
The Catecholaldehyde Hypothesis for the Pathogenesis of Catecholaminergic Neurodegeneration: What We Know and What We Do Not Know

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
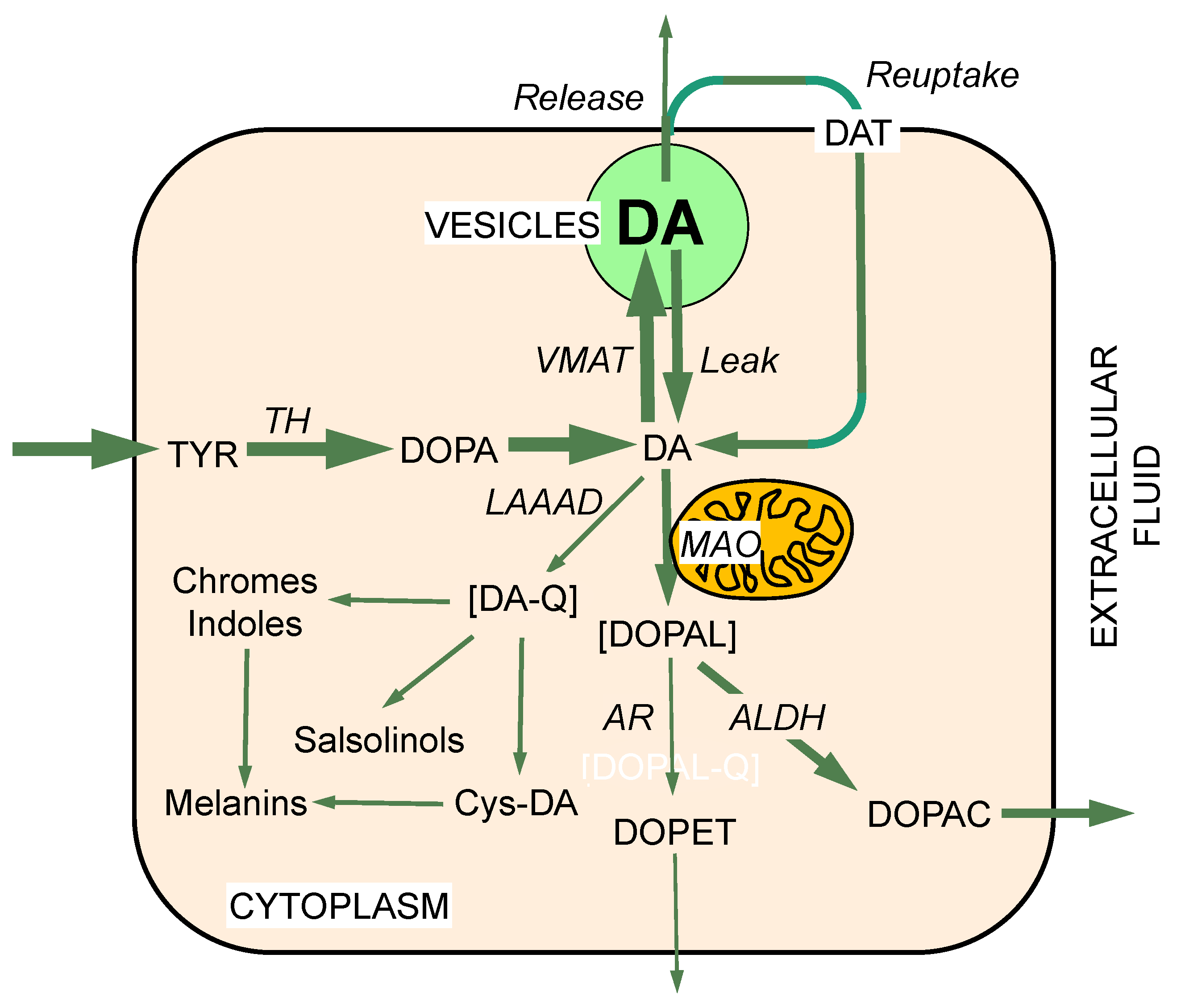
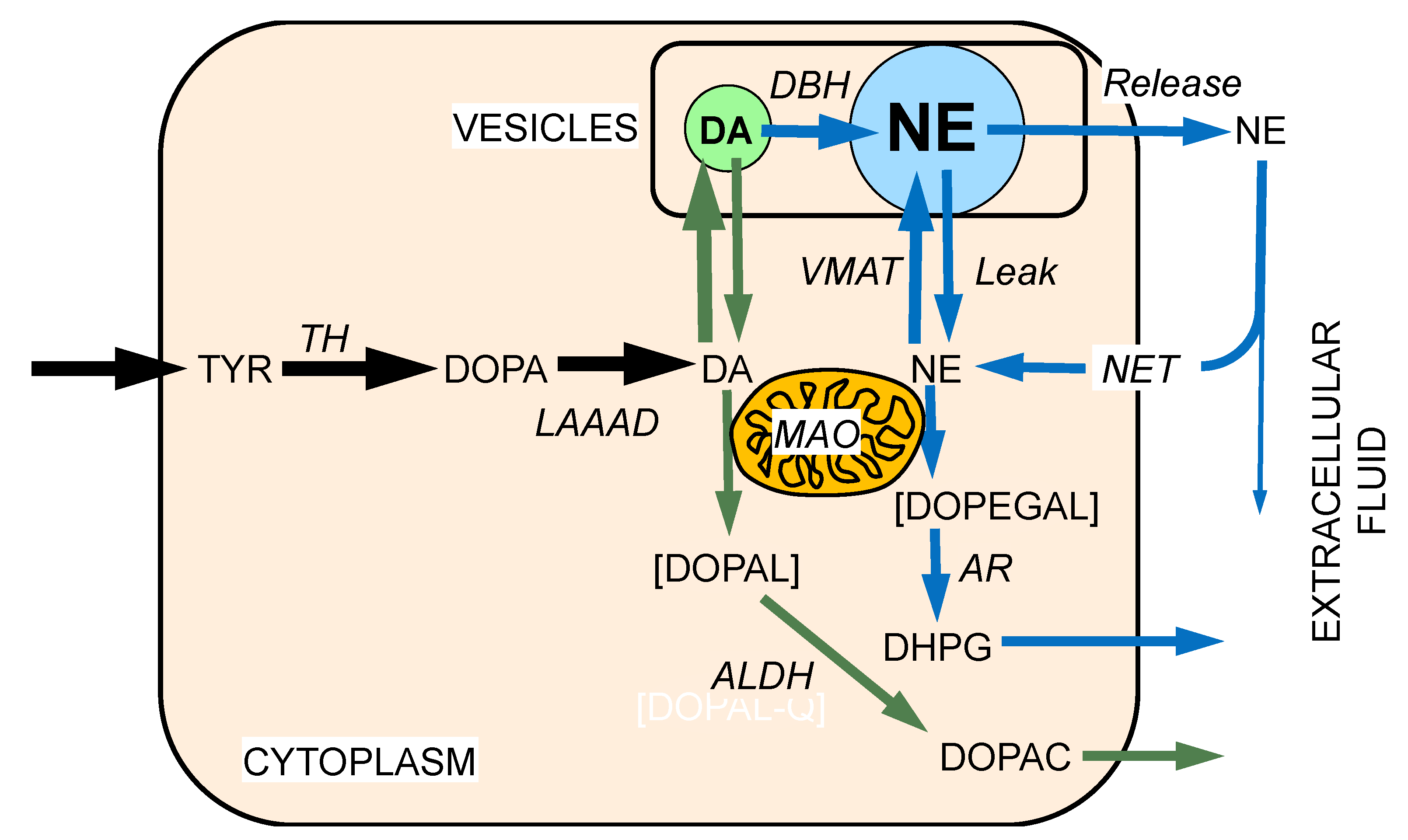
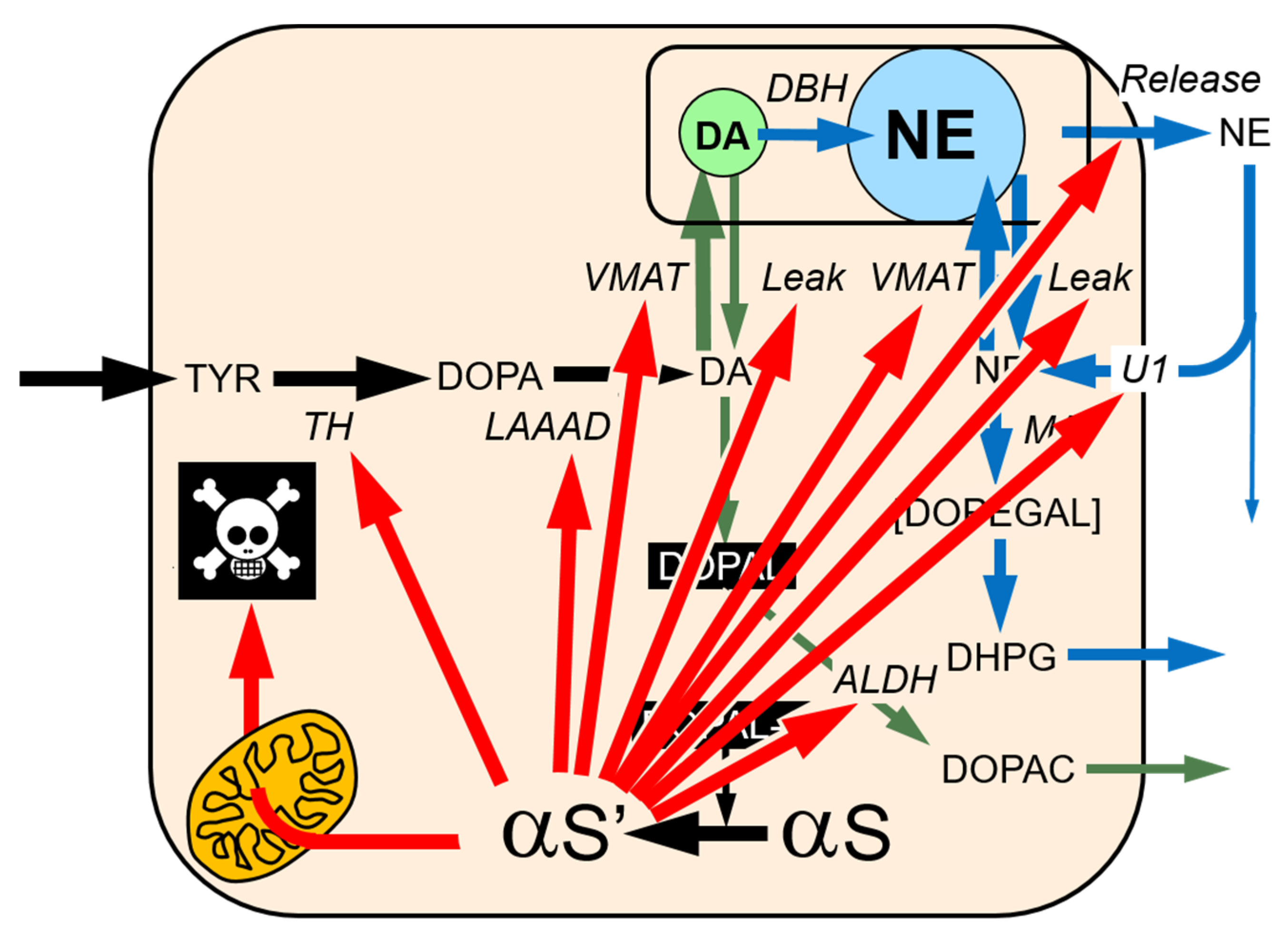
2. MAO and Intraneuronal Metabolism of Catecholamines
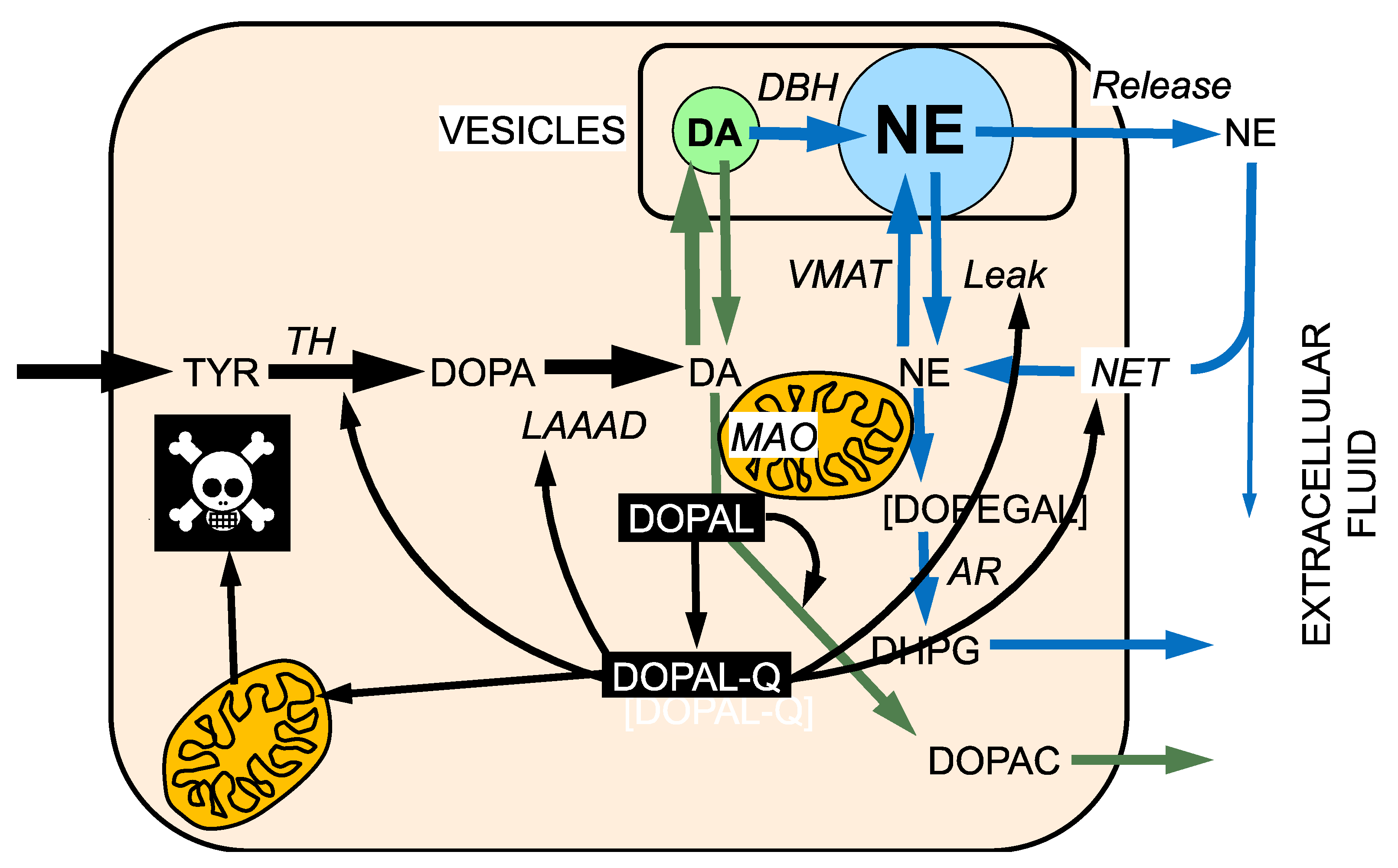
3. DOPAL Toxicity
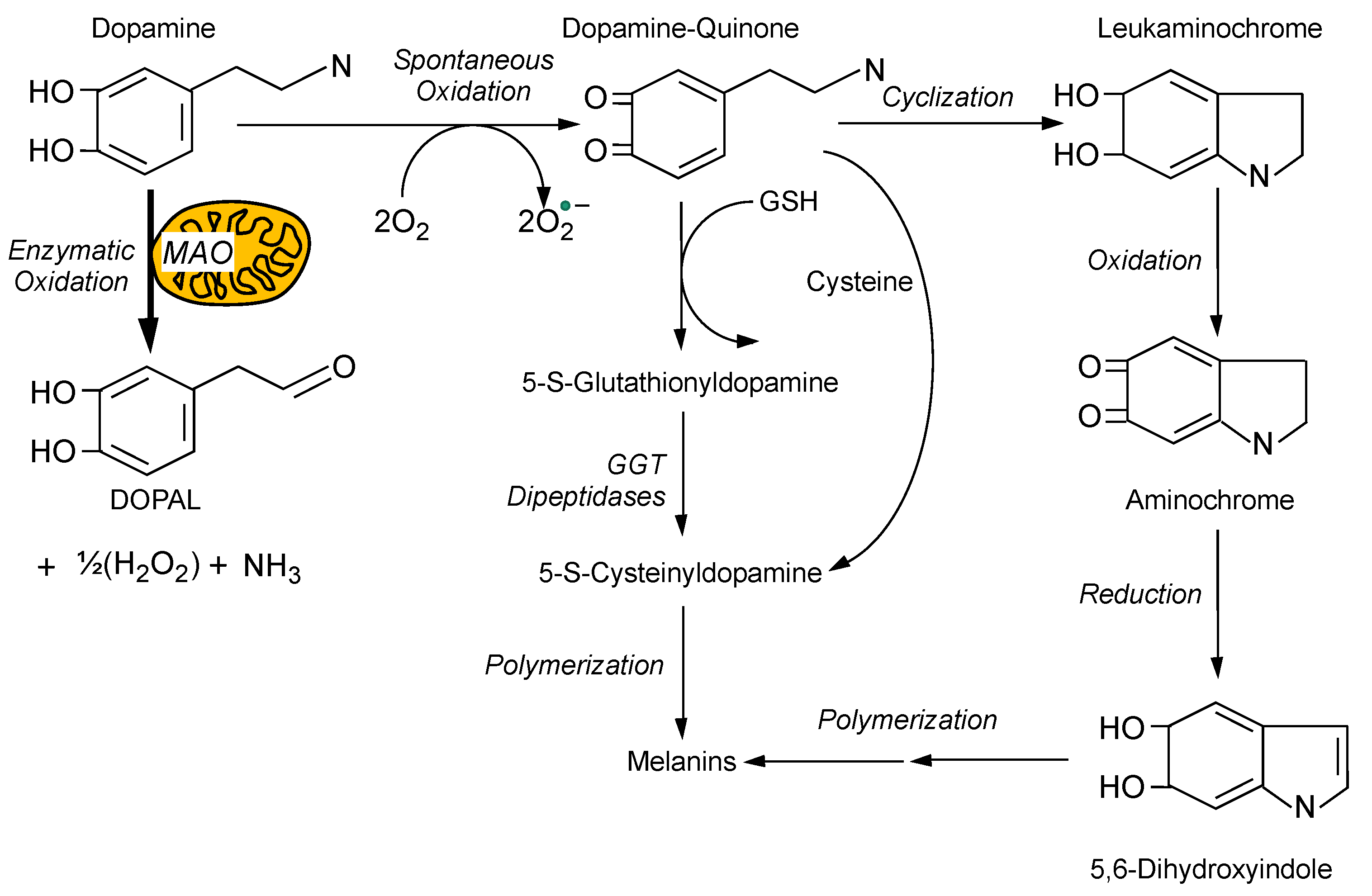
4. Toxicity from Spontaneous Oxidation of Cytoplasmic Dopamine
5. DOPAL Detoxification by Aldehyde Dehydrogenase (ALDH)
6. Vesicular Uptake as a Detoxification Mechanism
