation, and improved brain redox status [16,17,18,19,20].
Since exercise has a beneficial effect on brain function, it is thought to be able to slow the onset or progression of various NDs. In some studies, regular physical activity has less side effects, but better effects than currently used treatments for NDs [21,22,23]. The effect of exercise on NDs has been mostly studied on AD, one of the NDs with the highest incidence. Many systematic reviews and meta-analyses have shown that physical inactivity is one of the most common risk factors for AD development and that physically active elderly people have a lower risk of AD and dementia [23,24]. Moreover, beneficial effects of exercise on brain function and cognitive behavior have been repeatedly confirmed in AD mouse models [25]. In addition, physical exercise improved the physical functioning of people with PD [26] and it has been demonstrated that exercise protected the rodent models of PD from neurodegeneration [27,28,29,30,31]. Furthermore, studies on other NDs such as HD and ALS have also reported that exercise has a beneficial effect on these diseases [32,33]. However, some studies have failed to confirm the effect of exercise on NDs, and other studies have even shown that exercise has a rather harmful effect on NDs [34,35,36]. In addition, few studies have accurately proven the effect of exercise on NDs using appropriate biomarkers. Thus, verifying exercise’s effect through high-quality studies with large sample numbers is required in the future [32,35]. Nevertheless, it is clear that exercise is worth considering as an important therapeutic strategy if there are accurate data about the effect of exercise on each disease according to its type and amount.
Given the beneficial effect of exercise on neuronal health, various recent studies have revealed the importance of the muscle–brain axis in transmitting the effect of exercise to the brain as well as the role of muscles in secreting various factors for regulating brain function [37]. In this review, we examine the definition, function, and regulation of myokines released from skeletal muscles during exercise and their effects on neuronal health. In addition, molecular mechanisms underlying myokine’s effects on NDs are investigated. Finally, the potential of myokine as a therapeutic agent for NDs and the direction of future myokine research are discussed.
2. Myokines and Neuronal Health
Molecular mechanisms of how physical activity exerts protective effects against NDs have not been completely elucidated yet. However, inter-tissue communication by myokine is being proposed as a strong candidate for them [38]. Myokine was first postulated by Pederson in 2003 [39]. As a cytokine or a peptide produced and secreted by skeletal muscles, it exerts autocrine, paracrine, and endocrine effects [37,40]. It mediates crosstalk between muscles and other organs, promoting neurogenesis and vascularization in the brain [41,42], inducing browning of white adipose tissue (WAT) [43], accelerating hepatic glucose production, and stimulating insulin secretion by pancreatic β-cells [44,45]. In the following, we will review studies on factors secreted by muscles during exercise, which are known to affect neuronal health.
2.1. Apelin
Apelin, named APJ endogenous ligand, was first isolated in 1998 as a ligand for orphan G protein-coupled receptor APJ [46]. Apelin and APJ are widely distributed in the body and play an important role in cell protection in many organs [47]. The gene APLN encodes the pre-proprotein of apelin. Active apelin-13, pyroglutamate-apelin-13, apelin-17, and so on are produced by post-translational modification of the pre-proprotein [48].
Apelin was discovered as an exercise-induced myokine with an increased expression level after performing an 8-week endurance training program in 11 obese non-diabetic male subjects, and was confirmed to be secreted by human primary myotubes in vitro [49]. In addition, apelin level was decreased in an age-dependent manner in humans and rodents, and muscle function was decreased with aging in mice deficient in either apelin or its receptor APJ [50].
It is well-known that apelin contributes to conditions such as cardiovascular disease, obesity, and cancer [51]. In addition, apelins are distributed throughout the nervous system and have been reported to possess neuroprotective effects [52,53,54,55,56,57,58]. The role of apelin in NDs has only been studied recently. The results from these studies, which have been mainly focused on AD and PD using rodent models or cellular models, have suggested that apelin has beneficial effects on these diseases through various pathways [59,60,61,62,63,64,65,66,67]. One study has shown that apelin deficiency can accelerate ALS-like phenotype in an SOD1 (G93A) mouse model [68]. However, studies on changes in the expression level of apelin and its effects in human ND patients are insufficient. It is necessary to confirm the relationship of apelin with ND and its potential as a therapeutic agent through human studies in the future.
2.2. BDNF
BDNF is a member of the neurotrophin family, which regulates neural circuit development and function [69]. It was purified from porcine brain in 1982 as a neuronal survival factor [70]. Subsequently, numerous studies have shown that BDNF can perform a variety of functions. BDNF has been known to penetrate the blood–brain barrier (BBB) by a saturable transport system [71]. It can promote the survival and growth of a variety of neurons, affect synaptic transmission, enhance neurogenesis, and alter activity-dependent synaptic plasticity [72]. BDNF is initially synthesized as pre-pro-peptide that is cleaved to pro-BDNF, then the pro-BDNF is converted to mature BDNF by furin endopeptidase intracellularly, or by proteases such as plasmin or MMP7 extracellularly [73]. Mature BDNF is involved in neuronal plasticity such as neurogenesis, neurite arborization, and synaptogenesis by binding to tropomyosin receptor kinase B (TrkB) [74,75]. On the other hand, pro-BDNF functions in programmed neuronal death, neurite retraction, and synaptic pruning through a p75 neurotrophin receptor (p75NTR) [75].
According to previous studies, BDNF was increased in both acute aerobic and resistance exercises [76,77]. More recently, it has been shown that BDNF is expressed and secreted in muscles, and the muscle-derived BDNF acts as a hormone and affects whole-body metabolism and insulin secretion [78,79]. Additionally, increasing BDNF levels could mimic beneficial effects of exercise, including improving cognitive impairment and promoting combined adult hippocampal neurogenesis in 5×FAD mice [80]. In addition, TrkB inhibitor treatment blocked beneficial effects of exercise in a rat model of PD [81], suggesting that BDNF is an important factor mediating beneficial effects of exercise on ND.
Numerous studies have reported the importance of BDNF in ND pathology. In AD patients, hippocampal BDNF mRNA [82] and peripheral BDNF levels [83] were decreased compared to those in the control group, and serum BDNF levels were negatively correlated with future occurrence of dementia and AD [84], suggesting that BDNF plays a role in protecting the brain from AD. Consistently, studies using numerous in vitro and in vivo AD models have clearly shown that BDNF has neuroprotective effects [85,86,87,88,89]. In addition, genetic studies on human AD patients have reported that BDNFVal66Met SNP, known to reduce synaptic BDNF release [90], is associated with greater cognitive impairment and higher vulnerability of hippocampus-frontal connectivity to primary AD pathology [91,92]. Meanwhile, BDNF acts as a neurotrophic factor in dopaminergic neurons of the substantia nigra [93], and its neuroprotective effect has been verified in various PD models [94,95,96,97,98]. Recent studies have reported that α-synuclein, a PD-related protein, can block BDNF-TrkB signaling and induce dopaminergic cell death [99,100], suggesting that BDNF-TrkB signaling is implicated in PD pathology. The idea that degenerative diseases of the nervous system might be due to insufficient supply of neurotrophic factors has generated great interest in BDNF as a potential therapeutic agent.
2.3. CTSB
Cathepsin B (CTSB) is a lysosomal cysteine protease and is involved in catabolism of proteins in lysosome and autophagy [101,102]. Interestingly, it can be secreted from cells and plays a role in proteolysis of extracellular components, thus contributing to tumorigenic processes including apoptosis and invasion [103].
A recent study has demonstrated that CTSB is a myokine increased by aerobic exercise [41]. In the same study, treatment with AMP-kinase agonist induced CTSB secretion from cultured skeletal muscle cells, suggesting that CTSB expression is dependent on AMP kinase that has been suggested to mediate beneficial effects of exercise [41,104,105]. More recently, it has been reported that long-term exercise training (35 ± 15 years) significantly reduced resting serum levels of BDNF and plasma levels of CTSB in human, although they are beneficial for the brain and muscle health, which suggests that exercise may sensitize BDNF and CTSB signaling [106].
In CTSB-deficient condition, unlike in wild type (WT), beneficial effects of exercise, such as improvement of memory and adult neurogenesis, were not found, suggesting that CTSB mediates beneficial effects of exercise on brain health [41]. Furthermore, numerous studies using various models have suggested that CTSB would be beneficial to AD and PD by reducing Aβ and α-synuclein accumulation respectively, due to autophagy and lysosome-related functions of CTSB in the brain [107,108,109,110,111,112]. In the similar context, treatment with Z-Phe-Ala-diazomethylketone (PADK; also known as ZFAD), a CTSB activator, reduced Aβ deposition and improved synaptic and cognitive dysfunctions in an APP/presenilin-1 (PS1) and mild cognitive impairment (MCI) mouse model [112].
2.4. CX3CL1
C-X3-C Motif Chemokine Ligand 1 (CX3CL1), also called fractalkine or neurotactin, is a type of chemokines which are secreted proteins that play an important role in inflammation and trafficking of white blood cells during immune surveillance [113,114]. G-protein-coupled receptor C-X3-C Motif Chemokine Receptor 1 (CX3CR1) has been identified as the receptor for CX3CL1 and implicated in the function of leukocytes and microglia [115,116]. CX3CL1 has a cysteine signature motif, CX3C, which contains three unspecified amino acids between cysteine residues [113]. It is synthesized as a transmembrane molecule, and a chemo-attractive-soluble form containing CX3C motif is generated by metalloproteases ADAM 10 and ADAM 17 [117,118,119]. Based on its chemo-attractive role, CX3CL1 has been intensively associated with various inflammatory diseases [119].
CX3CL1 was identified as a protein secreted from skeletal muscles [120]. Several recent studies have found that CX3CL1 mRNA levels were increased in muscles and its protein levels were increased in plasma after acute or resistance exercise [121,122,123,124]. These findings suggest that CX3CL1 is an exercise-induced myokine that might be involved in communication between skeletal muscles and other organs.
In the brain, CX3CL1 might suppress neuroinflammation through activation of microglial CX3CR1 [125]. Given that neuroinflammation is an important factor in progressing NDs, the CX3CL1-CX3CR1 pathway is expected to have beneficial effects on NDs. However, to date, the role of the CX3CL1-CX3CR1 pathway in NDs is inconclusive and findings are controversial. First of all, there are conflicting reports about levels of CX3CL1 in AD patients. Some previous studies reported that CX3CL1 levels were decreased in brains and cerebrospinal fluid (CSF) of AD patients and in brains of AD model mice [126,127,128,129], while other studies reported that CX3CL1 was increased more in CSF or plasma of MCI and AD patients than in healthy people [130,131]. CX3CL1 expression is also inconsistent in other NDs. CX3CL1 levels in CSF of PD patients did not change compared to age-matched controls [132], whereas CX3CL1 levels in the putamen of HD patients were downregulated [133]. CX3CL1 mRNA levels in spinal cords of ALS model mice were reported to increase at 40 days of age but decrease at 90 and 120 days compared to those in WT mice [134]. In most ND-related studies, it was shown that the soluble form of CX3CL1 had beneficial effects [135,136,137,138,139,140,141], suggesting that increased CX3CL1 level in the blood after exercise might have beneficial effects on NDs. However, results of CX3CR1 deficiency are clearly contradictory. While CX3CR1 deficiency showed beneficial effects in some studies [142,143,144,145,146,147], it showed deteriorating effects in other studies [127,148,149,150,151]. The reason for these inconsistent results might be because the CX3CL1-CX3CR1 pathway is related to the function of microglia, which can play an opposing role in the ND process by acting on neuroinflammation and phagocytic clearance at the same time. To develop a therapeutic agent based on CX3CL1, in-depth studies on more detailed mechanisms should be conducted.
2.5. FGF2
Fibroblast growth factor 2 (FGF2), also known as basic fibroblast growth factor (bFGF) and FGF-β, is one of the growth factors that plays an essential role in neural development and proliferation of neural stem and progenitor cells [152]. FGF2 is ubiquitously expressed in various tissues, including brain and muscles, and aerobic exercise has been found to increase FGF2 expression levels in animal models [153,154,155]. However, it is unclear whether exercise can increase FGF2 expression in humans [156]. FGF2 is unconventionally secreted from cells, in which it forms lipidic membrane pores by binding to phosphoinositide PI(4,5)P2, and it is secreted by the separating action of membrane proximal heparan sulfates proteoglycans [157,158,159,160].
As a neurotrophic factor, FGF2 is known to stimulate neurogenesis and angiogenesis in adult brains and developing brains [161,162,163,164,165,166,167]. Accordingly, it exerted beneficial effects in some ND animal models [168,169]. Especially, FGF2 treatment inhibited Aβ production in primary cultured neurons of APP/PS1 mice and APPswe-HEK293 cells [170,171] and improved synaptic transduction, plasticity, and neurogenesis in an APP/PS1 mouse, while reducing hippocampal Aβ deposition and memory impairment [170]. However, effects of FGF2 on AD or ALS are more complex than expected. FGF2 levels in brains of AD patients and serum and CSF of ALS patients were reported to be elevated compared to those in normal controls [172,173,174]. FGF2 elevated the expression of tau, glycogen synthase kinase-3 (GSK-3) activity, and GSK-3-mediated tau phosphorylation [175]. Furthermore, it reduced neurogenesis in cultured neural progenitor cells derived from adult rat hippocampus [176] and induced dysregulation of dentate gyrus neurogenesis [177]. Contrary to the expectation that reduced FGF2 levels would worsen the phenotype of ALS, FGF2 deficiency significantly delayed disease onset and improved impaired motor performance in mutant SOD1 mice, a common ALS model [178].
2.6. FGF21
Fibroblast growth factor 21 (FGF21), a hormone belonging to the FGF superfamily, was first discovered in mouse embryos in 2000 [179]. Although this hormone is mainly expressed in the liver, it is also produced in various organs, including muscle, adipose tissue, pancreas, and heart, regulating energy homeostasis in an autocrine, paracrine, or endocrine manner [180,181]. As a result of preclinical studies, FGF21 has been attracting attention for its potential use as a treatment for metabolic syndromes such as diabetes by increasing insulin sensitivity, improving glucose tolerance, and reducing body weight [182]. Exercise elevated blood FGF21 levels mainly due to increased FGF21 expression in the liver [183,184,185]. In addition, expression levels of FGF receptor-1 (FGFR1) and β-Klotho (KLB), a co-receptor, were increased in adipose tissues during exercise, thus improving the sensitivity of adipose tissues to FGF21 [186] and promoting browning of white adipose tissues [187].
FGF21 was reported as a myokine regulated by the phosphoinositide 3-kinase (PI3K)/Akt axis [188], and its expression was increased in muscles during acute aerobic exercise [189]. Interestingly, FGF21 is also known as ’mitokine’, which is expressed and secreted in cells with mitochondrial damage due to autophagy dysfunction, endoplasmic reticulum stress, and mitochondrial gene abnormalities, and non-autonomously affects metabolism of other cells [190,191,192,193,194]. In particular, mitochondria damage in muscle cells induced an ATF4-dependent increase of FGF21 expression, thereby inducing systemic metabolic adaptation such as improved insulin sensitivity, increased energy expenditure, and enhanced lipid catabolism and WAT browning [190].
In addition to metabolic function, FGF21 can penetrate the BBB [195], and has been reported to improve cognitive performance in diabetes and trauma models [196,197,198,199]. In various brain damage models, FGF21 prevented inflammation and BBB disruption through PPAR-γ activation and induced neovascularization [196,197,198,200,201,202,203,204]. Although not many studies have been conducted on the effect of FGF21 on NDs, several recent preclinical studies have shown that FGF21 has a neuroprotective effect in ND models by affecting several signaling pathways. In both in vivo and in vitro AD models, FGF21 has shown anti-inflammatory and antioxidant effects, and prevented amyloid plaque formation, neurofibrillary tangle formation, and neurodegeneration [205,206,207,208]. Moreover, FGF21 treatment led to alleviated dopaminergic neuron loss, improved mitochondrial function and behavioral ability, and decreased inflammation in PD models [209,210,211,212]. Additionally, intraperitoneal injection of R1Mab1, a pharmacological agonist of FGF21 which is an IgG humanized monoclonal antibody with agonistic activity on FGFR1, improved the motility of the ALS model mice [213].
2.7. IGF-1
Insulin-like growth factor 1 (IGF-1), also called somatomedin C, is a secreted peptide with a structure similar to insulin and is involved in various physiological functions [214]. IGF-1 is composed of 70 amino acids with three disulfide bonds, the position of which is the same as the disulfide bond connecting A and B chains of insulin [215]. IGF-1 is a potent myoanabolic factor, which is expressed and secreted in muscle tissue, and muscle hypertrophy can increase IGF-1 expression [216]. According to previous studies, although there is some disagreement on the proportional relationship between exercise status and serum IGF-1 levels, many studies have shown that serum IGF-1 levels were increased in the elderly after aerobic and resistance exercise [217,218,219]. Moreover, a study revealed that IGF-1 is indispensable for exercise-induced neurogenesis [220].
A previous study showed that IGF-1 enters into the brain through the blood-CSF pathway [221]. Although it is known that IGF-1 plays an important role in brain development and neurogenesis and that large amounts of IGF-1 receptors are expressed in the brain [222], its role in cognitive function and NDs of the aging brain is still complex and controversial [214]. In some studies, serum IGF-1 levels in the elderly were positively correlated with cognitive function, whereas in other studies, those were not correlated or reversely correlated [223,224,225,226]. Results from studies on IGF-1 levels in ND patients are also complicated. A large-scale study on AD patients has reported that low serum IGF-1 levels are associated with an increased risk of developing AD dementia [227]. However, a meta-analysis based on results of nine studies comparing serum IGF-1 levels with normal subjects failed to find a significant difference between AD patients and normal subjects [228]. On the other hand, most studies on the association between PD and IGF-1 levels have reported higher IGF-1 levels in PD patients than in normal subjects [229]. More importantly, studies using rodent PD, HD, or ALS models have consistently shown that IGF-1 has beneficial effects on these diseases [230,231,232,233,234,235,236]. However, results from studies on the role of IGF-1 signaling in AD mouse models are not consistent with each other. In some studies, systemic infusion of IGF-1 reduced brain Aβ levels and toxicity [237,238]. However, in other studies, decreased IGF-1 signaling alleviated Aβ toxicity in AD mice [239,240,241]. In addition, a systematic review of the literature showed that it was unclear whether circulating or brain IGF-1 could reverse or slow the rate of decline in cognitive impairment in patients with dementia [242].
2.8. Irisin
Irisin, named after the Greek messenger goddess Iris, is a 112 amino acids-cleaved product of fibronectin type III domain-containing protein 5 (FNDC5), a type I transmembrane glycoprotein [43]. After being proteolytically cleaved from FNDC5, irisin is secreted and functions as a myokine [243]. Irisin was initially identified in skeletal muscles, but it was later found to also be expressed in a variety of tissues, including the brain [244]. Exercise elevated the expression of transcription factor PGC-1α in muscles [19], thus enhancing the expression of FNDC5 and consequently increasing the amount of irisin secreted into the blood by proteolytic cleavage of FNDC5, and irisin level was elevated in the plasma of individuals undergoing aerobic training [43,245].
Irisin has been shown to exert beneficial effects on various tissues including bone, fat, liver, and muscle [246,247,248]. One study showed that irisin could trigger cell proliferation in hippocampal cell lines [249] and the secretion of irisin during exercise enhanced the expression of BDNF and neurotrophic genes in mouse brains [250,251]. In addition, overexpression of irisin in the brain suppressed neuroplasticity defects and memory impairment in AD model mice, and intraperitoneal injection of anti-FNDC5 eliminated the beneficial effect of exercise on AD-like phenotype [252]. Moreover, co-treatment of irisin with bone marrow stem cells protected dopaminergic neurons from degeneration and apoptotic process in a MPTP-induced PD mouse model [253]. Interestingly, a study with 14 AD patients and 25 non-demented controls revealed that CSF irisin levels were positively correlated with levels of Aβ and BDNF in CSF and cognitive status of patients with AD [254]. Similarly, one study found that serum irisin levels in patients with ALS were higher than those in normal controls and that irisin levels were negatively correlated with the extent of functional and respiratory impairment in patients with ALS [255]. A study on the effect of irisin on ND is in its infancy, and many future studies are needed to reveal the potential of irisin as an ND therapeutic or diagnostic biomarker.
2.9. LIF
Leukemia inhibitory factor (LIF) is a member of the interleukin-6 family of cytokines with pleiotropic functions [256] and was first identified to be able to induce the differentiation of macrophages [257]. It plays an important role in promoting proliferation, differentiation, and survival of various types of cells, including neurons, myoblasts, hepatocytes, adipocytes, megakaryocyte progenitors, and myeloid cells [258].
LIF has also been identified as a myokine whose expression is increased by exercise in human and animal models [259,260]. It has a secretion signaling peptide that regulates its secretion from cells [261], and its secretory property was confirmed in cultured human myotubes and mouse skeletal muscles [262]. According to a study by Broholm et al., immediately after performing an aerobic exercise for 3 hours, LIF mRNA expression levels in muscles were increased up to 4 times and then gradually decreased [259]. In a more recent study, plasma LIF content was increased about 50% after a static exercise, although it was not increased after a dynamic exercise, which means that the regulation of LIF expression might differ depending on the type of exercise [263]. Meanwhile, treatment with ionomycin, a Ca2+ ionophore, elevated LIF mRNA and protein expression levels in human muscle cells [259], suggesting that oscillations of Ca2+ concentration following muscle contraction could affect LIF transcription. In addition, in cultured human myotubes, LIF was regulated by the PI3K-Akt pathway, and expression levels of JunB and c-Myc induced by LIF were also increased in skeletal muscles after a resistance exercise [264].
Since LIF is known to be able to pass through the BBB [265], it is expected that plasma LIF can affect the brain function. Expression levels of LIF and its receptor, LIFR, were increased in brains of AD and PD patients compared to healthy controls [266]. Furthermore, LIF was increased in skin samples of ALS patients [267], suggesting that LIF might be related to the pathophysiology of NDs. However, to date, research on the role of LIF in ND pathology is very limited. A recent study has shown that LIF reduced amyloid β-induced neurotoxicity in HT-22 mouse hippocampal cell lines and primary hippocampal cells through Akt/extracellular signal-regulated kinase (ERK)-mediated c-fos induction [268]. However, LIF treatment did not show a beneficial effect on the disease progression in ALS model mice [269]. Considering the effect of LIF on brain function as a neurogenesis- and inflammation-related factor, further studies are needed to determine the association of ND with LIF using various ND models.
3. Molecular Mechanisms Underlying Myokine Action in Neurodegenerative Diseases
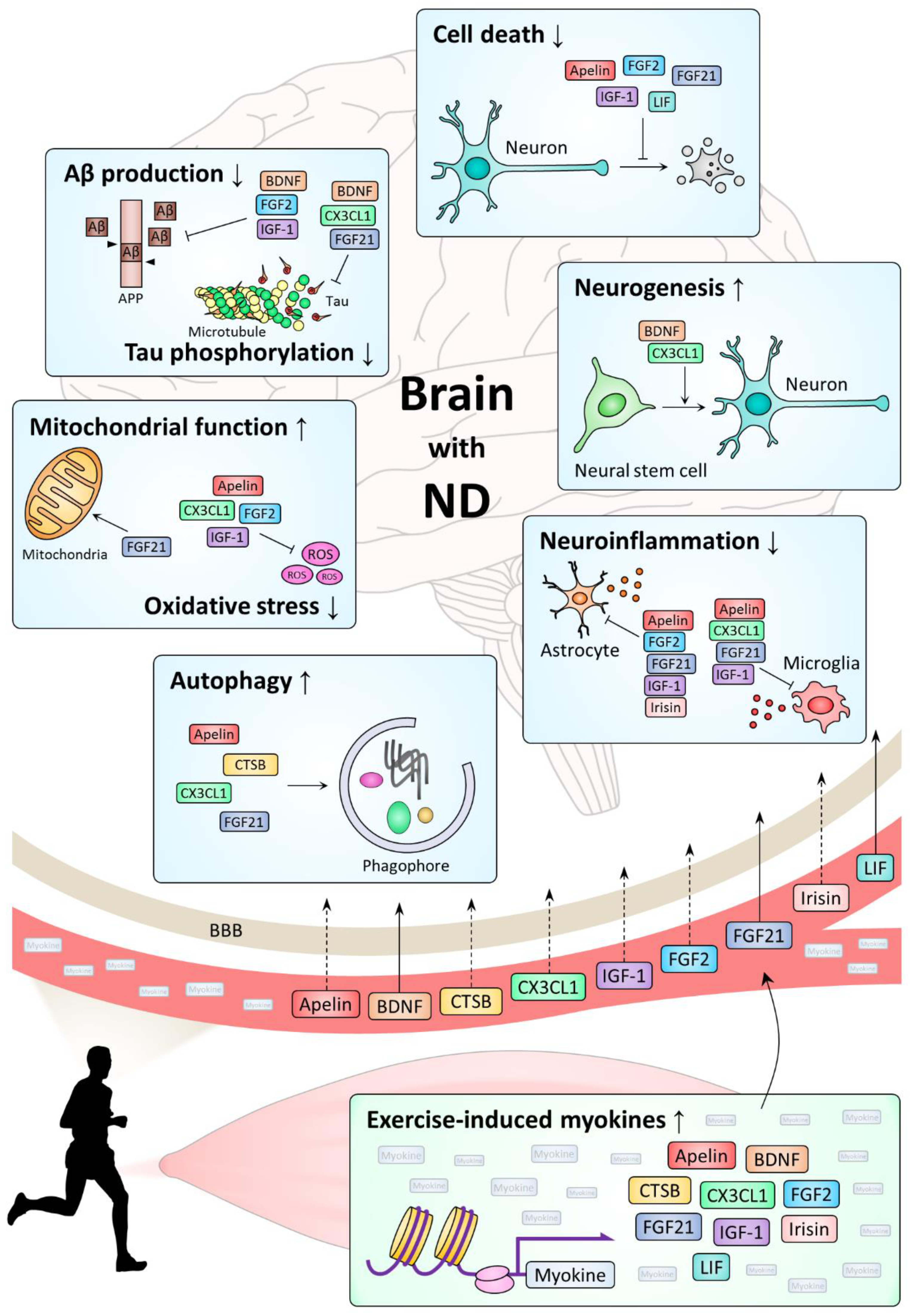
Most NDs, including AD, PD, HD, and ALS, share pathological changes such as progressive loss of neurons, accumulation of abnormal protein aggregates, and abnormally increased neuroinflammation [1]. Studies on exercise-induced myokines have shown that myokines play a beneficial role in NDs through a variety of mechanisms, including regulations of cell survival, neurogenesis, neuroinflammation, proteostasis, oxidative stress, and protein modification (Table 1). Here, we examined actions of myokines on NDs by each mechanism (Figure 1).
3.1. Cell Survival
Neuronal cell death is one of the most important features in the brain of patients during ND progression [270]. During ND, neurons die by activating the death signaling pathway, resulting in brain atrophy [271]. In particular, apoptosis is an important form of cell death in ND, which includes intrinsic pathways that occur inside cells damaged by stress and extrinsic pathways that are triggered by signals from other cells [272]. In apoptosis, cell death is induced by the activation of protease activity of a series of caspases, and then, various factors activate or inhibit caspase actions to form cell death or survival signals, respectively [271]. Numerous proteins including apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinases (JNK), B-cell lymphoma 2 (BCL2)-associated X protein (Bax), and apoptotic protease activating factor 1 (Apaf-1) are involved in cell death signals that activate apoptosis of neurons, while many factors including nerve growth factor (NGF), PI3K, Akt, and BCL2 are included in neuronal survival pathways [271].

{kind=link}
Table 1.
Myokines secreted during exercise and their beneficial effects on NDs. ↑, activated or increased; ↓, inactivated or decreased.
Table 1.
Myokines secreted during exercise and their beneficial effects on NDs. ↑, activated or increased; ↓, inactivated or decreased.
| Myokine | ND | Function | Mechanism | Model | Reference |
|---|---|---|---|---|---|
| Apelin | AD | Decreased cell death | Aβ-induced autophagy ↓ Caspase-3 activity ↓ mTOR phosphorylation ↑ | Rats injected with Aβ25–35 and apelin-13 | [62] |
| Decreased cell death | BDNF/TrkB signaling pathway ↑ | Rats injected with streptozotocin and apelin-13 | [64] | ||
| Anti-inflammation | Astrocyte and microglia activation ↓ IL-1β and TNF-α expression ↓ | ||||
| Decreased cell death | RIP1 and RIP3 expression ↓ | Rats injected with streptozotocin and apelin-13 | [67] | ||
| Anti-inflammation | TNF-α expression ↓ | ||||
| PD | Decreased cell death | ERK1/2 phosphorylation ↑ ER stress ↓ | SH-SY5Y cells treated with MPP+ and apelin-13 | [60] | |
| Decreased cell death | PI3K signaling pathway ↑ Cytoplasmic cytochrome c ↓ Cleaved caspase-3 ↓ | SH-SY5Y cells treated with 6-OHDA and apelin-13 | [61] | ||
| Increased α-synuclein clearance | PI3K/Akt/mTOR-autophagy signaling pathway ↑ | SH-SY5Y cells treated with MPP+ and apelin-36 | [65] | ||
| Decreased cell death | IRE1α/XBP1/CHOP signaling pathway ↓ | Mice injected with MPTP and apelin-13 | [273] | ||
| Increased α-synuclein clearance | Autophagy ↑ | ||||
| Increased α-synuclein clearance | AMPK/mTOR/ULK1-autophagy pathway ↑ | SH-SY5Y cells treated with rotenone and apelin-13 | [66] | ||
| Decreased cell death | ASK1/JNK signaling pathway ↓ | Mice injected with MPTP and apelin-36 | [274] | ||
| Caspase-3 activity ↓ | |||||
| Antioxidative stress | GSH and SOD ↑ | ||||
| ALS | Pro-inflammation | Microglia activation ↑ | SOD1-G93A mice crossed with apelin−/− mice | [68] | |
| BDNF | AD | Decreased Aβ production | BACE1 and PSEN1 ↓ | APPswe mice injected with TAT-BDNF peptide Rats injected with scopolamine and TAT-BDNF peptide | [275] |
| Decreased tau phosphorylation | GSK3β activation ↓ | ||||
| HD | Increased neurogenesis | TrkB phosphorylation ↑ | N171-82Q mice administered with 4′-DMA-7,8-DHF by oral gavage | [276] | |
| CTSB | AD | Increased Aβ clearance | Proteolytic activity of CTSB itself | hAPPJ20 mice injected with Lenti-CTSB Primary cortical neurons from hAPPJ20 mice infected with Lenti-CTSB In vitro cleavage assay using Aβ1–42 and CTSB | [107] |
| Increased Aβ clearance | Lamp1 expression ↑ | APP/PS1 mice injected with AAV-CTSB | [111] | ||
| CX3CL1 | AD | Pro-inflammation | IL-6 and TNF-α expression ↑ | hAPPJ20 mice crossed with CX3CR1−/− mice | [127] |
| Decreased tau phosphorylation | GSK3α/β activation ↓ | Tau P301L mice injected with AAV-CX3CL1 | [137] | ||
| Anti-inflammation | Microglia activation ↓ | ||||
| Pro-inflammation | NRF2/HO-1 signaling pathway ↓ | CX3CR1−/− mice injected with AAV-TAUP301L | [148] | ||
| Pro-oxidative stress | |||||
| Increased neurogenesis | TGF-β/Smad2 signaling pathway ↑ | Tau P301S mice crossed with Tg-CX3CL1 mice | [277] | ||
| Anti-inflammation | Microglia activation ↓ | APP/PS1 mice injected with MSCs carrying CX3CL1 | [141] | ||
| PD | Anti-inflammation | Microglia activation ↓ | Rats injected with 6-OHDA and CX3CL1 | [135] | |
| Anti-inflammation | Microglia activation ↓ | CX3CL1−/− mice injected with MPTP and CX3CL1 | [136] | ||
| TNF-α and IL-1β expression ↓ | |||||
| Pro-inflammation | Il-1β and IL-6 expression ↑ | CX3CR1−/− mice injected with AAV-α-SYN | [150] | ||
| ALS | Pro-inflammation | Microglial activation ↑ IL-1β, iNOS, and TNF-α expression ↑ Arginase 1 and TGF-β expression ↓ NF-κB signaling pathway ↑ | SOD1-G93A mice crossed with CX3CR1−/− mice | [151] | |
| Decreased SOD1 clearance | Autophagy ↓ | ||||
| FGF2 | AD | Decreased cell death | Akt phosphorylation ↑ | CVEC treated with Aβ1–40 and FGF2 | [278] |
| Decreased Aβ production | BACE1 expression ↓ | APP23 mice injected with FGF2 N2a cells transfected with APPswe and treated with FGF2 | [279] | ||
| Anti-inflammation | iNOS expression ↓ Astrocyte activation ↓ | ||||
| Decreased Aβ production | BACE1 expression ↓ | APPswe-HEK293 cells treated with GCM SH-SY5Y cells treated with FGF2 | [171] | ||
| PD | Antioxidative stress | GSH ↑ | Primary rat embryonic mesencephalic cultures treated with 6-OHDA and FGF2 | [280] | |
| Decreased cell death | MEK/ERK1/2 signaling pathway ↑ BAD phosphorylation ↑ AIF translocation ↓ PI3K/Akt signaling pathway ↑ | SH-SY5Y cells treated with rotenone and FGF2 Primary ventral mesencephalic cultures treated with rotenone and FGF2 | [281] | ||
| Decreased cell death | Caspase-3 expression ↓ | Rats injected with 6-OHDA and PEGylated FGF2 | [282] | ||
| Anti-inflammation | Astrocyte activation ↓ | PC12 cells treated with 6-OHDA and PEGylated FGF2 | |||
| Decreased cell death | MEK/ERK1/2 signaling pathway ↑ PI3K/Akt signaling pathway ↑ ER stress ↓ | Rats injected with 6-OHDA and FGF2 Primary hippocampal neurons treated with 6-OHDA and FGF2 | [283] | ||
| FGF21 | AD | Decreased cell death | Caspase-3 activity ↓ | SH-SY5Y cells treated with Aβ1–42 and FGF21 | [206] |
| Anti-inflammation | HSP90/TLR4/NF-kB signaling pathway ↓ | ||||
| Decreased cell death | Expression ratio of BCL2 to Bax (BCL2/Bax) ↑ Cleaved caspase-3 ↓ | Rats injected with Aβ25–35 and FGF21 SH-SY5Y cells treated with Aβ25–35 and FGF21 | [207] | ||
| Decreased tau phosphorylation | PP2A phosphorylation ↓ | ||||
| PD | Increased α-synuclein clearance | SIRT1-autophagy signaling pathway ↑ | Mice injected with MPTP and FGF21 SH-SY5Y cells treated with MPTP and FGF21 | [210] | |
| Decreased cell death | Cleaved caspase-3 and JNK phosphorylation ↓ Expression ratio of BCL2 to Bax (BCL2/Bax) ↑ | Mice injected with MPTP and treated with FGF21 via intranasal routine SH-SY5Y cells treated with MPP+ and FGF21 Primary dopaminergic neurons treated with MPP+ and FGF21 | [211] | ||
| Anti-inflammation | Astrocyte and microglia activation ↓ IL-1β, IL-12, IFN-γ, and TNF-α expression ↓ | ||||
| Enhanced mitochondrial function | AMPK/PGC-1α signaling pathway ↑ | ||||
| ALS | Anti-inflammation | Serum TNF-α, MCP-1 level ↓ | SOD1-G93A mice injected with R1Mab1 | [213] | |
| IGF-1 | AD | Decreased cell death | Akt phosphorylation ↑ | Rats infused with Aβ25–35 and IGF-1 via subcutaneous osmotic minipump | [237] |
| Increased Aβ clearance | Aβ carrier-mediated transport ↑ | APP/PS2 mice injected with IGF-1 Choroid plexus epithelial cell culture system treated with Aβ1–40 and IGF-1 | [238] | ||
| Anti-inflammation | Astrocyte activation ↓ | ||||
| Decreased cell death | Mitochondrial membrane potential ↑ Cytoplasmic cytochrome c ↓ Cleaved caspase-3 ↓ Expression ratio of BCL-XL to Bax (BCL-XL/Bax) ↑ | SH-SY5Y cells treated with Aβ25–35 and IGF-1 | [284] | ||
| Antioxidative stress | SOD and CAT activity ↑ PI3K/Akt/Nrf2/HO-1 signaling pathway ↑ | ||||
| Decreased cell death | C-myb expression ↑ | SH-SY5Y cells treated with Aβ25–35 and IGF-1 | [285] | ||
| Decreased tau phosphorylation | p25 protein production ↓ μ-Calpain expression ↓ | ||||