7. The Double Hit Concept
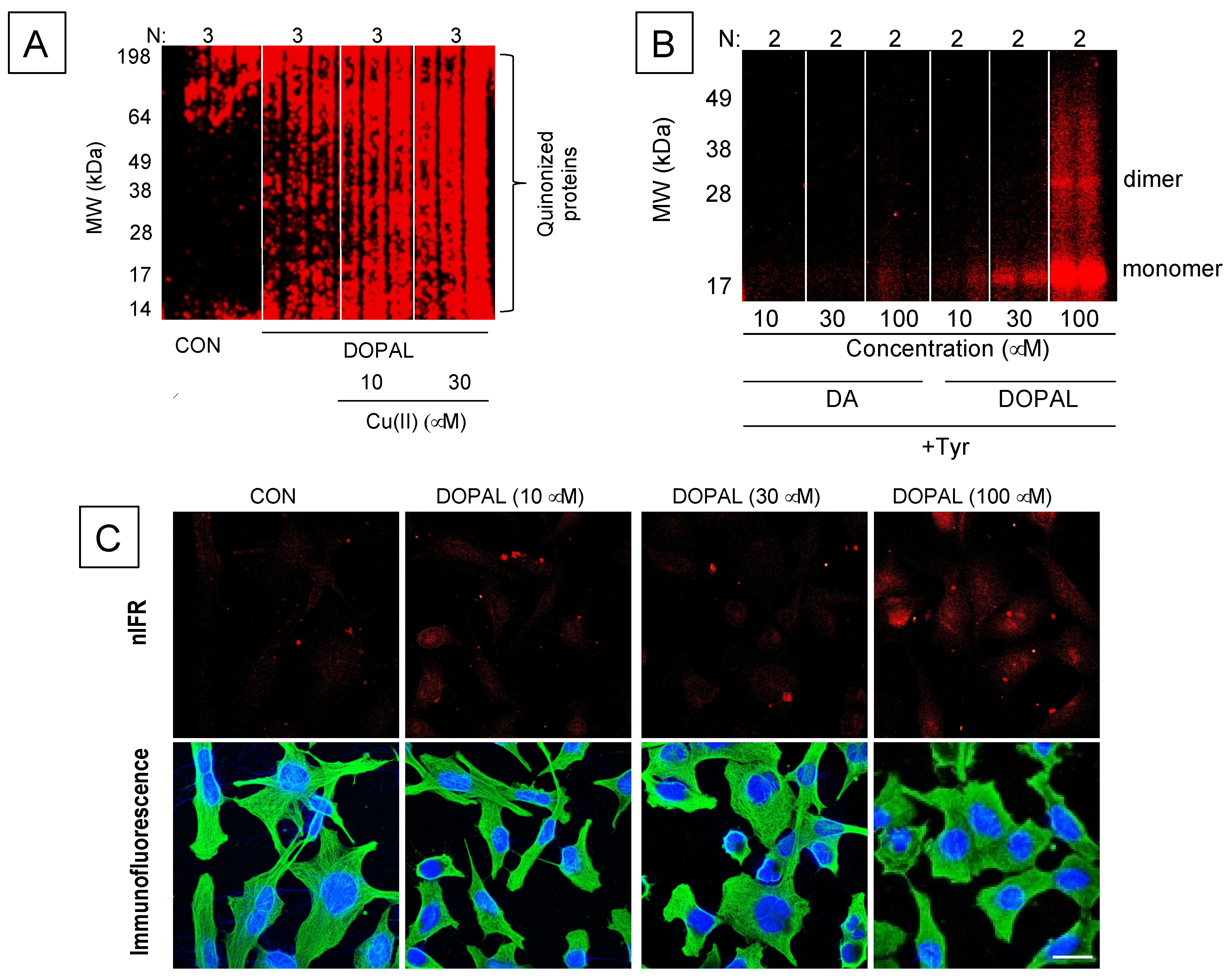
8. DOPAL-Protein Interactions, with Emphasis on Alpha-Synuclein
9. Stress and the Catecholaldehyde Hypothesis
10. MAO Inhibitor Trials and the Catecholaldehyde Hypothesis
11. Gaps in Knowledge and Goals for the Future
- (1)
- Regarding the metabolic fate of cytoplasmic dopamine, what proportions of cyto-plasmic dopamine undergo spontaneous vs. enzymatic oxidation? Why do MAO-B-selective drugs decrease levels of catecholamine metabolites thought to be produced within neurons that express MAO-A?
- (2)
- Regarding DOPAL-induced quinonization of intracellular proteins, what are the functional consequences for the proteins involved with reactions in catechola-minergic neurons? Does DOPAL quinonize intracellular proteins including αS in vivo in sympathetic noradrenergic neurons, such as in skin biopsies? Do Lewy bodies contain DOPAL-quinonized proteins?
- (3)
- Regarding DOPAL–αS interactions and disease pathogenesis, what are the relative contributions of DOPAL and alpha-synucleinopathy to catecholaminergic neuro-degeneration? In the absence of synucleinopathy is DOPAL accumulation patho-genic or a non-pathogenic biomarker? Do DOPAL and αS ascend together in the periphery in “body-first” PD or PAF?
- (4)
- At this point, there is insufficient literature about whether catecholaldehydes affect neurogenesis or neuroinflammation.
- (5)
- Diagnostic and therapeutic applications of the catecholaldehyde hypothesis would be especially valuable in preclinical disease. Cerebrospinal fluid indices of central catecholamine deficiency provide predictive biomarkers in individuals at risk of developing PD [171] and might be especially valuable in identifying appropriate candidates for clinical treatment or prevention trials.
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALDH | aldehyde dehydrogenase |
| AR | aldehyde/aldose reductase |
| αS | alpha-synuclein |
| BDNF | brain-derived neurotrophic factor |
| COMT | catechol-O-methyltransferase |
| Cys-DA | 5-S-cysteinyldopamine |
| DA | dopamine |
| DAT | cell membrane dopamine transporter |
| DLB | dementia with Lewy bodies |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| DOPAL | 3,4-dihydroxyphenylacetaldehyde |
| DOPAL-Q | DOPAL quinone |
| DOPEGAL | 3,4-dihydroxyphenylglycolaldehyde |
| EPI | epinephrine |
| GGT | gammaglutamyl transferase |
| LAAAD | L-aromatic-amino-acid decarboxylase |
| MAO | monoamine oxidase |
| MSA | multiple system atrophy |
| NAC | N-acetylcysteine |
| NE | norepinephrine |
| NET | cell membrane norepinephrine transporter |
| nIRF | near-infrared fluoroscopy |
| PD | Parkinson’s disease |
| TH | tyrosine hydroxylase |
| THP | tetrahydropapaveroline |
| TrkB | tyrosine kinase B |
| VMAT | vesicular monoamine transporter |
References
- Goldstein, D.S. The catecholaldehyde hypothesis: Where MAO fits in. J. Neural Transm. 2020, 127, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Hare, M.L. Tyramine oxidase: A new enzyme system in liver. Biochem. J. 1928, 22, 968–979. [Google Scholar] [CrossRef]
- Kohn, H.I. Tyramine oxidase. Biochem. J. 1937, 31, 1693–1704. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, J. Methylation reactions in the formation and metabolism of catecholamines and other biogenic amines. Pharmacol. Rev. 1966, 18, 95–113. [Google Scholar]
- Kopin, I.J. Storage and Metabolism of Catecholamines: The Role of Monoamine Oxidase. Pharmacol. Rev. 1964, 16, 179–191. [Google Scholar] [PubMed]
- Kopin, I.J. Monoamine oxidase and catecholamine metabolism. J. Neural Transm. Suppl. 1994, 41, 57–67. [Google Scholar] [CrossRef]
- Cotzias, G.C.; Dole, V.P. Metabolism of amines. II. Mitochondrial localization of monoamine oxidase. Proc. Soc. Exp. Biol. Med. 1951, 78, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020, 23, 15–20. [Google Scholar] [CrossRef]
- Collins, G.G.; Sandler, M.; Williams, E.D.; Youdim, M.B. Multiple forms of human brain mitochondrial monoamine oxidase. Nature 1970, 225, 817–820. [Google Scholar] [CrossRef]
- Youdim, M.B.; Collins, G.G.; Sandler, M. Multiple forms of rat brain monoamine oxidase. Nature 1969, 223, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.P. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem. Pharmacol. 1968, 17, 1285–1297. [Google Scholar] [CrossRef]
- Knoll, J.; Magyar, K. Some puzzling pharmacological effects of monoamine oxidase inhibitors. Adv. Biochem. Psychopharmacol. 1972, 5, 393–408. [Google Scholar] [PubMed]
- Pintar, J.E.; Barbosa, J.; Francke, U.; Castiglione, C.M.; Hawkins, M., Jr.; Breakefield, X.O. Gene for monoamine oxidase type A assigned to the human X chromosome. J. Neurosci. 1981, 1, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Kochersperger, L.M.; Parker, E.L.; Siciliano, M.; Darlington, G.J.; Denney, R.M. Assignment of genes for human monoamine oxidases A and B to the X chromosome. J. Neurosci Res. 1986, 16, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [Green Version]
- Finberg, J.P.M. The discovery and development of rasagiline as a new anti-Parkinson medication. J. Neural Transm. 2020, 127, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Demarest, K.T.; Moore, K.E. Type A monoamine oxidase catalyzes the intraneuronal deamination of dopamine within nigrostriatal, mesolimbic, tuberoinfundibular and tuberohypophyseal neurons in the rat. J. Neural Transm. 1981, 52, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, S.R.; Abercrombie, E.D. L-3,4-dihydroxyphenylalanine-induced dopamine release in the striatum of intact and 6-hydroxydopamine-treated rats: Differential effects of monoamine oxidase A and B inhibitors. J. Neurochem. 1994, 63, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Dyck, L.E.; Durden, D.A.; Boulton, A.A. Effects of monoamine oxidase inhibitors on the acid metabolites of some trace amines and of dopamine in the rat striatum. Biochem. Pharmacol. 1993, 45, 1317–1322. [Google Scholar] [CrossRef]
- Kumagae, Y.; Matsui, Y.; Iwata, N. Deamination of norepinephrine, dopamine, and serotonin by type A monoamine oxidase in discrete regions of the rat brain and inhibition by RS-8359. Jpn J. Pharmacol. 1991, 55, 121–128. [Google Scholar] [CrossRef]
- Colzi, A.; d’Agostini, F.; Kettler, R.; Borroni, E.; Da Prada, M. Effect of selective and reversible MAO inhibitors on dopamine outflow in rat striatum: A microdialysis study. J. Neural Transm. Suppl. 1990, 32, 79–84. [Google Scholar] [CrossRef]
- Jahng, J.W.; Houpt, T.A.; Wessel, T.C.; Chen, K.; Shih, J.C.; Joh, T.H. Localization of monoamine oxidase A and B mRNA in the rat brain by in situ hybridization. Synapse 1997, 25, 30–36. [Google Scholar] [CrossRef]
- Gershon, M.D.; Sherman, D.L.; Pintar, J.E. Type-specific localization of monoamine oxidase in the enteric nervous system: Relationship to 5-hydroxytryptamine, neuropeptides, and sympathetic nerves. J. Comp. Neurol. 1990, 301, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Goldstein, D.S.; Stull, R.; Keiser, H.R.; Sunderland, T.; Murphy, D.L.; Kopin, I.J. Simultaneous liquid-chromatographic determination of 3,4-dihydroxyphenylglycol, catecholamines, and 3,4-dihydroxyphenylalanine in plasma, and their responses to inhibition of monoamine oxidase. Clin. Chem. 1986, 32, 2030–2033. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Logan, J.; Volkow, N.D.; Shumay, E.; McCall-Perez, F.; Jayne, M.; Wang, G.J.; Alexoff, D.L.; Apelskog-Torres, K.; Hubbard, B.; et al. Evidence that formulations of the selective MAO-B inhibitor, selegiline, which bypass first-pass metabolism, also inhibit MAO-A in the human brain. Neuropsychopharmacology 2015, 40, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Riederer, P.; Maruyama, W. Modulation of monoamine oxidase (MAO) expression in neuropsychiatric disorders: Genetic and environmental factors involved in type A MAO expression. J. Neural Transm. 2016, 123, 91–106. [Google Scholar] [CrossRef]