| Decreased Aβ production | ADAM10 exprssion ↑ BACE1 expression ↓ | APP/PS1 mice injected with IGF-1 | [286] | ||
| PD | Decreased cell death | PI3K/Akt signaling pathway ↑ | Rats injected with 6-OHDA and IGF-1 | [232] | |
| Decreased cell death | Caspase-3 expression and activity ↓ PARP cleavage ↓ | PC12 cells treated with 6-OHDA and IGF-1 | [287] | ||
| Antioxidative stress | NRF2/HO-1 signaling pathway ↑ | ||||
| Decreased cell death | ERK1/2/CREB signaling pathway ↑ Akt/GSK3α/β/β-catenin signaling pathway ↑ | Rats injected with 6-OHDA and IGF-1 | [288] | ||
| HD | Decreased cell death | PI3K/Akt signaling pathway ↑ Huntingtin phosphorylation ↑ | Primary striatal neurons transfected with mutant huntingtin and treated with IGF-1 SH-SY5Y cells treated with IGF-1 HEK293T cells transfected with mutant huntingtin | [289] | |
| ALS | Decreased cell death | Akt/caspase-9/caspase-3 signaling pathway ↓ | SOD1-G93A mice injected with AAV-IGF-1 | [230] | |
| Anti-inflammation | Astrocyte activation ↓ | ||||
| Anti-inflammation | Astrocyte activation ↓ TNF-α expression ↓ | SOD1-G93A mice crossed with MLC/mIgf-1 transgenic mice | [233] | ||
| Decreased cell death | Cleaved caspase-9 ↓ | SOD1-G93A mice injected with AAV-IGF-1 SOD1-G93A astrocyte-neuron coculture system treated with IGF-1 | [290] | ||
| Anti-inflammation | Astrocyte and microglia activation ↓ NOS activity and peroxynitrite formation ↓ | ||||
| Anti-inflammation | Macrophage invasion ↓ TNF-α expression ↓ | SOD1-G93A mice injected with AAV-IGF-1 | [291] | ||
| Decreased cell death | JNK and p38 MAPK phosphorylation ↓ Bax expression ↓ BCL-2 expression ↑ Cleaved caspase-3 and cleaved caspase-9 ↓ | SOD1-G93A mice injected with AAV-IGF-1 | [292] | ||
| Anti-inflammation | Astrocyte and microglia activation ↓ | ||||
| Irisin | AD | Anti-inflammation | IL-1β and IL-6 level ↓ Akt/IκBα/NF-κB/COX-2 signaling pathway ↓ | Primary hippocampal astrocytes treated with Aβ25–35 and irisin | [293] |
| LIF | AD | Decreased cell death | Aβ-induced autophagy ↓ | HT-22 mouse hippocampal cells treated with Aβ1–42 and LIF | [268] |
IGF-1, a well-established activator of the PI3K-Akt pathway, has been reported to inhibit cell death by increasing Akt phosphorylation in various models [236,285,288]. Interestingly, in a human HD cell model, Akt activated by IGF-1 inhibited the toxicity of huntingtin by phosphorylating it [289]. Furthermore, IGF-1 treatment inhibited apoptosis in ND models by inactivating the apoptosis pathway, such as increasing the activity of NF-kB, expression of BCL2, or inhibiting caspase activity [230,284,287,290,292,294,295]. It was also found that FGF2 and FGF21 increased Akt phosphorylation and BCL2 expression in various ND models, while inhibiting apoptosis by lowering JNK and caspase activities [207,211,278,281,282,283]. BDNF and LIF could also reduce neuronal cell death in AD models by activating the PI3K-Akt pathway [268,296]. In addition, intranigral injection of apelin-36 in an MPTP-induced PD mouse model reduced cell death by inhibiting the ASK1/JNK/caspase-3 pathway [274].
3.2. Neurogenesis
Adult neurogenesis occurs actively in healthy brain subjects, but falls sharply in AD brains [7], suggesting that neurogenesis is deeply associated with the onset of ND [297]. Interestingly, exercise is known to promote adult neurogenesis [298], and as neurotrophins, some myokines are believed to have beneficial effects on NDs by mediating the promoting effect of exercise on neurogenesis. In particular, BDNF and CX3CL1 overexpression induced neurogenesis in AD model mice [277,299] and enhanced AD therapeutic effects of engrafted stem cells [141,300]. Moreover, neurogenesis was increased in an N171-82Q HD mouse model after they were treated with BDNF receptor agonists [276], and some myokines, including irisin, CTSB, and apelin-13, are thought to contribute to neurogenesis by increasing the expression of BDNF in brains of animal models [41,64,250]. In addition, FGF2 stimulated neurogenesis in various models of NDs including AD, PD and HD, although high concentrations of FGF2 rather inhibited neurogenesis [168,169,170,176,177]. Meanwhile, in mutant mice with low levels of serum IGF-I, adult hippocampal neurogenesis was lowered without showing a decrease in anxiety behavior by exercise, suggesting that IGF-1 is involved in exercise-induced neurogenesis [220].
3.3. Neuroinflammation
Inflammation is a host defense system by activating innate immune cells such as microglia against infection, tissue injury, and cellular insults, and various cytokines secreted from immune cells mediate or inhibit inflammation [301]. However, chronically activated neuroinflammation in the nervous system of ND patients plays a causal role in the pathogenesis of ND [302,303]. Various clinical and preclinical studies have reported that exercise reduces neuroinflammation by increasing the expression of anti-inflammatory cytokines and lowering levels of pro-inflammatory cytokines and activated microglia [304,305]. When apelin-13 was injected intracerebroventricularly, it inhibited the activation of microglia and astrocytes and reduced the expression of IL-1β and TNF-α in a streptozotocin-induced rat model of AD [64]. This effect of apelin-13 was inhibited by treatment with K252a, a TrkB antagonist, suggesting that apelin-13 inhibits neuroinflammation through the BDNF-TrkB pathway [64]. The anti-neuroinflammation function is best known in CX3CL1. As an inhibitor of microglial activation, CX3CL1 has been proven to be able to inhibit neuroinflammation in various models for ND [127,136,141,148,150,151]. Likewise, irisin showed an anti-inflammatory effect by reducing the secretion of cytokines, IL-6 and IL-1β, from cultured astrocytes, and irisin-pretreated astrocytes protected neurons from Aβ toxicity [293]. In addition, FGF21 exerted neuroprotective effects by suppressing the expression of NF-κB in Aβ42-treated SH-SY5Y cells and pro-inflammatory cytokines in the models of PD and ALS [206,211,213]. Furthermore, a series of studies showed that IGF-1 reduced the expression of TNF-α and the activity of astrocyte and microglia in an APP/PS2 AD mouse model and a SOD1(G93A) mouse model [230,233,238,290,291,292].
3.4. Proteostasis
The pathology of most NDs involves pathogenic protein aggregation and deposition. Aggregation and deposition of Aβ and tau in AD, α-synuclein in PD, and huntingtin in HD are examples. Neurons and glial cells internalize these proteins into cells through endocytosis and then degrade them using autophagy-lysosome and ubiquitin-proteasome systems, thereby protecting neuronal cells [306]. Exercise activates autophagy in the brain as well as muscles [307], and it has been reported that physical activity not only lowers levels of pathogenic protein aggregation in various ND models [308,309], but also tau in normal brains of the elderly [310]. However, there are insufficient data supporting these findings, and the specific mechanism is also unclear. Nevertheless, there are reports about the effect of some myokines in the maintenance of proteostasis. Intranigral apelin-13 injection promoted α-synuclein clearance by activating autophagy in MPTP-induced PD model mice [273]. In vitro and in vivo studies have demonstrated that CX3CR1-deficient microglia have poor phagocytosis ability against tau and α-synuclein proteins [146,149]. Moreover, CX3CR1 deficiency exacerbated SOD1 aggregation and impaired the autophagy-lysosome degradation pathway in the SOD1(G93A) ALS mouse model [151]. CTSB has been identified as a regulator of autophagy and lysosomal dynamics [102,311]. Accordingly, a series of studies have shown that CTSB plays an important role in the clearance of Aβ and α-synuclein [107,108,109,111,112]. Similarly, in the MPTP mouse model of PD, FGF21 promoted autophagic degradation of α-synuclein via SIRT1 [210].
3.5. Mitochondrial Function and Oxidative Damage
Oxidative stress due to increased mitochondrial damage and reactive oxygen stress (ROS) plays an important role in the pathophysiology of ND [312]. Therefore, one of the possible mechanisms by which myokine exerts beneficial effects on NDs is to act as an antioxidant scavenging ROS or to protect mitochondria in the ND brain. In fact, it has been shown that some myokines have antioxidant or mitochondrial protective functions in several ND models. Most importantly, as a mitokine that is induced by mitochondrial dysfunction, FGF21 is intensively related to oxidative stress [313]. FGF21 treatment reduced oxidative stress in an Aβ-injected rat model and an Aβ-treated cell model by inhibiting HSP90-TLR4-NF-κB or PP2A-MAPKs-HIF-1α pathways [206,207]. Furthermore, FGF21 treatment not only enhanced mitochondrial functions through PGC-1α activation in human dopamine neurons [209], but also showed neuroprotective effects by stimulating the AMPK/PGC-1α axis to promote mitochondrial functions in MPTP-treated PD models [211]. Apelin-13 and FGF2 also showed neuroprotective effects in cellular PD models treated with 6-OHDA, an oxidative injury inducer [61,280]. In addition, IGF-1 promoted the survival of rat primary neurons and hypothalamic rat GT1-7 cells treated with hydrogen peroxide by the PI3K-NF-κB pathway [294].
3.6. Aβ Production and Tau Phosphorylation
Aβ production through APP processing and hyperphosphorylation of tau are important in the pathophysiology of AD [314]. The amyloidogenic process that produces Aβ is catalyzed by beta- and gamma-secretase and takes place in the intracellular endosome [315]. It has been reported that intracranial injection of BDNF into the hippocampus reduced Aβ production in the brain of wild-type mice by upregulating gene expression of sorting protein-related receptor with A-type repeats (SORLA), which acts as a sorting receptor for APP and downregulates its processing into Aβ [316]. In addition, in vitro and in vivo studies have demonstrated that FGF2 reduced Aβ production in part by lowering BACE1 expression level [171,279]. Meanwhile, IGF-1 treatment increased α-secretase activity in a PI3K-dependent manner, and subcutaneous injection of IGF-1 increased the expression of ADAM 10 and decreased the expression of BACE1 in the cortex of APP/PS1 mice, which suppressed Aβ production by precluding the amyloidogenic pathway [286,317]. However, other studies showed that neuronal insulin receptor deficiency rather reduces Aβ deposition in APPswe mice [318]. Thus, further studies on detailed functions of IGF-1 in APP processing are needed.
Neurofibrillary tangles composed of highly phosphorylated tau proteins is a characteristic pathological feature of AD brain [319]. It is known that insoluble tau present in AD brain is phosphorylated at more than 45 residues by various kinases, including glycogen synthase kinase-3 (GSK-3), cyclin-dependent kinase-5 (cdk5), casein kinase 1 (CK1), and cyclic AMP-dependent protein kinase (PKA), while it is dephosphorylated by phosphatases such as PP2A [319]. Interestingly, BDNF stimulation of neuronally differentiated mouse embryonic cells resulted in a rapid decrease in tau phosphorylation in a PI3K-GSK-3β-dependent manner [320]. Given that the PI3K-Akt pathway negatively regulates GSK-3β [321], the BDNF-TrkB pathway might activate PI3K to inhibit GSK-3β, thereby lowering tau phosphorylation. Moreover, intraperitoneal injection of mature BDNF reduced Aβ and tau pathologies by inhibiting GSK-3β in the hippocampi of APPswe mice [275]. Similarly, when mesenchymal stem cells carrying CX3CL1 and Wnt3a were transplanted into brains of APP/PS1 mice, CX3CL1 activated PI3K-Akt signaling to inhibit GSK-3β [141]. In addition, tau phosphorylation was decreased in the hippocampi of APP23 transgenic mice when FGF2 was injected subcutaneously, but some studies showed that FGF2 rather elevates tau expression and phosphorylation by increasing GSK-3β activity in adult rat hippocampal progenitor cells [175,177,279]. Meanwhile, FGF21 induced by calorie restriction reduced tau phosphorylation through the mTOR axis, and tau pathology was ameliorated by FGF21 in Aβ-treated SH-SY5Y cells [205,207]. Furthermore, astrocyte-neuron lactate shuttle (ANLS) was implicated in decreased tau phosphorylation by FGF21 in a transwell co-culture system with C6 astrocytes and PC12 neurons [208].
4. Conclusions and Perspectives
In this review, we examined the beneficial effects of exercise-induced myokines in NDs and their molecular mechanisms. In particular, we focused on the direct mechanism of action of myokine on the brain. However, myokines not only act directly on the brain, but also affect systemic metabolism, and consequently can have beneficial effects on NDs. Exercise regulated the remodeling of adipose tissue, reducing lipid content and controlling lipid browning [322]. In addition, muscles were closely related to systemic lipid homeostasis [323,324]. Moreover, some myokines have been known to affect lipid homeostasis [38,325]. For example, BDNF polymorphism was associated with type 2 diabetes mellitus in Caucasian females with obesity [326], and serum BDNF level was associated with obesity in female patients with type 2 diabetes mellitus [327]. Furthermore, BDNF treatment improved the lipid metabolism of a mouse model of type 2 diabetes [328], and intracerebroventricular injection of BDNF in rats increased lipolysis in adipose tissue [329]. In addition, FGF21 was identified as a key mediator of hepatic lipid metabolism in a high-fat, low-carbohydrate ketogenic diet [330]. A recent study showed that exercise sensitizes the action of FGF21 in adipose tissues, while long-term high-fat diet-fed obese mice exhibited compromised effects of exogenous FGF21 on alleviation of hyperglycemia, hyperinsulinemia, and hyperlipidemia [186]. Meanwhile, it has been suggested that dysregulation of lipid homeostasis is associated with NDs such as AD and PD [331,332]. Several previous studies have provided evidence that diverse types of lipids influence AD pathogenesis through various mechanisms, including mitochondrial dysfunction, BBB destruction, amyloidogenesis, inflammation, and oxidative stress [331]. Although there are few definitive studies on the effects of systemic regulation of lipid homeostasis by myokine on NDs so far, considering the function of some myokines in maintaining lipid homeostasis and the importance of lipid in the pathogenesis of NDs, myokines not only act directly on the brain, but may exert beneficial effects on NDs by affecting systemic metabolism.
Myokines may have indirect beneficial effects on NDs by affecting the composition of gut microbiota. Gut microbiota composition is affected by exercise, aging, diet, etc., and they not only affect the profile of myokines [333], but are closely related to NDs [334]. For example, according to a recent study, the gut microbiota of PD patients was different from that of normal people, which was associated with the motor phenotype [335]. Furthermore, a recent study reported that gut microbiota modulated motor deficits and neuroinflammation in a mouse model of PD [336]. Interestingly, it was reported that muscle mass decreased in germ-free mice [337], suggesting a close association between microbiota and muscle. Considering the effect of exercise on the gut microbiota, it is possible that myokine might affect the microbiota. However, few studies have been carried out on how myokines affect the composition of the microbiota. Further studies on this point are needed in the future, and the results of the studies are expected to provide more information about the role of myokine as a systemic regulator.