- Bartl, J.; Muller, T.; Grunblatt, E.; Gerlach, M.; Riederer, P. Chronic monoamine oxidase-B inhibitor treatment blocks monoamine oxidase-A enzyme activity. J. Neural Transm. 2014, 121, 379–383. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Holmes, C.; Kopin, I.J.; Sharabi, Y. Comparison of monoamine oxidase inhibitors in decreasing production of the autotoxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 cells. J. Pharmacol. Exp. Ther. 2016, 356, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Blaschko, H. Amine oxidase and amine metabolism. Pharmacol. Rev. 1952, 4, 415–458. [Google Scholar]
- Mattammal, M.B.; Haring, J.H.; Chung, H.D.; Raghu, G.; Strong, R. An endogenous dopaminergic neurotoxin: Implication for Parkinson’s disease. Neurodegeneration 1995, 4, 271–281. [Google Scholar] [CrossRef]
- Mattammal, M.B.; Chung, H.D.; Strong, R.; Hsu, F.F. Confirmation of a dopamine metabolite in parkinsonian brain tissue by gas chromatography-mass spectrometry. J. Chromatogr. 1993, 614, 205–212. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Sharabi, Y.; Sullivan, P.; Isonaka, R.; Goldstein, D.S. 3,4-Dihydroxyphenylacetaldehyde-induced protein modifications and their mitigation by N-acetylcysteine. J. Pharmacol. Exp. Ther. 2018, 366, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Kopin, I.J.; Sharabi, Y. Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacol. Ther. 2014, 144, 268–282. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Kristal, B.S.; Conway, A.D.; Brown, A.M.; Jain, J.C.; Ulluci, P.A.; Li, S.W.; Burke, W.J. Selective dopaminergic vulnerability: 3,4-dihydroxyphenylacetaldehyde targets mitochondria. Free. Radic. Biol. Med. 2001, 30, 924–931. [Google Scholar] [CrossRef]
- Li, S.W.; Lin, T.S.; Minteer, S.; Burke, W.J. 3,4-Dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: Possible role in Parkinson’s disease pathogenesis. Brain Res. Mol. Brain Res. 2001, 93, 1–7. [Google Scholar] [CrossRef]
- Burke, W.J.; Li, S.W.; Williams, E.A.; Nonneman, R.; Zahm, D.S. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: Implications for Parkinson’s disease pathogenesis. Brain Res. 2003, 989, 205–213. [Google Scholar] [CrossRef]
- Panneton, W.M.; Kumar, V.B.; Gan, Q.; Burke, W.J.; Galvin, J.E. The neurotoxicity of DOPAL: Behavioral and stereological evidence for its role in Parkinson disease pathogenesis. PLoS ONE 2010, 5, e15251. [Google Scholar] [CrossRef]
- Fornstedt, B.; Carlsson, A. Effects of inhibition of monoamine oxidase on the levels of 5-S-cysteinyl adducts of catechols in dopaminergic regions of the brain of the guinea pig. Neuropharmacology 1991, 30, 463–468. [Google Scholar] [CrossRef]
- Carlsson, A.; Fornstedt, B. Possible mechanisms underlying the special vulnerability of dopaminergic neurons. Acta Neurol. Scand. Suppl. 1991, 136, 16–18. [Google Scholar] [CrossRef]
- Weingarten, P.; Zhou, Q.Y. Protection of intracellular dopamine cytotoxicity by dopamine disposition and metabolism factors. J. Neurochem. 2001, 77, 776–785. [Google Scholar] [CrossRef]
- Dukes, A.A.; Korwek, K.M.; Hastings, T.G. The effect of endogenous dopamine in rotenone-induced toxicity in PC12 cells. Antioxid. Redox. Signal. 2005, 7, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.H.; Sen, T.; Maiti, A.K.; Jana, S.; Chatterjee, U.; Chakrabarti, S. Inhibition of rat brain mitochondrial electron transport chain activity by dopamine oxidation products during extended in vitro incubation: Implications for Parkinson’s disease. Biochim. Biophys. Acta 2005, 1741, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, T.; Matsuzaki-Kobayashi, M.; Takeda, A.; Sugeno, N.; Kikuchi, A.; Furukawa, K.; Perry, G.; Smith, M.A.; Itoyama, Y. Alpha-synuclein facilitates the toxicity of oxidized catechol metabolites: Implications for selective neurodegeneration in Parkinson’s disease. FEBS Lett. 2006, 580, 2147–2152. [Google Scholar] [CrossRef] [Green Version]
- Bisaglia, M.; Mammi, S.; Bubacco, L. Kinetic and structural analysis of the early oxidation products of dopamine: Analysis of the interactions with alpha-synuclein. J. Biol. Chem. 2007, 282, 15597–15605. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Ding, Y.; Cagniard, B.; Van Laar, A.D.; Mortimer, A.; Chi, W.; Hastings, T.G.; Kang, U.J.; Zhuang, X. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 2008, 28, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Paris, I.; Lozano, J.; Perez-Pastene, C.; Munoz, P.; Segura-Aguilar, J. Molecular and neurochemical mechanisms in PD pathogenesis. Neurotoxic. Res. 2009, 16, 271–279. [Google Scholar] [CrossRef]
- Leong, S.L.; Cappai, R.; Barnham, K.J.; Pham, C.L. Modulation of alpha-synuclein aggregation by dopamine: A review. Neurochem. Res. 2009, 34, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Hastings, T.G. The role of dopamine oxidation in mitochondrial dysfunction: Implications for Parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 469–472. [Google Scholar] [CrossRef]
- Bisaglia, M.; Greggio, E.; Maric, D.; Miller, D.W.; Cookson, M.R.; Bubacco, L. Alpha-synuclein overexpression increases dopamine toxicity in BE2-M17 cells. BMC Neurosci. 2010, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J.; Goldberg, J.A. The origins of oxidant stress in Parkinson’s disease and therapeutic strategies. Antioxid. Redox Signal. 2011, 14, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.N.; Johnson, S.W. Dopamine oxidation facilitates rotenone-dependent potentiation of N-methyl-D-aspartate currents in rat substantia nigra dopamine neurons. Neuroscience 2011, 195, 138–144. [Google Scholar] [CrossRef]
- Jana, S.; Sinha, M.; Chanda, D.; Roy, T.; Banerjee, K.; Munshi, S.; Patro, B.S.; Chakrabarti, S. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Surh, Y.J.; Kim, H.J. Neurotoxic effects of tetrahydroisoquinolines and underlying mechanisms. Exp. Neurobiol. 2010, 19, 63–70. [Google Scholar] [CrossRef]
- Lee, H.J.; Baek, S.M.; Ho, D.H.; Suk, J.E.; Cho, E.D.; Lee, S.J. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp. Mol. Med. 2011, 43, 216–222. [Google Scholar] [CrossRef]
- Gautam, A.H.; Zeevalk, G.D. Characterization of reduced and oxidized dopamine and 3,4-dihydrophenylacetic acid, on brain mitochondrial electron transport chain activities. Biochim. Biophys. Acta 2011, 1807, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Munoz, P.; Paris, I.; Sanders, L.H.; Greenamyre, J.T.; Segura-Aguilar, J. Overexpression of VMAT-2 and DT-diaphorase protects substantia nigra-derived cells against aminochrome neurotoxicity. Biochim. Biophys. Acta 2012, 1822, 1125–1136. [Google Scholar] [CrossRef] [Green Version]