Since many myokines are simultaneously expressed and secreted by the muscles during exercise, the beneficial effects of exercise are manifested not only through each myokine, but also through the interactions between them. In this respect, it is noteworthy that some myokines promote the expression of others and act on the same molecular mechanism. For example, it has been reported that apelin, IGF-1, and irisin can maximize their effects by increasing the expression of BDNF [250,338,339]. In addition, as shown in Table 1, various myokines influence NDs by modulating similar molecular pathways: for example, both FGF2 and IGF-1 activate the PI3K/AKT signaling pathway. Therefore, when their expression is increased by exercise, it can be expected that they will create synergy. Advanced studies on the interactions between them are needed, and these studies will broaden our knowledge about the benefits of exercise manifested by myokine.
Based on the results of previous studies over the past 20 years, myokine is emerging as a promising treatment for NDs. However, studies about the effects of myokines on NDs are currently in their infancy, for several reasons. First, studies to date have focused on only a few myokines. In this review, we have examined nine myokines that are increased by exercise with relatively clear evidence for their direct effects on NDs. However, they only account for a small fraction of total myokines released from skeletal muscles during exercise. In fact, previous proteomics studies have shown that muscles secrete more than 600 proteins [340]. Some myokines, including adiponectin, β-aminoisobutyric acid, bone morphogenetic protein- and retinoic acid-inducible neural-specific protein-3, ciliary neurotrophic factor, CXCL10, CXCL12, follistatin-like-1, IL-6, IL-7, IL-15, matrix metalloproteinases-2, meteorin 1, musclin, myonectin, myostatin, osteoglycin, and secreted protein acidic and rich in cysteine (SPARC), are well-known for their effects on other organs [341,342], while their effects on the nervous system remain unclear. Therefore, more myokines with effects on NDs should be newly discovered and studied in the future. Second, even for known myokines studied up to date, their efficacies for NDs have not been fully established yet. For example, as we have seen in this review, IGF-1 showed beneficial and sometimes harmful effects. This means that effects of myokines may appear differently depending on physiological conditions. To use them safely as therapeutic agents, their effects under various conditions must be accurately grasped. Third, data about detailed mechanisms of action of myokines are currently lacking. With the exception of very few myokines, the exact mechanism of action is unknown for most myokines. Moreover, despite many studies showing the beneficial effects of myokines on ND, there are few studies on the ability of myokines to penetrate the BBB. The ability to penetrate the BBB has been experimentally demonstrated only for some of the myokines, such as BDNF, FGF21, and LIF. Although there is no direct experimental evidence yet, there is no reason to rule out the possibility that the remaining myokines will penetrate the BBB. In the future, detailed studies on this issue are needed. In addition, it should be considered that different types of physical exercise can release different profiles of myokines. Since the expression level of each myokine may differ depending on the type of exercise, the effects of exercise on NDs may vary depending on it. These limitations are important obstacles that should be overcome to develop myokines as therapeutic agents.
Despite these limitations, results on beneficial effects of exercise and myokines on NDs emphasize that it is important to study myokines induced by exercise for developing treatments for NDs. Each myokines’ expression condition is diverse depending on exercise type so difference of exercise type could bring different beneficial effects by expressing different myokines. In the absence of clear treatments for most NDs, studies of myokines known to mediate beneficial effects of exercise on NDs could provide a breakthrough for the development of novel treatments for these diseases.
Author Contributions
Conceptualization, K.S.C.; Formal analysis, B.L., M.S., Y.P., and S.-Y.W.; Data curation, B.L. and M.S.; Writing—original draft preparation, K.S.C., B.L., M.S., Y.P., and S.-Y.W.; Writing—review and editing, K.S.C., S.-Y.W., and B.L.; Supervision, K.S.C. and S.-Y.W.; Project administration, K.S.C. and S.-Y.W.; Funding acquisition, K.S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Konkuk University Research Fund in 2017 (2017-A019-0582, to K.S.C.).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| α-SYN | Alpha-synuclein |
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| AIF | Apoptosis-inducing factor |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| APP | Amyloid beta precursor protein |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| BACE1 | Beta-site APP cleaving enzyme 1 |
| BAD | BCL2-associated agonist of cell death |
| BAX | BCL2-associated X protein |
| BCL2 | B-cell lymphoma 2 |
| BCL-XL | B-cell lymphoma-extra large |
| BDNF | Brain-derived neurotrophic factor |
| CAT | Catalase |
| CHOP | C/EBP homologous protein |
| c-myb | Cellular myeloblastosis |
| COX-2 | Cyclooxygenase 2 |
| CREB | cAMP responsive element binding protein |
| CTSB | Cathepsin B |
| CVEC | Post-capillary venular endothelial cell |
| CX3CL1 | C-X3-C motif chemokine ligand 1 |
| CX3CR1 | C-X3-C motif chemokine receptor 1 |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FGF2 | Fibroblast growth factor 2 |
| FGF21 | Fibroblast growth factor 21 |
| 4′-DMA-7,8-DHF | 4′-Dimethylamino-7,8- dihydroxyflavone |
| GCM | Glioma cell-conditioned medium |
| GSH | Glutathione |
| GSK3 | Glycogen synthase kinase 3 |
| HD | Huntington’s disease |
| HO-1 | Heme oxygenase 1 |
| HSP90 | Heat shock protein 90 |
| IFN-γ | Interferon gamma |
| IGF-1 | Insulin-like growth factor 1 |
| IκBα | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| IL-12 | Interleukin 12 |
| iNOS | Inducible nitric oxide synthase |
| IRE1α | Inositol-requiring enzyme 1 alpha |
| JNK | c-Jun N-terminal kinase |
| Lamp1 | Lysosomal-associated membrane protein 1 |
| LIF | Leukemia inhibitory factor |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemo-attractant protein 1 |
| MEK | Mitogen-activated protein kinase kinase |
| MPP+ | 1-Methyl-4-phenylpyridinium |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MSC | Mesenchymal stem cell |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa B |
| NOS | Nitric oxide synthase |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PARP | Poly (ADP-ribose) polymerase |
| PD | Parkinson’s disease |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | Phosphoinositide 3-kinase |
| PP2A | Protein phosphatase 2 |
| PSEN1 | Presenilin 1 |
| RIP1 | Receptor-interacting protein kinase 1 |
| RIP3 | Receptor-interacting protein kinase 3 |
| SIRT1 | Sirtuin 1 |
| 6-OHDA | 6-hydroxydopamine |
| Smad2 | Mothers against decapentaplegic homolog 1 |
| SOD | Superoxide dismutase |
| TAT | Transactivator of transcription |
| TGF-β | Transforming growth factor beta |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor alpha |
| TrkB | Tropomyosin receptor kinase B |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| XBP1 | X-box binding protein 1 |
Notes
| APP/PS1 mice | APP/PS1 mice overexpress human APP with Swedish mutation (K670N and M671L) and presenilin 1, bearing an L166P mutation in neurons driven by thymocyte differentiation antigen 1 (Thy-1) promoter [343]. |
| APP/PS2 mice | APP/PS2 mice overexpress human APP with the Swedish mutation and presenilin 2, bearing an N141I mutation driven by the Thy-1 promoter and prion protein (Prnp) promoter, respectively [344,345]. |
| APPswe mice | APPswe mice overexpress human APP with the Swedish mutation driven by the Prnp promoter [346]. |
| APP23 mice | APP23 mice overexpress human APP with the Swedish mutation in neurons driven by the Thy-1 promoter [347]. |
| hAPPJ20 mice | hAPPJ20 mice overexpress human APP with the Swedish mutation and Indiana mutation (V717F) in neurons driven by platelet-derived growth factor (PDGF)-β promoter [348]. |
| MLC/mIGF-1 | MLC/mIGF-1 mice express rat mIGF-1, an isoform of IGF-1, cDNA driven by skeletal muscle-specific regulatory elements from the rat myosin light chain (MLC)-1/3 locus [349]. |
| N171-82Q mice | N171-82Q mice express an N-terminally truncated huntingtin cDNA that contains 82 glutamines and encompasses the first 171 amino acids of huntingtin (N171-82Q) driven by the Prnp promoter [350]. |
| PS19 mice | PS19 mice overexpress human tau with a mutation (P301S) driven by the Prnp promoter [351] |
| R1Mab1 | R1Mab1 is an IgG humanized monoclonal antibody with agonistic activity on the fibroblast growth factor receptor 1 (FGFR1) [213]. |
| SOD-G93A mice | SOD-G93A mice overexpress human SOD1 with a mutation (G93A) and develop adult-onset motor neuron loss [352]. |
| Tau P301L mice | Tau P301L mice overexpress human tau with a mutation (P301L) in neurons driven by the Thy-1 promoter [353]. |
References
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Shaw, P.J.; De Vos, K.J. Protein homeostasis in amyotrophic lateral sclerosis: Therapeutic opportunities? Front. Mol. Neurosci. 2017, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Cotman, C.W.; Berchtold, N.C.; Christie, L.-A. Exercise builds brain health: Key roles of growth factor cascades and inflammation. Trends Neurosci. 2007, 30, 464–472. [Google Scholar] [CrossRef]
- Cabral, D.F.; Rice, J.; Morris, T.P.; Rundek, T.; Pascual-Leone, A.; Gomes-Osman, J. Exercise for brain health: An investigation into the underlying mechanisms guided by dose. Neurotherapeutics 2019, 16, 580–599. [Google Scholar] [CrossRef]
- Neeper, S.A.; Gomez-Pinilla, F.; Choi, J.; Cotman, C. Exercise and brain neurotrophins. Nature 1995, 373, 109. [Google Scholar] [CrossRef]
- Cotman, C.W.; Berchtold, N.C. Exercise: A behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002, 25, 295–301. [Google Scholar] [CrossRef]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 2004, 20, 2580–2590. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1α in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 101108. [Google Scholar] [CrossRef]
- Ahlskog, J.E.; Geda, Y.E.; Graff-Radford, N.R.; Petersen, R.C. Physical exercise as a preventive or disease-modifying treatment of dementia and brain aging. Mayo Clin. Proc. 2011, 86, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cass, S.P. Alzheimer’s disease and exercise: A literature review. Curr. Sports Med. Rep. 2017, 16, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Lin, M.-S.; Tzeng, I. Relationship between exercise and Alzheimer’s disease: A narrative literature review. Front. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, C.; O’Sullivan, R.; Caserotti, P.; Tully, M.A. Consequences of physical inactivity in older adults: A systematic review of reviews and meta-analyses. Scand. J. Med. Sci. Sports 2020, 30, 816–827. [Google Scholar] [CrossRef]
- Da Costa Daniele, T.M.; de Bruin, P.F.C.; de Matos, R.S.; de Bruin, G.S.; Junior, C.M.C.; de Bruin, V.M.S. Exercise effects on brain and behavior in healthy mice, Alzheimer’s disease and Parkinson’s disease model—A systematic review and meta-analysis. Behav. Brain Res. 2020, 383, 112488. [Google Scholar] [CrossRef]
- Goodwin, V.A.; Richards, S.H.; Taylor, R.S.; Taylor, A.H.; Campbell, J.L. The effectiveness of exercise interventions for people with Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2008, 23, 631–640. [Google Scholar] [CrossRef]
- Mabandla, M.; Kellaway, L.; Gibson, A.S.C.; Russell, V.A. Voluntary running provides neuroprotection in rats after 6-hydroxydopamine injection into the medial forebrain bundle. Metab. Brain Dis. 2004, 19, 43–50. [Google Scholar] [CrossRef]
- Howells, F.M.; Russell, V.A.; Mabandla, M.V.; Kellaway, L.A. Stress reduces the neuroprotective effect of exercise in a rat model for Parkinson’s disease. Behav. Brain Res. 2005, 165, 210–220. [Google Scholar] [CrossRef]
- Poulton, N.P.; Muir, G.D. Treadmill training ameliorates dopamine loss but not behavioral deficits in hemi-parkinsonian rats. Exp. Neurol. 2005, 193, 181–197. [Google Scholar] [CrossRef]
- O’dell, S.J.; Gross, N.B.; Fricks, A.N.; Casiano, B.D.; Nguyen, T.B.; Marshall, J.F. Running wheel exercise enhances recovery from nigrostriatal dopamine injury without inducing neuroprotection. Neuroscience 2007, 144, 1141–1151. [Google Scholar] [CrossRef]
- Mabandla, M.V.; Kellaway, L.A.; Daniels, W.M.U.; Russell, V.A. Effect of exercise on dopamine neuron survival in prenatally stressed rats. Metab. Brain Dis. 2009, 24, 525–539. [Google Scholar] [CrossRef] [Green Version]
- Fritz, N.E.; Rao, A.K.; Kegelmeyer, D.; Kloos, A.; Busse, M.; Hartel, L.; Carrier, J.; Quinn, L. Physical therapy and exercise interventions in Huntington’s disease: A mixed methods systematic review. J. Huntingt. Dis. 2017, 6, 217–235. [Google Scholar] [CrossRef] [Green Version]
- Tsitkanou, S.; Della Gatta, P.; Foletta, V.; Russell, A. The role of exercise as a non-pharmacological therapeutic approach for amyotrophic lateral sclerosis: Beneficial or detrimental? Front. Neurol. 2019, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Corrochano, S.; Blanco, G.; Williams, D.; Wettstein, J.; Simon, M.; Kumar, S.; Moir, L.; Agnew, T.; Stewart, M.; Landman, A.; et al. A genetic modifier suggests that endurance exercise exacerbates Huntington’s disease. Hum. Mol. Genet. 2018, 27, 1723–1731. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, K.S.; Gjerum, L.; Waldemar, G.; Hasselbalch, S.G. Effects of physical exercise on Alzheimer’s disease biomarkers: A systematic review of intervention studies. J. Alzheimers Dis. 2018, 61, 359–372. [Google Scholar] [CrossRef]
- Playle, R.; Dimitropoulou, P.; Kelson, M.; Quinn, L.; Busse, M. Exercise Interventions in Huntington’s Disease: An Individual Patient Data Meta-Analysis. Mov. Disord. Clin. Pract. 2019, 6, 567–575. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical activity and muscle-brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Leal, L.G.; Lopes, M.A.; Batista, M.L., Jr. Physical exercise-induced myokines and muscle-adipose tissue crosstalk: A review of current knowledge and the implications for health and metabolic diseases. Front. Physiol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Febbraio, M.; Saltin, B. Searching for the exercise factor: Is IL-6 a candidate? J. Muscle Res. Cell Motil. 2003, 24, 113–119. [Google Scholar] [CrossRef]
- Whitham, M.; Febbraio, M.A. The ever-expanding myokinome: Discovery challenges and therapeutic implications. Nat. Rev. Drug. Discov. 2016, 15, 719. [Google Scholar] [CrossRef]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.T.; Greig, N.H.; Mattison, J.A.; et al. Running-Induced systemic cathepsin B secretion is associated with memory function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Morland, C.; Andersson, K.A.; Haugen, Ø.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor HCAR1. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Hiscock, N.; Sacchetti, M.; Fischer, C.P.; Pedersen, B.K. Interleukin-6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes 2004, 53, 1643–1648. [Google Scholar] [CrossRef] [Green Version]
- Mizgier, M.L.; Fernández-Verdejo, R.; Cherfan, J.; Pinget, M.; Bouzakri, K.; Galgani, J.E. Insights on the role of putative muscle-derived factors on pancreatic beta cell function. Front. Physiol. 2019, 10, 1024. [Google Scholar] [CrossRef] [Green Version]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.-X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef]
- Antushevich, H.; Wójcik, M. Apelin in disease. Clin. Chim. Acta 2018, 483, 241–248. [Google Scholar] [CrossRef]