- Bisaglia, M.; Greggio, E.; Beltramini, M.; Bubacco, L. Dysfunction of dopamine homeostasis: Clues in the hunt for novel Parkinson’s disease therapies. FASEB J. 2013, 27, 2101–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Duan, J.; Ying, Z.; Hou, Y.; Zhang, Y.; Wang, R.; Deng, Y. Increased vulnerability of parkin knock down PC12 cells to hydrogen peroxide toxicity: The role of salsolinol and NM-salsolinol. Neuroscience 2013, 233, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Munshi, S.; Sen, O.; Pramanik, V.; Roy Mukherjee, T.; Chakrabarti, S. Dopamine cytotoxicity involves both oxidative and nonoxidative pathways in SH-SY5Y cells: Potential role of alpha-synuclein overexpression and proteasomal inhibition in the etiopathogenesis of Parkinson’s disease. Parkinsons Dis. 2014, 2014, 878935. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Liu, G.; Sun, L.; Ding, J. Aldehyde Dehydrogenase 1 making molecular inroads into the differential vulnerability of nigrostriatal dopaminergic neuron subtypes in Parkinson’s disease. Transl. Neurodegener. 2014, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.; Munoz, P.; Steinbusch, H.W.M.; Segura-Aguilar, J. Are Dopamine Oxidation Metabolites Involved in the Loss of Dopaminergic Neurons in the Nigrostriatal System in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef]
- Badillo-Ramirez, I.; Saniger, J.M.; Rivas-Arancibia, S. 5-S-cysteinyl-dopamine, a neurotoxic endogenous metabolite of dopamine: Implications for Parkinson’s disease. Neurochem. Int. 2019, 129, 104514. [Google Scholar] [CrossRef]
- Linsenbardt, A.J.; Wilken, G.H.; Westfall, T.C.; Macarthur, H. Cytotoxicity of dopaminochrome in the mesencephalic cell line, MN9D, is dependent upon oxidative stress. Neurotoxicology 2009, 30, 1030–1035. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J. On the role of aminochrome in mitochondrial dysfunction and endoplasmic reticulum stress in Parkinson’s disease. Front. Neurosci. 2019, 13, 271. [Google Scholar] [CrossRef]
- Montine, T.J.; Picklo, M.J.; Amarnath, V.; Whetsell, W.O., Jr.; Graham, D.G. Neurotoxicity of endogenous cysteinylcatechols. Exp. Neurol. 1997, 148, 26–33. [Google Scholar] [CrossRef]
- Storch, A.; Ott, S.; Hwang, Y.I.; Ortmann, R.; Hein, A.; Frenzel, S.; Matsubara, K.; Ohta, S.; Wolf, H.U.; Schwarz, J. Selective dopaminergic neurotoxicity of isoquinoline derivatives related to Parkinson’s disease: Studies using heterologous expression systems of the dopamine transporter. Biochem. Pharmacol. 2002, 63, 909–920. [Google Scholar] [CrossRef]
- Nagatsu, T. Isoquinoline neurotoxins in the brain and Parkinson’s disease. Neurosci. Res. 1997, 29, 99–111. [Google Scholar] [CrossRef]
- Blaschko, H. Metabolism and storage of biogenic amines. Experientia 1957, 13, 9–13. [Google Scholar] [CrossRef]
- Lamensdorf, I.; Hrycyna, C.; He, L.P.; Nechushtan, A.; Tjurmina, O.; Harvey-White, J.; Eisenhofer, G.; Rojas, E.; Kopin, I.J. Acidic dopamine metabolites are actively extruded from PC12 cells by a novel sulfonylurea-sensitive transporter. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 361, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Sandler, M. Activation of monoamine oxidase and inhibition of aldehyde dehydrogenase by reserpine. Eur. J. Pharmacol. 1968, 4, 105–107. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Pietruszko, R. Chemical modification of human aldehyde dehydrogenase by physiological substrate. Biochim. Biophys. Acta 1987, 911, 306–317. [Google Scholar] [CrossRef]
- Wey, M.; Fernandez, E.; Martinez, P.A.; Sullivan, P.; Goldstein, D.S.; Strong, R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: Implications for Parkinson’s disease. PLoS ONE 2012, 7, e31522. [Google Scholar] [CrossRef] [PubMed]
- Casida, J.E.; Ford, B.; Jinsmaa, Y.; Sullivan, P.; Cooney, A.; Goldstein, D.S. Benomyl, aldehyde dehydrogenase, DOPAL, and the catecholaldehyde hypothesis for the pathogenesis of Parkinson’s disease. Chem. Res. Toxicol. 2014, 27, 1359–1361. [Google Scholar] [CrossRef]
- Fitzmaurice, A.G.; Rhodes, S.L.; Lulla, A.; Murphy, N.P.; Lam, H.A.; O’Donnell, K.C.; Barnhill, L.; Casida, J.E.; Cockburn, M.; Sagasti, A.; et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc. Natl. Acad. Sci. USA 2013, 110, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Fitzmaurice, A.G.; Rhodes, S.L.; Cockburn, M.; Ritz, B.; Bronstein, J.M. Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology 2014, 82, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Ritz, B.R.; Paul, K.C.; Bronstein, J.M. Of pesticides and men: A California story of genes and environment in Parkinson’s disease. Curr. Environ. Health Rep. 2016, 3, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Molochnikov, L.; Rabey, J.M.; Dobronevsky, E.; Bonucelli, U.; Ceravolo, R.; Frosini, D.; Grunblatt, E.; Riederer, P.; Jacob, C.; Aharon-Peretz, J.; et al. A molecular signature in blood identifies early Parkinson’s disease. Molec. Neurodegen. 2012, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunblatt, E.; Ruder, J.; Monoranu, C.M.; Riederer, P.; Youdim, M.B.; Mandel, S.A. Differential Alterations in Metabolism and Proteolysis-Related Proteins in Human Parkinson’s Disease Substantia Nigra. Neurotox. Res. 2018, 33, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Grunblatt, E.; Riederer, P.; Amariglio, N.; Jacob-Hirsch, J.; Rechavi, G.; Youdim, M.B. Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann. N. Y. Acad. Sci. 2005, 1053, 356–375. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Sullivan, P.; Cooney, A.; Jinsmaa, Y.; Kopin, I.J.; Sharabi, Y. Rotenone decreases intracellular aldehyde dehydrogenase activity: Implications for the pathogenesis of Parkinson’s disease. J. Neurochem. 2015, 133, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamensdorf, I.; Eisenhofer, G.; Harvey-White, J.; Hayakawa, Y.; Kirk, K.; Kopin, I.J. Metabolic stress in PC12 cells induces the formation of the endogenous dopaminergic neurotoxin, 3,4-dihydroxyphenylacetaldehyde. J. Neurosci. Res. 2000, 60, 552–558. [Google Scholar] [CrossRef]