- Kurowska, P.; Barbe, A.; Różycka, M.; Chmielińska, J.; Dupont, J.; Rak, A. Apelin in reproductive physiology and pathology of different species: A critical review. Int. J. Endocrinol. 2018, 2018. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Montastier, E.; Vinel, C.; Castan-Laurell, I.; Louche, K.; Dray, C.; Daviaud, D.; Mir, L.; Marques, M.; Thalamas, C.; et al. Effect of endurance training on skeletal muscle myokine expression in obese men: Identification of apelin as a novel myokine. Int. J. Obes. 2014, 38, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Vinel, C.; Lukjanenko, L.; Batut, A.; Deleruyelle, S.; Pradere, J.-P.; Le Gonidec, S.; Dortignac, A.; Geoffre, N.; Pereira, O.; Karaz, S.; et al. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018, 24, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The role of apelin in cardiovascular diseases, obesity and cancer. Front. Physiol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.A.; Agrawal, A.; Sabnekar, P.; Dichter, M.A.; Lynch, D.R.; Kolson, D.L. Apelin, an endogenous neuronal peptide, protects hippocampal neurons against excitotoxic injury. J. Neurochem. 2007, 102, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.J.; Yu, S.P.; Zhang, L.; Wei, L. Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Exp. Cell Res. 2010, 316, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, B.; Chen, J.; Bai, B.; Xin, Q. Neuroprotection of apelin and its signaling pathway. Peptides 2012, 37, 171–173. [Google Scholar] [CrossRef]
- Xin, Q.; Cheng, B.; Pan, Y.; Liu, H.; Yang, C.; Chen, J.; Bai, B. Neuroprotective effects of apelin-13 on experimental ischemic stroke through suppression of inflammation. Peptides 2015, 63, 55–62. [Google Scholar] [CrossRef]
- Bao, H.; Yang, X.; Huang, Y.; Qiu, H.; Huang, G.; Xiao, H.; Kuai, J. The neuroprotective effect of apelin-13 in a mouse model of intracerebral hemorrhage. Neurosci. Lett. 2016, 628, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Kalantaripour, T.P.; Esmaeili-Mahani, S.; Sheibani, V.; Asadi-Shekaari, M.; Pasban-Aliabadi, H. Anticonvulsant and neuroprotective effects of apelin-13 on pentylenetetrazole-induced seizures in male rats. Biomed. Pharmacother. 2016, 84, 258–263. [Google Scholar] [CrossRef]
- Xu, W.; Li, T.; Gao, L.; Zheng, J.; Yan, J.; Zhang, J.; Shao, A. Apelin-13/APJ system attenuates early brain injury via suppression of endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation and oxidative stress in a AMPK-dependent manner after subarachnoid hemorrhage in rats. J. Neuroinflamm. 2019, 16, 1–14. [Google Scholar] [CrossRef]
- Haghparast, E.; Esmaeili-Mahani, S.; Abbasnejad, M.; Sheibani, V. Apelin-13 ameliorates cognitive impairments in 6-hydroxydopamine-induced substantia nigra lesion in rats. Neuropeptides 2018, 68, 28–35. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, H.; Ji, B.; Wang, Z.; Wang, C.; Yang, C.; Pan, Y.; Chen, J.; Cheng, B.; Bai, B. Apelin-13 attenuates ER stress-associated apoptosis induced by MPP+ in SH-SY5Y cells. Int. J. Mol. Med. 2018, 42, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Pouresmaeili-Babaki, E.; Esmaeili-Mahani, S.; Abbasnejad, M.; Ravan, H. Protective effect of neuropeptide apelin-13 on 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y dopaminergic cells: Involvement of its antioxidant and antiapoptotic properties. Rejuvenation Res. 2018, 21, 162–167. [Google Scholar] [CrossRef]
- Aminyavari, S.; Zahmatkesh, M.; Farahmandfar, M.; Khodagholi, F.; Dargahi, L.; Zarrindast, M.-R. Protective role of Apelin-13 on amyloid β25–35-induced memory deficit; Involvement of autophagy and apoptosis process. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 322–334. [Google Scholar] [CrossRef]
- Aminyavari, S.; Zahmatkesh, M.; Khodagholi, F.; Sanati, M. Anxiolytic impact of Apelin-13 in a rat model of Alzheimer’s disease: Involvement of glucocorticoid receptor and FKBP5. Peptides 2019, 118, 170102. [Google Scholar] [CrossRef]
- Luo, H.; Xiang, Y.; Qu, X.; Liu, H.; Liu, C.; Li, G.; Han, L.; Qin, X. Apelin-13 suppresses neuroinflammation against cognitive deficit in a streptozotocin-induced rat model of Alzheimer’s disease through activation of BDNF-TrkB signaling pathway. Front. Pharmacol. 2019, 10, 395. [Google Scholar] [CrossRef]
- Zhu, J.; Dou, S.; Jiang, Y.; Bai, B.; Chen, J.; Wang, C.; Cheng, B. Apelin-36 exerts the cytoprotective effect against MPP+-induced cytotoxicity in SH-SY5Y cells through PI3K/Akt/mTOR autophagy pathway. Life Sci. 2019, 224, 95–108. [Google Scholar] [CrossRef]
- Chen, P.; Wang, Y.; Chen, L.; Song, N.; Xie, J. Apelin-13 Protects Dopaminergic Neurons against Rotenone—Induced Neurotoxicity through the AMPK/mTOR/ULK-1 Mediated Autophagy Activation. Int. J. Mol. Sci. 2020, 21, 8376. [Google Scholar] [CrossRef]
- Nasseri, B.; Zareian, P.; Alizade, H. Apelin attenuates streptozotocin-induced learning and memory impairment by modulating necroptosis signaling pathway. Int. Immunopharmacol. 2020, 84, 106546. [Google Scholar] [CrossRef]
- Kasai, A.; Kinjo, T.; Ishihara, R.; Sakai, I.; Ishimaru, Y.; Yoshioka, Y.; Yamamuro, A.; Ishige, K.; Ito, Y.; Maeda, S. Apelin deficiency accelerates the progression of amyotrophic lateral sclerosis. PLoS ONE 2011, 6, e23968. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Barde, Y.-A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef]
- Binder, D.K.; Scharfman, H.E. Brain-Derived neurotrophic factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Hing, B.; Sathyaputri, L.; Potash, J.B. A comprehensive review of genetic and epigenetic mechanisms that regulate BDNF expression and function with relevance to major depressive disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 143–167. [Google Scholar] [CrossRef] [Green Version]
- Squinto, S.P.; Stitt, T.N.; Aldrich, T.H.; Davis, S.; Blanco, S.M.; RadzieJewski, C.; Glass, D.J.; Masiakowski, P.; Furth, M.E.; Valenzuela, D.M.; et al. trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell 1991, 65, 885–893. [Google Scholar] [CrossRef]
- Castrén, E.; Rantamäki, T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev. Neurobiol. 2010, 70, 289–297. [Google Scholar] [CrossRef]
- Etnier, J.L.; Wideman, L.; Labban, J.D.; Piepmeier, A.T.; Pendleton, D.M.; Dvorak, K.K.; Becofsky, K. The effects of acute exercise on memory and brain-derived neurotrophic factor (BDNF). J. Sport Exerc. Psychol. 2016, 38, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.J.; Tschakovsky, M.E. Exercise and circulating BDNF: Mechanisms of release and implications for the design of exercise interventions. Appl. Physiol. Nutr. Metab. 2018, 43, 1095–1104. [Google Scholar] [CrossRef]
- Yang, X.; Brobst, D.; Chan, W.S.; Tse, M.C.L.; Herlea-Pana, O.; Ahuja, P.; Bi, X.; Zaw, A.M.; Kwong, Z.S.W.; Jia, W.-H.; et al. Muscle-Generated BDNF is a sexually dimorphic myokine that controls metabolic flexibility. Sci. Signal. 2019, 12, eaau1468. [Google Scholar] [CrossRef]
- Fulgenzi, G.; Hong, Z.; Tomassoni-Ardori, F.; Barella, L.F.; Becker, J.; Barrick, C.; Swing, D.; Yanpallewar, S.; St Croix, B.; Wess, J.; et al. Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361. [Google Scholar] [CrossRef] [Green Version]
- Real, C.C.; Ferreira, A.F.B.; Chaves-Kirsten, G.P.; Torrao, A.S.; Pires, R.S.; Britto, L.R.G. BDNF receptor blockade hinders the beneficial effects of exercise in a rat model of Parkinson’s disease. Neuroscience 2013, 237, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Qin, X.-Y.; Cao, C.; Cawley, N.X.; Liu, T.-T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N = 7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Beiser, A.S.; Choi, S.H.; Preis, S.R.; Chen, T.C.; Vorgas, D.; Au, R.; Pikula, A.; Wolf, P.A.; DeStefano, A.L.; et al. Serum brain-derived neurotrophic factor and the risk for dementia: The Framingham Heart Study. JAMA Neurol. 2014, 71, 55–61. [Google Scholar] [CrossRef]
- Arancibia, S.; Silhol, M.; Mouliere, F.; Meffre, J.; Höllinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Müller, F.-J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 2013, 33, 15596–15602. [Google Scholar] [CrossRef] [Green Version]
- de Pins, B.; Cifuentes-Díaz, C.; Farah, A.T.; López-Molina, L.; Montalban, E.; Sancho-Balsells, A.; López, A.; Ginés, S.; Delgado-García, J.M.; Alberch, J.; et al. Conditional BDNF delivery from astrocytes rescues memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of Alzheimer disease. J. Neurosci. 2019, 39, 2441–2458. [Google Scholar] [CrossRef] [Green Version]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.Y.; Villemagne, V.L.; Laws, S.M.; Pietrzak, R.H.; Snyder, P.J.; Ames, D.; Ellis, K.A.; Harrington, K.; Rembach, A.; Martins, R.N.; et al. APOE and BDNF polymorphisms moderate amyloid β-related cognitive decline in preclinical Alzheimer’s disease. Mol. Psychiatry 2015, 20, 1322–1328. [Google Scholar] [CrossRef]
- Franzmeier, N.; Ren, J.; Damm, A.; Monté-Rubio, G.; Boada, M.; Ruiz, A.; Ramirez, A.; Jessen, F.; Düzel, E.; Gómez, O.R.; et al. The BDNF Val66Met SNP modulates the association between β-amyloid and hippocampal disconnection in Alzheimer’s disease. Mol. Psychiatry 2021, 26, 614–628. [Google Scholar] [CrossRef]
- Hyman, C.; Hofer, M.; Barde, Y.-A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef]
- Altar, C.A.; Boylan, C.B.; Jackson, C.; Hershenson, S.; Miller, J.; Wiegand, S.J.; Lindsay, R.M.; Hyman, C. Brain-Derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 11347–11351. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Negro, A.; Facci, L.; Toso, R.D. Brain-Derived neurotrophic factor selectively rescues mesencephalic dopaminergic neurons from 2, 4, 5-trihydroxyphenylalanine-induced injury. J. Neurosci. Res. 1993, 34, 478–487. [Google Scholar] [CrossRef]
- Frim, D.M.; Uhler, T.A.; Galpern, W.R.; Beal, M.F.; Breakefield, X.O.; Isacson, O. Implanted fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevent 1-methyl-4-phenylpyridinium toxicity to dopaminergic neurons in the rat. Proc. Natl. Acad. Sci. USA 1994, 91, 5104–5108. [Google Scholar] [CrossRef] [Green Version]
- Levivier, M.; Przedborski, S.; Bencsics, C.; Kang, U.J. Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson’s disease. J. Neurosci. 1995, 15, 7810–7820. [Google Scholar] [CrossRef] [Green Version]
- Galpern, W.R.; Frim, D.M.; Tatter, S.B.; Altar, C.A.; Beal, M.F.; Isacson, O. Cell-Mediated delivery of brain-derived neurotrophic factor enhances dopamine levels in an MPP+ rat model of substantia nigra degeneration. Cell Transplant. 1996, 5, 225–232. [Google Scholar] [CrossRef]
- Fang, F.; Yang, W.; Florio, J.B.; Rockenstein, E.; Spencer, B.; Orain, X.M.; Dong, S.X.; Li, H.; Chen, X.; Sung, K.; et al. Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson’s disease. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J.; Sun, Y.E.; Ye, K. TrkB neurotrophic activities are blocked by α-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 10773–10778. [Google Scholar] [CrossRef] [Green Version]
- Mort, J.S.; Buttle, D.J. Cathepsin b. Int. J. Biochem. Cell Biol. 1997, 29, 715–720. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin B. Autophagy 2016, 12, 2504–2505. [Google Scholar] [CrossRef] [Green Version]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, H.C.; Fujita, S.; Cadenas, J.G.; Chinkes, D.L.; Volpi, E.; Rasmussen, B.B. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J. Physiol. 2006, 576, 613–624. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Holloway, G.P.; Steinberg, G.R. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: Implications for obesity. Mol. Cell. Endocrinol. 2013, 366, 135–151. [Google Scholar] [CrossRef]
- De la Rosa, A.; Solana, E.; Corpas, R.; Bartrés-Faz, D.; Pallàs, M.; Vina, J.; Sanfeliu, C.; Gomez-Cabrera, M.C. Long-Term exercise training improves memory in middle-aged men and modulates peripheral levels of BDNF and Cathepsin, B. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.-H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-cathepsin B axis regulates amyloid β levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sun, B.; Zhou, Y.; Grubb, A.; Gan, L. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 2012, 287, 39834–39841. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.-G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef] [Green Version]
- Embury, C.M.; Dyavarshetty, B.; Lu, Y.; Wiederin, J.L.; Ciborowski, P.; Gendelman, H.E.; Kiyota, T. Cathepsin B improves ß-amyloidosis and learning and memory in models of Alzheimer’s disease. J. Neuroimmune Pharmacol. 2017, 12, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Estick, C.M.; Ikonne, U.S.; Butler, D.; Pait, M.C.; Elliott, L.H.; Ruiz, S.; Smith, K.; Rentschler, K.M.; Mundell, C.; et al. The role of lysosomes in a broad disease-modifying approach evaluated across transgenic mouse models of Alzheimer’s disease and Parkinson’s disease and models of mild cognitive impairment. Int. J. Mol. Sci. 2019, 20, 4432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX 3 C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Lloyd, C.; Zhou, H.; Dolich, S.; Deeds, J.; Gonzalo, J.-A.; Vath, J.; Gosselin, M.; Ma, J.; Dussault, B.; et al. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature 1997, 387, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor necrosis factor-α-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [CrossRef] [PubMed]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A potential new target for inflammatory diseases. Mol. Interv. 2010, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Peake, J.M.; Della Gatta, P.; Suzuki, K.; Nieman, D.C. Cytokine expression and secretion by skeletal muscle cells: Regulatory mechanisms and exercise effects. Exerc. Immunol. Rev. 2015, 21, 8–25. [Google Scholar]
- Catoire, M.; Mensink, M.; Kalkhoven, E.; Schrauwen, P.; Kersten, S. Identification of human exercise-induced myokines using secretome analysis. Physiol. Genom. 2014, 46, 256–267. [Google Scholar] [CrossRef]
- Della Gatta, P.A.; Cameron-Smith, D.; Peake, J.M. Acute resistance exercise increases the expression of chemotactic factors within skeletal muscle. Eur. J. Appl. Physiol. 2014, 114, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Catoire, M.; Kersten, S. The search for exercise factors in humans. FASEB J. 2015, 29, 1615–1628. [Google Scholar] [CrossRef]
- Strömberg, A.; Olsson, K.; Dijksterhuis, J.P.; Rullman, E.; Schulte, G.; Gustafsson, T. CX3CL1—A macrophage chemoattractant induced by a single bout of exercise in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R297–R304. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Liu, L.; Cardona, A.E. Chemokines and chemokine receptors: Multipurpose players in neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 187–204. [Google Scholar] [CrossRef]
- Duan, R.-S.; Yang, X.; Chen, Z.-G.; Lu, M.-O.; Morris, C.; Winblad, B.; Zhu, J. Decreased Fractalkine and Increased IP-10 Expression in Aged Brain of APP swe Transgenic Mice. Neurochem. Res. 2008, 33, 1085–1089. [Google Scholar] [CrossRef]
- Cho, S.-H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef] [Green Version]
- Perea, J.R.; Lleó, A.; Alcolea, D.; Fortea, J.; Ávila, J.; Bolós, M. Decreased CX3CL1 levels in the cerebrospinal fluid of patients with Alzheimer’s disease. Front. Neurosci. 2018, 12, 609. [Google Scholar] [CrossRef]
- González-Prieto, M.; Gutiérrez, I.L.; García-Bueno, B.; Caso, J.R.; Leza, J.C.; Ortega-Hernández, A.; Gómez-Garre, D.; Madrigal, J.L.M. Microglial CX3CR1 production increases in Alzheimer’s disease and is regulated by noradrenaline. Glia 2021, 69, 73–90. [Google Scholar] [CrossRef]
- Kim, T.-S.; Lim, H.-K.; Lee, J.Y.; Kim, D.-J.; Park, S.; Lee, C.; Lee, C.-U. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2008, 436, 196–200. [Google Scholar] [CrossRef]
- Kulczyńska-Przybik, A.; Słowik, A.; Mroczko, P.; Borawski, B.; Groblewska, M.; Borawska, R.; Mroczko, B. Cerebrospinal Fluid and Blood CX3CL1 as a Potential Biomarker in Early Diagnosis and Prognosis of Dementia. Curr. Alzheimer Res. 2020, 17, 709–721. [Google Scholar] [CrossRef]
- Shi, M.; Bradner, J.; Hancock, A.M.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; Jankovic, J.; Zabetian, C.P.; Kim, H.M.; et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann. Neurol. 2011, 69, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; García-García, E.; Straccia, M.; Comella-Bolla, A.; Miguez, A.; Masana, M.; Alberch, J.; Canals, J.M.; Rodríguez, M.J. Reduced Fractalkine Levels Lead to Striatal Synaptic Plasticity Deficits in Huntington’s Disease. Front. Cell. Neurosci. 2020, 14, 163. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Liu, X.; Li, S.; Cheng, C.; Chen, S.; Le, W. Dynamic changes of CX3CL1/CX3CR1 axis during microglial activation and motor neuron loss in the spinal cord of ALS mouse model. Transl. Neurodegener. 2018, 7, 1–14. [Google Scholar] [CrossRef]
- Pabon, M.M.; Bachstetter, A.D.; Hudson, C.E.; Gemma, C.; Bickford, P.C. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflamm. 2011, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Morganti, J.M.; Nash, K.R.; Grimmig, B.A.; Ranjit, S.; Small, B.; Bickford, P.C.; Gemma, C. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 2012, 32, 14592–14601. [Google Scholar] [CrossRef] [Green Version]
- Nash, K.R.; Lee, D.C.; Hunt, J.B., Jr.; Morganti, J.M.; Selenica, M.-L.; Moran, P.; Reid, P.; Brownlow, M.; Yang, C.G.-Y.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef]
- Nash, K.R.; Moran, P.; Finneran, D.J.; Hudson, C.; Robinson, J.; Morgan, D.; Bickford, P.C. Fractalkine over expression suppresses α-synuclein-mediated neurodegeneration. Mol. Ther. 2015, 23, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Merino, J.J.; Muñetón-Gómez, V.; Alvárez, M.-I.; Toledano-Díaz, A. Effects of cx3cr1 and fractalkine chemokines in amyloid β clearance and p-tau accumulation in Alzheimer, s disease (ad) rodent models: Is fractalkine a systemic biomarker for ad? Curr. Alzheimer Res. 2016, 13, 403–412. [Google Scholar] [CrossRef]
- Finneran, D.J.; Morgan, D.; Gordon, M.N.; Nash, K.R. CNS-Wide over expression of fractalkine improves cognitive functioning in a tauopathy model. J. Neuroimmune Pharmacol. 2019, 14, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Zhao, J.; Fan, C.; Zhu, L.; Huang, C.; Li, Q.; Gan, D.; Wen, C.; Chen, M.; Lu, D. Delivery of exogenous proteins by mesenchymal stem cells attenuates early memory deficits in a murine model of Alzheimer’s disease. Neurobiol. Aging 2020, 86, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces β-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Hong-Min, T.; Yi, F.; Jun-Peng, G.; Yue, F.; Yan-Hong, T.; Yun-Ke, Y.; Wen-Wei, L.; Xiang-Yu, W.; Jun, M.; et al. New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson’s disease. Neurobiol. Aging 2011, 32, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Dworzak, J.; Renvoisé, B.; Habchi, J.; Yates, E.V.; Combadière, C.; Knowles, T.P.; Dobson, C.M.; Blackstone, C.; Paulsen, O.; Murphy, P.M. Neuronal Cx3cr1 deficiency protects against amyloid β-induced neurotoxicity. PLoS ONE 2015, 10, e0127730. [Google Scholar] [CrossRef] [Green Version]
- Thome, A.D.; Standaert, D.G.; Harms, A.S. Fractalkine signaling regulates the inflammatory response in an α-synuclein model of Parkinson disease. PLoS ONE 2015, 10, e0140566. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; Coleman, U.; Kingery-Gallagher, N.D.; El Khoury, J. Heterozygous CX3CR1 deficiency in microglia restores neuronal β-Amyloid clearance pathways and slows progression of Alzheimer’s Like-Disease in PS1-APP Mice. Front. Immunol. 2019, 10, 2780. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rabano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Perea, J.R.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; López-Royo, T.; Casarejos, M.; Lanciego, J.L.; Lastres-Becker, I. Cx3cr1-deficiency exacerbates alpha-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia 2018, 66, 1752–1762. [Google Scholar] [CrossRef]
- Liu, C.; Hong, K.; Chen, H.; Niu, Y.; Duan, W.; Liu, Y.; Ji, Y.; Deng, B.; Li, Y.; Li, Z.; et al. Evidence for a protective role of the CX3CL1/CX3CR1 axis in a model of amyotrophic lateral sclerosis. Biol. Chem. 2019, 400, 651–661. [Google Scholar] [CrossRef]
- Cattaneo, E.; McKay, R. Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature 1990, 347, 762–765. [Google Scholar] [CrossRef]
- Breen, E.C.; Johnson, E.C.; Wagner, H.; Tseng, H.M.; Sung, L.A.; Wagner, P.D. Angiogenic growth factor mRNA responses in muscle to a single bout of exercise. J. Appl. Physiol. 1996, 81, 355–361. [Google Scholar] [CrossRef]
- Morrow, N.G.; Kraus, W.E.; Moore, J.W.; Williams, R.S.; Swain, J.L. Increased expression of fibroblast growth factors in a rabbit skeletal muscle model of exercise conditioning. J. Clin. Investig. 1990, 85, 1816–1820. [Google Scholar] [CrossRef]
- Gómez-Pinilla, F.; Dao, L.; So, V. Physical exercise induces FGF-2 and its mRNA in the hippocampus. Brain Res. 1997, 764, 1–8. [Google Scholar] [CrossRef]
- Brenner, D.R.; Ruan, Y.; Adams, S.C.; Courneya, K.S.; Friedenreich, C.M. The impact of exercise on growth factors (VEGF and FGF2): Results from a 12-month randomized intervention trial. Eur. Rev. Aging Phys. Act. 2019, 16, 1–6. [Google Scholar] [CrossRef]
- Temmerman, K.; Ebert, A.D.; Müller, H.-M.; Sinning, I.; Tews, I.; Nickel, W. A direct role for phosphatidylinositol-4, 5-bisphosphate in unconventional secretion of fibroblast growth factor 2. Traffic 2008, 9, 1204–1217. [Google Scholar] [CrossRef]
- Zehe, C.; Engling, A.; Wegehingel, S.; Schäfer, T.; Nickel, W. Cell-Surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc. Natl. Acad. Sci. USA 2006, 103, 15479–15484. [Google Scholar] [CrossRef] [Green Version]
- Steringer, J.P.; Bleicken, S.; Andreas, H.; Zacherl, S.; Laussmann, M.; Temmerman, K.; Contreras, F.X.; Bharat, T.A.M.; Lechner, J.; Müller, H.-M.; et al. Phosphatidylinositol 4, 5-bisphosphate (PI (4, 5) P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J. Biol. Chem. 2012, 287, 27659–27669. [Google Scholar] [CrossRef] [Green Version]
- Steringer, J.P.; Lange, S.; Čujová, S.; Šachl, R.; Poojari, C.; Lolicato, F.; Beutel, O.; Müller, H.-M.; Unger, S.; Coskun, Ü.; et al. Key steps in unconventional secretion of fibroblast growth factor 2 reconstituted with purified components. eLife 2017, 6, e28985. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Davis, A.A.; Goderie, S.K.; Temple, S. FGF2 concentration regulates the generation of neurons and glia from multipotent cortical stem cells. Neuron 1997, 18, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Vaccarino, F.M.; Schwartz, M.L.; Raballo, R.; Nilsen, J.; Rhee, J.; Zhou, M.; Doetschman, T.; Coffin, J.D.; Wyland, J.J.; Hung, Y.-T.E. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat. Neurosci. 1999, 2, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, S.; Takagi, Y.; Harada, J.; Teramoto, T.; Thomas, S.S.; Waeber, C.; Bakowska, J.C.; Breakefield, X.O.; Moskowitz, M.A. FGF-2 regulation of neurogenesis in adult hippocampus after brain injury. Proc. Natl. Acad. Sci. USA 2001, 98, 5874–5879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.; Sun, Y.; Xie, L.; Batteur, S.; Mao, X.O.; Smelick, C.; Logvinova, A.; Greenberg, D.A. Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell 2003, 2, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Bonomo, A.; Di Liberto, V.; Frinchi, M.; Fuxe, K.; Belluardo, N. The FGF-2/FGFRs neurotrophic system promotes neurogenesis in the adult brain. J. Neural Transm. 2009, 116, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Andrés, G.; Leali, D.; Dell’Era, P.; Ronca, R. Inflammatory cells and chemokines sustain FGF2-induced angiogenesis. Eur. Cytokine Netw. 2009, 20, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Kakkar, A.; Sharma, R.; Kharbanda, O.P.; Monga, N.; Kumar, M.; Chowdhary, S.; Airan, B.; Mohanty, S. Synergistic effect of BDNF and FGF2 in efficient generation of functional dopaminergic neurons from human mesenchymal stem cells. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; LaFevre-Bernt, M.; Sun, Y.; Chen, S.; Gafni, J.; Crippen, D.; Logvinova, A.; Ross, C.A.; Greenberg, D.A.; Ellerby, L.M. FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 18189–18194. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Xie, L.; Jin, K.; Greenberg, D.A.; Andersen, J.K. Fibroblast growth factor 2 enhances striatal and nigral neurogenesis in the acute 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine model of Parkinson’s disease. Neuroscience 2008, 153, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Kiyota, T.; Ingraham, K.L.; Jacobsen, M.T.; Xiong, H.; Ikezu, T. FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc. Natl. Acad. Sci. USA 2011, 108, E1339–E1348. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Xiao, Z.; Huang, J. C6 glioma-secreted NGF and FGF2 regulate neuronal APP processing through up-regulation of ADAM10 and down-regulation of BACE1, respectively. J. Mol. Neurosci. 2016, 59, 334–342. [Google Scholar] [CrossRef]
- Stopa, E.G.; Gonzalez, A.-M.; Chorsky, R.; Corona, R.J.; Alvarez, J.; Bird, E.D.; Baird, A. Basic fibroblast growth factor in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1990, 171, 690–696. [Google Scholar] [CrossRef]
- Cummings, B.J.; Su, J.H.; Cotman, C.W. Neuritic involvement within bFGF immunopositive plaques of Alzheimer’s disease. Exp. Neurol. 1993, 124, 315–325. [Google Scholar] [CrossRef]
- Johansson, A.; Larsson, A.; Nygren, I.; Blennow, K.; Askmark, H. Increased serum and cerebrospinal fluid FGF-2 levels in amyotrophic lateral sclerosis. Neuroreport 2003, 14, 1867–1869. [Google Scholar] [CrossRef]
- Tatebayashi, Y.; Iqbal, K.; Grundke-Iqbal, I. Dynamic regulation of expression and phosphorylation of tau by fibroblast growth factor-2 in neural progenitor cells from adult rat hippocampus. J. Neurosci. 1999, 19, 5245–5254. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Tung, Y.-C.; Li, B.; Iqbal, K.; Grundke-Iqbal, I. Trophic factors counteract elevated FGF-2-induced inhibition of adult neurogenesis. Neurobiol. Aging 2007, 28, 1148–1162. [Google Scholar] [CrossRef]
- Tatebayashi, Y.; Lee, M.H.; Li, L.; Iqbal, K.; Grundke-Iqbal, I. The dentate gyrus neurogenesis: A therapeutic target for Alzheimer’s disease. Acta Neuropathol. 2003, 105, 225–232. [Google Scholar] [CrossRef]
- Thau, N.; Jungnickel, J.; Knippenberg, S.; Ratzka, A.; Dengler, R.; Petri, S.; Grothe, C. Prolonged survival and milder impairment of motor function in the SOD1 ALS mouse model devoid of fibroblast growth factor 2. Neurobiol. Dis. 2012, 47, 248–257. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Lewis, J.E.; Ebling, F.J.P.; Samms, R.J.; Tsintzas, K. Going back to the biology of FGF21: New insights. Trends Endocrinol. Metab. 2019, 30, 491–504. [Google Scholar] [CrossRef]
- Cuevas-Ramos, D.; Almeda-Valdés, P.; Meza-Arana, C.E.; Brito-Córdova, G.; Gómez-Pérez, F.J.; Mehta, R.; Oseguera-Moguel, J.; Aguilar-Salinas, C.A. Exercise increases serum fibroblast growth factor 21 (FGF21) levels. PLoS ONE 2012, 7, e38022. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, S.H.; Min, Y.-K.; Yang, H.-M.; Lee, J.-B.; Lee, M.-S. Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS ONE 2013, 8, e63517. [Google Scholar] [CrossRef]
- Hansen, J.S.; Clemmesen, J.O.; Secher, N.H.; Hoene, M.; Drescher, A.; Weigert, C.; Pedersen, B.K.; Plomgaard, P. Glucagon-to-insulin ratio is pivotal for splanchnic regulation of FGF-21 in humans. Mol. Metab. 2015, 4, 551–560. [Google Scholar] [CrossRef]
- Geng, L.; Liao, B.; Jin, L.; Huang, Z.; Triggle, C.R.; Ding, H.; Zhang, J.; Huang, Y.; Lin, Z.; Xu, A. Exercise alleviates obesity-induced metabolic dysfunction via enhancing FGF21 sensitivity in adipose tissues. Cell Rep. 2019, 26, 2738–2752.e4. [Google Scholar] [CrossRef] [Green Version]
- Douris, N.; Stevanovic, D.M.; Fisher, F.M.; Cisu, T.I.; Chee, M.J.; Nguyen, N.L.; Zarebidaki, E.; Adams, A.C.; Kharitonenkov, A.; Flier, J.S.; et al. Bart Central fibroblast growth factor 21 browns white fat via sympathetic action in male mice. Endocrinology 2015, 156, 2470–2481. [Google Scholar] [CrossRef]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008, 582, 3805–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimura, Y.; Aoi, W.; Takanami, Y.; Kawai, Y.; Mizushima, K.; Naito, Y.; Yoshikawa, T. Acute exercise increases fibroblast growth factor 21 in metabolic organs and circulation. Physiol. Rep. 2016, 4, e12828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.-N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keipert, S.; Ost, M.; Johann, K.; Imber, F.; Jastroch, M.; van Schothorst, E.M.; Keijer, J.; Klaus, S. Skeletal muscle mitochondrial uncoupling drives endocrine cross-talk through the induction of FGF21 as a myokine. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E469–E482. [Google Scholar] [CrossRef]
- Ji, K.; Zheng, J.; Lv, J.; Xu, J.; Ji, X.; Luo, Y.-B.; Li, W.; Zhao, Y.; Yan, C. Skeletal muscle increases FGF21 expression in mitochondrial disorders to compensate for energy metabolic insufficiency by activating the mTOR-YY1-PGC1α pathway. Free Radic. Biol. Med. 2015, 84, 161–170. [Google Scholar] [CrossRef]
- Pereira, R.O.; Tadinada, S.M.; Zasadny, F.M.; Oliveira, K.J.; Pires, K.M.P.; Olvera, A.; Jeffers, J.; Souvenir, R.; Mcglauflin, R.; Seei, A.; et al. OPA 1 deficiency promotes secretion of FGF 21 from muscle that prevents obesity and insulin resistance. EMBO J. 2017, 36, 2126–2145. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Mokhtar, R.; Bayliss, J.; Parkington, H.C.; Suturin, V.M.; Bruce, C.R.; Watt, M.J. Perilipin 5 deletion unmasks an endoplasmic reticulum stress-fibroblast growth factor 21 axis in skeletal muscle. Diabetes 2018, 67, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Hsuchou, H.; Pan, W.; Kastin, A.J. The fasting polypeptide FGF21 can enter brain from blood. Peptides 2007, 28, 2382–2386. [Google Scholar] [CrossRef] [Green Version]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Satjaritanun, P.; Wang, X.; Liang, G.; Li, X.; Jiang, C.; Pratchayasakul, W.; Chattipakorn, N.; et al. FGF21 improves cognition by restored synaptic plasticity, dendritic spine density, brain mitochondrial function and cell apoptosis in obese-insulin resistant male rats. Horm. Behav. 2016, 85, 86–95. [Google Scholar] [CrossRef]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Wang, X.; Liang, G.; Li, X.; Jiang, C.; Pratchayasakul, W.; Chattipakorn, N.; et al. FGF21 and DPP-4 inhibitor equally prevents cognitive decline in obese rats. Biomed. Pharmacother. 2018, 97, 1663–1672. [Google Scholar] [CrossRef]
- Wang, Q.; Yuan, J.; Yu, Z.; Lin, L.; Jiang, Y.; Cao, Z.; Zhuang, P.; Whalen, M.J.; Song, B.; Wang, X.-J.; et al. FGF21 attenuates high-fat diet-induced cognitive impairment via metabolic regulation and anti-inflammation of obese mice. Mol. Neurobiol. 2018, 55, 4702–4717. [Google Scholar] [CrossRef]
- Shahror, R.A.; Linares, G.R.; Wang, Y.; Hsueh, S.-C.; Wu, C.-C.; Chuang, D.-M.; Chiang, Y.-H.; Chen, K.-Y. Transplantation of mesenchymal stem cells overexpressing fibroblast growth factor 21 facilitates cognitive recovery and enhances neurogenesis in a mouse model of traumatic brain injury. J. Neurotrauma 2020, 37, 14–26. [Google Scholar] [CrossRef]
- Chen, J.; Hu, J.; Liu, H.; Xiong, Y.; Zou, Y.; Huang, W.; Shao, M.; Wu, J.; Yu, L.; Wang, X.; et al. FGF21 protects the blood-brain barrier by upregulating PPARγ via FGFR1/β-klotho after traumatic brain injury. J. Neurotrauma 2018, 35, 2091–2103. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, N.; Wang, Q.; Yu, Z.; Lin, L.; Yuan, J.; Guo, S.; Ahn, B.J.; Wang, X.-J.; Li, X.; et al. Endocrine regulator rFGF21 (recombinant human fibroblast growth factor 21) improves neurological outcomes following focal ischemic stroke of type 2 diabetes mellitus male mice. Stroke 2018, 49, 3039–3049. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lin, L.; Liu, N.; Wang, Q.; Yuan, J.; Li, Y.; Chung, K.K.; Guo, S.; Yu, Z.; Wang, X. FGF21 protects against aggravated blood-brain barrier disruption after ischemic focal stroke in diabetic db/db male mice via cerebrovascular PPARγ activation. Int. J. Mol. Sci. 2020, 21, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Lin, L.; Jiang, Y.; Chin, I.; Wang, X.; Li, X.; Lo, E.H.; Wang, X. Recombinant FGF21 protects against blood-brain barrier leakage through Nrf2 upregulation in type 2 diabetes mice. Mol. Neurobiol. 2019, 56, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, F.; Zhu, L.; Lin, P.; Han, F.; Wang, X.; Tan, X.; Lin, L.; Xiong, Y. FGF21 alleviates neuroinflammation following ischemic stroke by modulating the temporal and spatial dynamics of microglia/macrophages. J. Neuroinflamm. 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Rühlmann, C.; Wölk, T.; Blümel, T.; Stahn, L.; Vollmar, B.; Kuhla, A. Long-Term caloric restriction in ApoE-deficient mice results in neuroprotection via Fgf21-induced AMPK/mTOR pathway. Aging 2016, 8, 2777. [Google Scholar] [CrossRef] [Green Version]
- Amiri, M.; Braidy, N.; Aminzadeh, M. Protective Effects of Fibroblast Growth Factor 21 against Amyloid-β 1–42-Induced Toxicity in SH-SY5Y Cells. Neurotox. Res. 2018, 34, 574–583. [Google Scholar] [CrossRef]
- Chen, S.; Chen, S.-T.; Sun, Y.; Xu, Z.; Wang, Y.; Yao, S.-Y.; Yao, W.-B.; Gao, X.-D. Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer’s disease. Redox Biol. 2019, 22, 101133. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Chen, S.-T.; Chen, Y.-J.; Shen, J.; Yao, W.-B.; Gao, X.-D.; Chen, S. Modulation of the Astrocyte-Neuron Lactate Shuttle System contributes to Neuroprotective action of Fibroblast Growth Factor 21. Theranostics 2020, 10, 8430. [Google Scholar] [CrossRef]
- Mäkelä, J.; Tselykh, T.V.; Maiorana, F.; Eriksson, O.; Do, H.T.; Mudò, G.; Korhonen, L.T.; Belluardo, N.; Lindholm, D. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. SpringerPlus 2014, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Shen, J.; Qi, G.; Zha, Q.; Zhang, C.; Yao, W.; Gao, X.; Chen, S. Potential therapeutic role of fibroblast growth factor 21 in neurodegeneration: Evidence for ameliorating parkinsonism via silent information regulator 2 homolog 1 and implication for gene therapy. Neuropharmacology 2020, 181, 108335. [Google Scholar] [CrossRef]
- Fang, X.; Ma, J.; Mu, D.; Li, B.; Lian, B.; Sun, C. FGF21 Protects Dopaminergic Neurons in Parkinson’s Disease Models Via Repression of Neuroinflammation. Neurotox. Res. 2020, 37, 616–627. [Google Scholar] [CrossRef]
- Kakoty, V.; Sarathlal, K.; Tang, R.-D.; Yang, C.H.; Dubey, S.K.; Taliyan, R. Fibroblast growth factor 21 and autophagy: A complex interplay in Parkinson disease. Biomed. Pharmacother. 2020, 127, 110145. [Google Scholar] [CrossRef]
- Delaye, J.B.; Lanznaster, D.; Veyrat-Durebex, C.; Fontaine, A.; Bacle, G.; Lefevre, A.; Hergesheimer, R.; Lecron, J.C.; Vourc’h, P.; Andres, C.R.; et al. Behavioral, Hormonal, Inflammatory, and Metabolic Effects Associated with FGF21-Pathway Activation in an ALS Mouse Model. Neurotherapeutics 2020, 1–12. [Google Scholar] [CrossRef]
- Gubbi, S.; Quipildor, G.F.; Barzilai, N.; Huffman, D.M.; Milman, S. 40 YEARS of IGF1: IGF1: The Jekyll and Hyde of the aging brain. J. Mol. Endocrinol. 2018, 61, T171–T185. [Google Scholar] [CrossRef]
- Perdue, J.F. Chemistry, structure, and function of insulin-like growth factors and their receptors: A review. Can. J. Biochem. Cell. Biol. 1984, 62, 1237–1245. [Google Scholar] [CrossRef]
- Turner, J.D.; Rotwein, P.; Novakofski, J.; Bechtel, P.J. Induction of mRNA for IGF-I and-II during growth hormone-stimulated muscle hypertrophy. Am. J. Physiol. 1988, 255, E513–E517. [Google Scholar] [CrossRef]
- Voss, M.W.; Erickson, K.I.; Prakash, R.S.; Chaddock, L.; Kim, J.S.; Alves, H.; Szabo, A.; Phillips, S.M.; Wójcicki, T.R.; Mailey, E.L.; et al. Neurobiological markers of exercise-related brain plasticity in older adults. Brain Behav. Immun. 2013, 28, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-L.; Wang, C.-H.; Pan, C.-Y.; Chen, F.-C. The effects of long-term resistance exercise on the relationship between neurocognitive performance and GH, IGF-1, and homocysteine levels in the elderly. Front. Behav. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-T.; Chung, Y.-C.; Chen, Y.-J.; Ho, S.-Y.; Wu, H.-J. Effects of different types of exercise on body composition, muscle strength, and IGF-1 in the elderly with sarcopenic obesity. J. Am. Geriatr. Soc. 2017, 65, 827–832. [Google Scholar] [CrossRef]
- Trejo, J.L.; Llorens-Martin, M.V.; Torres-Alemán, I. The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol. Cell. Neurosci. 2008, 37, 402–411. [Google Scholar] [CrossRef]
- Carro, E.; Nuñez, A.; Busiguina, S.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J. Neurosci. 2000, 20, 2926–2933. [Google Scholar] [CrossRef]
- O’Kusky, J.; Ye, P. Neurodevelopmental effects of insulin-like growth factor signaling. Front. Neuroendocrinol. 2012, 33, 230–251. [Google Scholar] [CrossRef] [Green Version]
- Al-Delaimy, W.K.; Von Muhlen, D.; Barrett-Connor, E. Insulinlike Growth Factor-1, Insulinlike Growth Factor Binding Protein-1, and Cognitive Function in Older Men and Women. J. Am. Geriatr. Soc. 2009, 57, 1441–1446. [Google Scholar] [CrossRef]
- Doi, T.; Shimada, H.; Makizako, H.; Tsutsumimoto, K.; Hotta, R.; Nakakubo, S.; Suzuki, T. Association of insulin-like growth factor-1 with mild cognitive impairment and slow gait speed. Neurobiol. Aging 2015, 36, 942–947. [Google Scholar] [CrossRef]
- Tumati, S.; Burger, H.; Martens, S.; van der Schouw, Y.T.; Aleman, A. Association between cognition and serum insulin-like growth factor-1 in middle-aged & older men: An 8 year follow-up study. PLoS ONE 2016, 11, e0154450. [Google Scholar] [CrossRef]
- Wennberg, A.M.V.; Hagen, C.E.; Machulda, M.M.; Hollman, J.H.; Roberts, R.O.; Knopman, D.S.; Petersen, R.C.; Mielke, M.M. The association between peripheral total IGF-1, IGFBP-3, and IGF-1/IGFBP-3 and functional and cognitive outcomes in the Mayo Clinic Study of Aging. Neurobiol. Aging 2018, 66, 68–74. [Google Scholar] [CrossRef]
- Westwood, A.J.; Beiser, A.; DeCarli, C.; Harris, T.B.; Chen, T.C.; He, X.-M.; Roubenoff, R.; Pikula, A.; Au, R.; Braverman, L.E.; et al. Insulin-Like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 2014, 82, 1613–1619. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.-P. Meta-Analysis of serum insulin-like growth factor 1 in Alzheimer’s disease. PLoS ONE 2016, 11, e0155733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilla-Cortázar, I.; Aguirre, G.A.; Femat-Roldán, G.; Martín-Estal, I.; Espinosa, L. Is insulin-like growth factor-1 involved in Parkinson’s disease development? J. Transl. Med. 2020, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspar, B.K.; Lladó, J.; Sherkat, N.; Rothstein, J.D.; Gage, F.H. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 2003, 301, 839–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesada, A.; Micevych, P.E. Estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxdopamine lesions. J. Neurosci. Res. 2004, 75, 107–116. [Google Scholar] [CrossRef]
- Quesada, A.; Lee, B.Y.; Micevych, P.E. PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson’s disease. Dev. Neurobiol. 2008, 68, 632–644. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musarò, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Ebert, A.D.; Beres, A.J.; Barber, A.E.; Svendsen, C.N. Human neural progenitor cells over-expressing IGF-1 protect dopamine neurons and restore function in a rat model of Parkinson’s disease. Exp. Neurol. 2008, 209, 213–223. [Google Scholar] [CrossRef]
- Duarte, A.I.; Petit, G.H.; Ranganathan, S.; Li, J.-Y.; Oliveira, C.R.; Brundin, P.; Björkqvist, M.; Rego, A.C. IGF-1 protects against diabetic features in an in vivo model of Huntington’s disease. Exp. Neurol. 2011, 231, 314–319. [Google Scholar] [CrossRef]
- Lopes, C.; Ribeiro, M.; Duarte, A.I.; Humbert, S.; Saudou, F.; De Almeida, L.P.; Hayden, M.; Rego, A.C. IGF-1 intranasal administration rescues Huntington’s disease phenotypes in YAC128 mice. Mol. Neurobiol. 2014, 49, 1126–1142. [Google Scholar] [CrossRef]
- Aguado-Llera, D.; Arilla-Ferreiro, E.; Campos-Barros, A.; Puebla-Jiménez, L.; Barrios, V. Protective effects of insulin-like growth factor-I on the somatostatinergic system in the temporal cortex of β-amyloid-treated rats. J. Neurochem. 2005, 92, 607–615. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Gerber, A.; Loetscher, H.; Torrado, J.; Metzger, F.; Torres-Aleman, I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol. Aging 2006, 27, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.; Paulsson, J.F.; Blinder, P.; Burstyn-Cohen, T.; Du, D.; Estepa, G.; Adame, A.; Pham, H.M.; Holzenberger, M.; Kelly, J.W.; et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 2009, 139, 1157–1169. [Google Scholar] [CrossRef] [Green Version]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Aβ accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [CrossRef]
- Gontier, G.; George, C.; Chaker, Z.; Holzenberger, M.; Aïd, S. Blocking IGF signaling in adult neurons alleviates Alzheimer’s disease pathology through amyloid-β clearance. J. Neurosci. 2015, 35, 11500–11513. [Google Scholar] [CrossRef] [PubMed]
- Frater, J.; Lie, D.; Bartlett, P.; McGrath, J.J. Insulin-Like Growth Factor 1 (IGF-1) as a marker of cognitive decline in normal ageing: A review. Ageing Res. Rev. 2018, 42, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Panati, K.; Suneetha, Y.; Narala, V.R. Irisin/FNDC5–An updated review. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 689–697. [Google Scholar] [PubMed]
- Aydin, S.; Kuloglu, T.; Aydin, S.; Kalayci, M.; Yilmaz, M.; Cakmak, T.; Albayrak, S.; Gungor, S.; Colakoglu, N.; Ozercan, İ.H. A comprehensive immunohistochemical examination of the distribution of the fat-burning protein irisin in biological tissues. Peptides 2014, 61, 130–136. [Google Scholar] [CrossRef]
- Jedrychowski, M.P.; Wrann, C.D.; Paulo, J.A.; Gerber, K.K.; Szpyt, J.; Robinson, M.M.; Nair, K.S.; Gygi, S.P.; Spiegelman, B.M. Detection and quantitation of circulating human irisin by tandem mass spectrometry. Cell Metab. 2015, 22, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Lee, J.O.; Kim, N.; Kim, J.K.; Kim, H.I.; Lee, Y.W.; Kim, S.J.; Choi, J.-I.; Oh, Y.; Kim, J.H.; et al. Irisin, a novel myokine, regulates glucose uptake in skeletal muscle cells via AMPK. Mol. Endocrinol. 2015, 29, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Xin, C.; Liu, J.; Zhang, J.; Zhu, D.; Wang, H.; Xiong, L.; Lee, Y.; Ye, J.; Lian, K.; Xu, C.; et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int. J. Obes. 2016, 40, 443–451. [Google Scholar] [CrossRef]
- Kim, H.; Wrann, C.D.; Jedrychowski, M.; Vidoni, S.; Kitase, Y.; Nagano, K.; Zhou, C.; Chou, J.; Parkman, V.-J.A.; Novick, S.J.; et al. Irisin mediates effects on bone and fat via αV integrin receptors. Cell 2018, 175, 1756–1768. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.-S.; Dincer, F.; Mantzoros, C.S. Pharmacological concentrations of irisin increase cell proliferation without influencing markers of neurite outgrowth and synaptogenesis in mouse H19-7 hippocampal cell lines. Metabolism 2013, 62, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Dipaola, L.; Spagnuolo, R.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F. Irisin increases the expression of anorexigenic and neurotrophic genes in mouse brain. Diabetes Metab. Res. Rev. 2020, 36, e3238. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Zarbakhsh, S.; Safari, M.; Aldaghi, M.R.; Sameni, H.R.; Ghahari, L.; Lagmouj, Y.K.; Jaberi, K.R.; Parsaie, H. Irisin protects the substantia nigra dopaminergic neurons in the rat model of Parkinson’s disease. Iran. J. Basic Med. Sci. 2019, 22, 722. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Ribeiro, F.C.; Sudo, F.K.; Drummond, C.; Assunção, N.; Vanderborght, B.; Tovar-Moll, F.; Mattos, P.; De Felice, F.G.; Ferreira, S.T. Cerebrospinal fluid irisin correlates with amyloid-β, BDNF, and cognition in Alzheimer’s disease. Alzheimers Dement. 2020, 12, e12034. [Google Scholar] [CrossRef]
- Lunetta, C.; Lizio, A.; Tremolizzo, L.; Ruscica, M.; Macchi, C.; Riva, N.; Weydt, P.; Corradi, E.; Magni, P.; Sansone, V. Serum irisin is upregulated in patients affected by amyotrophic lateral sclerosis and correlates with functional and metabolic status. J. Neurol. 2018, 265, 3001–3008. [Google Scholar] [CrossRef]
- Nicola, N.A.; Babon, J.J. Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev. 2015, 26, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Gearing, D.P.; Gough, N.M.; King, J.A.; Hilton, D.J.; Nicola, N.A.; Simpson, R.J.; Nice, E.C.; Kelso, A.; Metcalf, D. Molecular cloning and expression of cDNA encoding a murine myeloid leukaemia inhibitory factor (LIF). EMBO J. 1987, 6, 3995–4002. [Google Scholar] [CrossRef]
- Hilton, D.J. LIF: Lots of interesting functions. Trends Biochem. Sci. 1992, 17, 72–76. [Google Scholar] [CrossRef]
- Broholm, C.; Mortensen, O.H.; Nielsen, S.; Akerstrom, T.; Zankari, A.; Dahl, B.; Pedersen, B.K. Exercise induces expression of leukaemia inhibitory factor in human skeletal muscle. J. Physiol. 2008, 586, 2195–2201. [Google Scholar] [CrossRef]
- Jia, D.; Cai, M.; Xi, Y.; Du, S.; ZhenjunTian. Interval exercise training increases LIF expression and prevents myocardial infarction-induced skeletal muscle atrophy in rats. Life Sci. 2018, 193, 77–86. [Google Scholar] [CrossRef]
- Hill, E.J.; Vernallis, A.B. Polarized secretion of leukemia inhibitory factor. BMC Cell Biol. 2008, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broholm, C.; Pedersen, B.K. Leukaemia inhibitory factor-an exercise-induced myokine. Exerc. Immunol. Rev. 2010, 16, 77–85. [Google Scholar] [PubMed]
- Kapilevich, L.V.; Zakharova, A.N.; Kabachkova, A.V.; Kironenko, T.A.; Orlov, S.N. Dynamic and static exercises differentially affect plasma cytokine content in elite endurance-and strength-trained athletes and untrained volunteers. Front. Physiol. 2017, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broholm, C.; Laye, M.J.; Brandt, C.; Vadalasetty, R.; Pilegaard, H.; Pedersen, B.K.; Scheele, C. LIF is a contraction-induced myokine stimulating human myocyte proliferation. J. Appl. Physiol. 2011, 111, 251–259. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J.; Brennan, J.M. Saturable entry of leukemia inhibitory factor from blood to the central nervous system. J. Neuroimmunol. 2000, 106, 172–180. [Google Scholar] [CrossRef]
- Soilu-Hänninen, M.; Broberg, E.; Röyttä, M.; Mattila, P.; Rinne, J.; Hukkanen, V. Expression of LIF and LIF receptor beta in Alzheimer’s and Parkinson’s diseases. Acta Neurol. Scand. 2010, 121, 44–50. [Google Scholar] [CrossRef]
- Hu, J.; Imai, T.; Shimizu, N.; Nakagawa, H.; Ono, S. Expression of leukaemia inhibitory factor in skin of patients with amyotrophic lateral sclerosis. Lancet 1999, 353, 2126–2127. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.O.; Lee, Y.W.; Kim, S.A.; Seo, I.H.; Han, J.A.; Kang, M.J.; Kim, S.J.; Cho, Y.-H.; Park, J.-J.; et al. Lif, a novel myokine, protects against amyloid-β-induced neurotoxicity via Akt-mediated autophagy signaling in hippocampal cells. Int. J. Neuropsychopharmacol. 2019, 22, 402–414. [Google Scholar] [CrossRef]
- Feeney, S.J.; Austin, L.; Bennett, T.M.; Kurek, J.B.; Jean-Francois, M.J.B.; Muldoon, C.; Byrne, E. The effect of leukaemia inhibitory factor on SOD1 G93A murine amyotrophic lateral sclerosis. Cytokine 2003, 23, 108–118. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–130. [Google Scholar] [CrossRef]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Zhu, J.; Dou, S.; Jiang, Y.; Chen, J.; Wang, C.; Cheng, B. Apelin-13 protects dopaminergic neurons in MPTP-induced Parkinson’s disease model mice through inhibiting endoplasmic reticulum stress and promoting autophagy. Brain Res. 2019, 1715, 203–212. [Google Scholar] [CrossRef]
- Zhu, J.; Gao, W.; Shan, X.; Wang, C.; Wang, H.; Shao, Z.; Dou, S.; Jiang, Y.; Wang, C.; Cheng, B. Apelin-36 mediates neuroprotective effects by regulating oxidative stress, autophagy and apoptosis in MPTP-induced Parkinson’s disease model mice. Brain Res. 2020, 1726, 146493. [Google Scholar] [CrossRef]
- Wu, Y.; Luo, X.; Liu, X.; Liu, D.; Wang, X.; Guo, Z.; Zhu, L.; Tian, Q.; Yang, X.; Wang, J.-Z. Intraperitoneal administration of a novel TAT-BDNF peptide ameliorates cognitive impairments via modulating multiple pathways in two Alzheimer’s rodent models. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Peng, Q.; Liu, X.; Jin, J.; Hou, Z.; Zhang, J.; Mori, S.; Ross, C.A.; Ye, K.; Duan, W. Small-Molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 2462–2470. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; He, W.; Gayen, M.; Benoit, M.R.; Luo, X.; Hu, X.; Yan, R. Activated CX3CL1/Smad2 signals prevent neuronal loss and Alzheimer’s tau pathology-mediated cognitive dysfunction. J. Neurosci. 2020, 40, 1133–1144. [Google Scholar] [CrossRef] [Green Version]
- Donnini, S.; Cantara, S.; Morbidelli, L.; Giachetti, A.; Ziche, M. FGF-2 overexpression opposes the β amyloid toxic injuries to the vascular endothelium. Cell Death Differ. 2006, 13, 1088–1096. [Google Scholar] [CrossRef] [Green Version]
- Katsouri, L.; Ashraf, A.; Birch, A.M.; Lee, K.K.; Mirzaei, N.; Sastre, M. Systemic administration of fibroblast growth factor-2 (FGF2) reduces BACE1 expression and amyloid pathology in APP23 mice. Neurobiol. Aging 2015, 36, 821–831. [Google Scholar] [CrossRef]
- Hou, J.G.; Cohen, G.; Mytilineou, C. Basic fibroblast growth factor stimulation of glial cells protects dopamine neurons from 6-hydroxydopamine toxicity: Involvement of the glutathione system. J. Neurochem. 1997, 69, 76–83. [Google Scholar] [CrossRef]
- Hsuan, S.-L.; Klintworth, H.M.; Xia, Z. Basic fibroblast growth factor protects against rotenone-induced dopaminergic cell death through activation of extracellular signal-regulated kinases 1/2 and phosphatidylinositol-3 kinase pathways. J. Neurosci. 2006, 26, 4481–4491. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Chen, G.; Shi, L.; Feng, J.; Wang, Y.; Ye, C.; Feng, W.; Niu, J.; Huang, Z. PEGylated rhFGF-2 conveys long-term neuroprotection and improves neuronal function in a rat model of Parkinson’s disease. Mol. Neurobiol. 2015, 51, 32–42. [Google Scholar] [CrossRef]
- Cai, P.; Ye, J.; Zhu, J.; Liu, D.; Chen, D.; Wei, X.; Johnson, N.R.; Wang, Z.; Zhang, H.; Cao, G.; et al. Inhibition of endoplasmic reticulum stress is involved in the neuroprotective effect of bFGF in the 6-OHDA-induced Parkinson’s disease model. Aging Dis. 2016, 7, 336. [Google Scholar] [CrossRef]
- Wang, Z.; Xiong, L.; Wang, G.; Wan, W.; Zhong, C.; Zu, H. Insulin-Like growth factor-1 protects SH-SY5Y cells against β-amyloid-induced apoptosis via the PI3K/Akt-Nrf2 pathway. Exp. Gerontol. 2017, 87, 23–32. [Google Scholar] [CrossRef]
- Zhang, J.; Shu, Y.; Qu, Y.; Zhang, L.; Chu, T.; Zheng, Y.; Zhao, H. C-myb Plays an Essential Role in the Protective Function of IGF-1 on Cytotoxicity Induced by Aβ 25–35 via the PI3K/Akt Pathway. J. Mol. Neurosci. 2017, 63, 412–418. [Google Scholar] [CrossRef]
- Song, F.; Liu, T.; Meng, S.; Li, F.; Zhang, Y.; Jiang, L. Insulin-Like Growth Factor-1 Alleviates Expression of Aβ 1–40 and α-, β-, and γ-Secretases in the Cortex and Hippocampus of APP/PS1 Double Transgenic Mice. J. Mol. Neurosci. 2018, 66, 595–603. [Google Scholar] [CrossRef]
- Kim, Y.; Li, E.; Park, S. Insulin-Like growth factor-1 inhibits 6-hydroxydopamine-mediated endoplasmic reticulum stress-induced apoptosis via regulation of heme oxygenase-1 and Nrf2 expression in PC12 cells. Int. J. Neurosci. 2012, 122, 641–649. [Google Scholar] [CrossRef] [PubMed]
- El Ayadi, A.; Zigmond, M.J.; Smith, A.D. IGF-1 protects dopamine neurons against oxidative stress: Association with changes in phosphokinases. Exp. Brain Res. 2016, 234, 1863–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, S.; Bryson, E.A.; Cordelières, F.P.; Connors, N.C.; Datta, S.R.; Finkbeiner, S.; Greenberg, M.E.; Saudou, F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2002, 2, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Dodge, J.C.; Haidet, A.M.; Yang, W.; Passini, M.A.; Hester, M.; Clarke, J.; Roskelley, E.M.; Treleaven, C.M.; Rizo, L.; Martin, H.; et al. Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol. Ther. 2008, 16, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Duan, W.; Liu, Y.; Liu, Y.; Liu, C.; Li, Y.; Wen, D.; Li, Z.; Li, C. IGF1 affects macrophage invasion and activation and TNF-α production in the sciatic nerves of female SOD1G93A mice. Neurosci. Lett. 2018, 668, 1–6. [Google Scholar] [CrossRef]
- Wang, W.; Wen, D.; Duan, W.; Yin, J.; Cui, C.; Wang, Y.; Li, Z.; Liu, Y.; Li, C. Systemic administration of scAAV9-IGF1 extends survival in SOD1G93A ALS mice via inhibiting p38 MAPK and the JNK-mediated apoptosis pathway. Brain Res. Bull. 2018, 139, 203–210. [Google Scholar] [CrossRef]
- Wang, K.; Li, H.; Wang, H.; Wang, J.-H.; Song, F.; Sun, Y. Irisin exerts neuroprotective effects on cultured neurons by regulating astrocytes. Mediat. Inflamm. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Heck, S.; Lezoualc’h, F.; Engert, S.; Behl, C. Insulin-Like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor κB. J. Biol. Chem. 1999, 274, 9828–9835. [Google Scholar] [CrossRef] [Green Version]
- Offen, D.; Shtaif, B.; Hadad, D.; Weizman, A.; Melamed, E.; Gil-Ad, I. Protective effect of insulin-like-growth-factor-1 against dopamine-induced neurotoxicity in human and rodent neuronal cultures: Possible implications for Parkinson’s disease. Neurosci. Lett. 2001, 316, 129–132. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Melo, C.V.; Gomes, J.R.; Mendes, C.S.; Grãos, M.M.; Carvalho, R.F.; Carvalho, A.P.; Duarte, C.B. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005, 12, 1329–1343. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [Green Version]
- van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef]
- Hsiao, Y.-H.; Hung, H.-C.; Chen, S.-H.; Gean, P.-W. Social interaction rescues memory deficit in an animal model of Alzheimer’s disease by increasing BDNF-dependent hippocampal neurogenesis. J. Neurosci. 2014, 34, 16207–16219. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Lien, C.-C.; Hou, W.-H.; Chiang, P.-M.; Tsai, K.-J. Gain of BDNF function in engrafted neural stem cells promotes the therapeutic potential for Alzheimer’s disease. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef]
- Cho, K.S.; Lee, J.H.; Cho, J.; Cha, G.-H.; Song, G.J. Autophagy modulators and neuroinflammation. Curr. Med. Chem. 2020, 27, 955–982. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Lexell, J.; Deierborg, T. Effects of physical exercise on neuroinflammation, neuroplasticity, neurodegeneration, and behavior: What we can learn from animal models in clinical settings. Neurorehabil. Neural Repair 2015, 29, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Little, J.P.; Klegeris, A. Physical activity and exercise attenuate neuroinflammation in neurological diseases. Brain Res. Bull. 2016, 125, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Sumpter, R., Jr.; Levine, B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuede, C.M.; Zimmerman, S.D.; Dong, H.; Kling, M.J.; Bero, A.W.; Holtzman, D.M.; Timson, B.F.; Csernansky, J.G. Effects of voluntary and forced exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 35, 426–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Barkow, J.C.; Freed, C.R. Running wheel exercise reduces α-synuclein aggregation and improves motor and cognitive function in a transgenic mouse model of Parkinson’s disease. PLoS ONE 2017, 12, e0190160. [Google Scholar] [CrossRef]
- Liang, K.Y.; Mintun, M.A.; Fagan, A.M.; Goate, A.M.; Bugg, J.M.; Holtzman, D.M.; Morris, J.C.; Head, D. Exercise and Alzheimer’s disease biomarkers in cognitively normal older adults. Ann. Neurol. 2010, 68, 311–318. [Google Scholar] [CrossRef]
- Jiang, Y.; Woosley, A.N.; Sivalingam, N.; Natarajan, S.; Howe, P.H. Cathepsin-B-mediated cleavage of Disabled-2 regulates TGF-β-induced autophagy. Nat. Cell Biol. 2016, 18, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325. [Google Scholar] [CrossRef]
- Gómez-Sámano, M.Á.; Grajales-Gómez, M.; Zuarth-Vázquez, J.M.; Navarro-Flores, M.F.; Martínez-Saavedra, M.; Juárez-León, Ó.A.; Morales-García, M.G.; Enríquez-Estrada, V.M.; Gómez-Pérez, F.J.; Cuevas-Ramos, D. Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. 2017, 11, 335–341. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Campion, D.; Pottier, C.; Nicolas, G.; Le Guennec, K.; Rovelet-Lecrux, A. Alzheimer disease: Modeling an Aβ-centered biological network. Mol. Psychiatry 2016, 21, 861–871. [Google Scholar] [CrossRef]
- Rohe, M.; Synowitz, M.; Glass, R.; Paul, S.M.; Nykjaer, A.; Willnow, T.E. Brain-Derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. J. Neurosci. 2009, 29, 15472–15478. [Google Scholar] [CrossRef]
- Jacobsen, K.T.; Adlerz, L.; Multhaup, G.; Iverfeldt, K. Insulin-like growth factor-1 (IGF-1)-induced processing of amyloid-β precursor protein (APP) and APP-like protein 2 is mediated by different metalloproteinases. J. Biol. Chem. 2010, 285, 10223–10231. [Google Scholar] [CrossRef] [Green Version]
- Stöhr, O.; Schilbach, K.; Moll, L.; Hettich, M.M.; Freude, S.; Wunderlich, F.T.; Ernst, M.; Zemva, J.; Brüning, J.C.; Krone, W.; et al. Insulin receptor signaling mediates APP processing and β-amyloid accumulation without altering survival in a transgenic mouse model of Alzheimer’s disease. Age 2013, 35, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Atlas, R.; Lange, A.; Ginzburg, I. Brain-Derived neurotrophic factor induces a rapid dephosphorylation of tau protein through a PI-3Kinase signalling mechanism. Eur. J. Neurosci. 2005, 22, 1081–1089. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Middelbeek, R.J.W.; Goodyear, L.J. Exercise effects on white adipose tissue: Beiging and metabolic adaptations. Diabetes 2015, 64, 2361–2368. [Google Scholar] [CrossRef] [Green Version]
- Seldin, M.M.; Peterson, J.M.; Byerly, M.S.; Wei, Z.; Wong, G.W. Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J. Biol. Chem. 2012, 287, 11968–11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Bassel-Duby, R.; Olson, E.N. Heart-and muscle-derived signaling system dependent on MED13 and Wingless controls obesity in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, 9491–9496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K. The diseasome of physical inactivity-and the role of myokines in muscle-fat cross talk. J. Physiol. 2009, 587, 5559–5568. [Google Scholar] [CrossRef] [PubMed]
- de Luis, D.A.; Aller, R.; Izaola, O.; Primo, D.; Romero, E. rs10767664 gene variant in brain-derived neurotrophic factor is associated with diabetes mellitus type 2 in Caucasian females with obesity. Ann. Nutr. Metab. 2017, 70, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Suwa, M.; Kishimoto, H.; Nofuji, Y.; Nakano, H.; Sasaki, H.; Radak, Z.; Kumagai, S. Serum brain-derived neurotrophic factor level is increased and associated with obesity in newly diagnosed female patients with type 2 diabetes mellitus. Metabolism 2006, 55, 852–857. [Google Scholar] [CrossRef]
- Tsuchida, A.; Nonomura, T.; Nakagawa, T.; Itakura, Y.; Ono-Kishino, M.; Yamanaka, M.; Sugaru, E.; Taiji, M.; Noguchi, H. Brain-Derived neurotrophic factor ameliorates lipid metabolism in diabetic mice. Diabetes Obes. Metab. 2002, 4, 262–269. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, X.; Glazier, B.J.; Krolick, K.N.; Yang, S.; He, J.; Lo, C.C.; Shi, H. Differential sympathetic activation of adipose tissues by brain-derived neurotrophic factor. Biomolecules 2019, 9, 452. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.-C.; Ho, P.-C.; Tu, Y.-K.; Jou, I.; Tsai, K.-J. Lipids and Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 1505. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The role of lipids in Parkinson’s disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Suriano, F.; Van Hul, M.; Cani, P.D. Gut microbiota and regulation of myokine-adipokine function. Curr. Opin. Pharmacol. 2020, 52, 9–17. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, S.; Kim, H.; Garcia-Perez, I.; Reza, M.M.; Martin, K.A.; Kundu, P.; Cox, L.M.; Selkrig, J.; Posma, J.M.; Zhang, H.; et al. The gut microbiota influences skeletal muscle mass and function in mice. Sci. Transl. Med. 2019, 11, eaan5662. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; Russo-Neustadt, A.A. Running exercise-and antidepressant-induced increases in growth and survival-associated signaling molecules are IGF-dependent. Growth Factors 2007, 25, 118–131. [Google Scholar] [CrossRef]
- Dai, T.-T.; Wang, B.; Xiao, Z.-Y.; You, Y.; Tian, S.-W. Apelin-13 upregulates BDNF against chronic stress-induced depression-like phenotypes by ameliorating HPA axis and hippocampal glucocorticoid receptor dysfunctions. Neuroscience 2018, 390, 151–159. [Google Scholar] [CrossRef]
- Henningsen, J.; Rigbolt, K.T.G.; Blagoev, B.; Pedersen, B.K.; Kratchmarova, I. Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol. Cell. Proteom. 2010, 9, 2482–2496. [Google Scholar] [CrossRef] [Green Version]
- Nelke, C.; Dziewas, R.; Minnerup, J.; Meuth, S.G.; Ruck, T. Skeletal muscle as potential central link between sarcopenia and immune senescence. EBioMedicine 2019, 49, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wang, L.; You, W.; Shan, T. Myokines mediate the cross talk between skeletal muscle and other organs. J. Cell. Physiol. 2021, 236, 2393–2412. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-Driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.-M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [Green Version]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of transgenic APP mRNA is the key determinant for β-amyloid deposition in PS2APP transgenic mice. Neurodegener. Dis. 2009, 6, 29–36. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef] [Green Version]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-Level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Musarò, A.; McCullagh, K.; Paul, A.; Houghton, L.; Dobrowolny, G.; Molinaro, M.; Barton, E.R.; Sweeney, H.L.; Rosenthal, N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 2001, 27, 195–200. [Google Scholar] [CrossRef]
- Schilling, G.; Becher, M.W.; Sharp, A.H.; Jinnah, H.A.; Duan, K.; Kotzuk, J.A.; Slunt, H.H.; Ratovitski, T.; Cooper, J.K.; Jenkins, N.A.; et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999, 8, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.-X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Lasrado, R.; Snauwaert, J.; Vandeweert, E.; Van Haesendonck, C.; Borghgraef, P.; Van Leuven, F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J. Biol. Chem. 2005, 280, 3963–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mechanism of action of representative exercise-induced myokines in the brain with ND. While there is experimental evidence that some myokines penetrate the BBB (solid arrows from blood vessels through the BBB to the brain), others do not so far (dashed arrows from the blood vessels through the BBB to the brain).
Figure 1.
Mechanism of action of representative exercise-induced myokines in the brain with ND. While there is experimental evidence that some myokines penetrate the BBB (solid arrows from blood vessels through the BBB to the brain), others do not so far (dashed arrows from the blood vessels through the BBB to the brain).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, B.; Shin, M.; Park, Y.; Won, S.-Y.; Cho, K.S. Physical Exercise-Induced Myokines in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 5795. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115795
AMA Style
Lee B, Shin M, Park Y, Won S-Y, Cho KS. Physical Exercise-Induced Myokines in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(11):5795. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115795
Chicago/Turabian StyleLee, Banseok, Myeongcheol Shin, Youngjae Park, So-Yoon Won, and Kyoung Sang Cho. 2021. "Physical Exercise-Induced Myokines in Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 11: 5795. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115795
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.