- Chiu, C.C.; Yeh, T.H.; Lai, S.C.; Wu-Chou, Y.H.; Chen, C.H.; Mochly-Rosen, D.; Huang, Y.C.; Chen, Y.J.; Chen, C.L.; Chang, Y.M.; et al. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp. Neurol. 2015, 263, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genomics 2011, 5, 283–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florang, V.R.; Rees, J.N.; Brogden, N.K.; Anderson, D.G.; Hurley, T.D.; Doorn, J.A. Inhibition of the oxidative metabolism of 3,4-dihydroxyphenylacetaldehyde, a reactive intermediate of dopamine metabolism, by 4-hydroxy-2-nonenal. Neurotoxicology 2007, 28, 76–82. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Florang, V.R.; Rees, J.N.; Anderson, D.G.; Strack, S.; Doorn, J.A. Products of oxidative stress inhibit aldehyde oxidation and reduction pathways in dopamine catabolism yielding elevated levels of a reactive intermediate. Chem. Res. Toxicol. 2009, 22, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Gainetdinov, R.R.; Fumagalli, F.; Wang, Y.M.; Jones, S.R.; Levey, A.I.; Miller, G.W.; Caron, M.G. Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. J. Neurochem. 1998, 70, 1973–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, F.; Gainetdinov, R.R.; Wang, Y.M.; Valenzano, K.J.; Miller, G.W.; Caron, M.G. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J. Neurosci. 1999, 19, 2424–2431. [Google Scholar] [CrossRef] [Green Version]
- Staal, R.G.; Sonsalla, P.K. Inhibition of brain vesicular monoamine transporter (VMAT2) enhances 1-methyl-4-phenylpyridinium neurotoxicity in vivo in rat striata. J. Pharmacol. Exp. Ther. 2000, 293, 336–342. [Google Scholar]
- Guillot, T.S.; Miller, G.W. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol. Neurobiol. 2009, 39, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M.; Richardson, J.R.; Wang, M.Z.; Taylor, T.N.; Guillot, T.S.; McCormack, A.L.; Colebrooke, R.E.; Di Monte, D.A.; Emson, P.C.; Miller, G.W. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 2007, 27, 8138–8148. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.N.; Alter, S.P.; Wang, M.; Goldstein, D.S.; Miller, G.W. Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology 2014, 76, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, K.M.; Bernstein, A.I.; Stout, K.A.; Dunn, A.R.; Lazo, C.R.; Alter, S.P.; Wang, M.; Li, Y.; Fan, X.; Hess, E.J.; et al. Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 9977–9982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Miller, G.W.; Sharabi, Y.; Kopin, I.J. A vesicular sequestration to oxidative deamination shift in myocardial sympathetic nerves in Parkinson disease. J. Neurochem. 2014, 131, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Sullivan, P.; Cooney, A.; Jinsmaa, Y.; Sullivan, R.; Gross, D.J.; Holmes, C.; Kopin, I.J.; Sharabi, Y. Vesicular uptake blockade generates the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 cells: Relevance to the pathogenesis of Parkinson’s disease. J. Neurochem. 2012, 123, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Sullivan, P.; Holmes, C.; Miller, G.W.; Alter, S.; Strong, R.; Mash, D.C.; Kopin, I.J.; Sharabi, Y. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J. Neurochem. 2013, 126, 591–603. [Google Scholar] [CrossRef]
- Mexas, L.M.; Florang, V.R.; Doorn, J.A. Inhibition and covalent modification of tyrosine hydroxylase by 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Neurotoxicology 2011, 32, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine Hydroxylase. The Initial Step in Norepinephrine Biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar] [CrossRef]
- Vermeer, L.M.; Florang, V.R.; Doorn, J.A. Catechol and aldehyde moieties of 3,4-dihydroxyphenylacetaldehyde contribute to tyrosine hydroxylase inhibition and neurotoxicity. Brain Res. 2012, 1474, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Arai, K.; Kato, N.; Kashiwado, K.; Hattori, T. Pure autonomic failure in association with human alpha-synucleinopathy. Neurosci. Lett. 2000, 296, 171–173. [Google Scholar] [CrossRef]
- Kaufmann, H.; Hague, K.; Perl, D. Accumulation of alpha-synuclein in autonomic nerves in pure autonomic failure. Neurology 2001, 56, 980–981. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Armakola, M.; Dumoulin, M.; Parastatidis, I.; Ischiropoulos, H. Cellular oligomerization of alpha-synuclein is determined by the interaction of oxidized catechols with a C-terminal sequence. J. Biol. Chem. 2007, 282, 31621–31630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, D.E.; Daniels, M.J.; Ischiropoulos, H. The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov. Disord. 2019, 34, 167–179. [Google Scholar] [CrossRef]
- Saha, S.; Khan, M.A.I.; Mudhara, D.; Deep, S. Tuning the Balance between Fibrillation and Oligomerization of alpha-Synuclein in the Presence of Dopamine. ACS Omega 2018, 3, 14213–14224. [Google Scholar] [CrossRef] [Green Version]
- Huenchuguala, S.; Sjodin, B.; Mannervik, B.; Segura-Aguilar, J. Novel Alpha-Synuclein Oligomers Formed with the Aminochrome-Glutathione Conjugate Are Not Neurotoxic. Neurotoxic. Res. 2019, 35, 432–440. [Google Scholar] [CrossRef]
- Munoz, P.; Cardenas, S.; Huenchuguala, S.; Briceno, A.; Couve, E.; Paris, I.; Segura-Aguilar, J. DT-Diaphorase Prevents Aminochrome-Induced Alpha-Synuclein Oligomer Formation and Neurotoxicity. Toxicol. Sci. 2015, 145, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.L.; Leong, S.L.; Ali, F.E.; Kenche, V.B.; Hill, A.F.; Gras, S.L.; Barnham, K.J.; Cappai, R. Dopamine and the dopamine oxidation product 5,6-dihydroxylindole promote distinct on-pathway and off-pathway aggregation of alpha-synuclein in a pH-dependent manner. J. Mol. Biol. 2009, 387, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Follmer, C.; Coelho-Cerqueira, E.; Yatabe-Franco, D.Y.; Araujo, G.D.; Pinheiro, A.S.; Domont, G.B.; Eliezer, D. Oligomerization and membrane-binding properties of covalent adducts formed by the interaction of alpha-synuclein with the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL). J. Biol. Chem. 2015, 290, 27660–27679. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.G.; Mariappan, S.V.; Buettner, G.R.; Doorn, J.A. Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J. Biol. Chem. 2011, 286, 26978–26986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, W.J.; Kumar, V.B.; Pandey, N.; Panneton, W.M.; Gan, Q.; Franko, M.W.; O’Dell, M.; Li, S.W.; Pan, Y.; Chung, H.D.; et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008, 115, 193–203. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, G.; Lindstrom, V.; Rostami, J.; Nordstrom, E.; Lannfelt, L.; Bergstrom, J.; Ingelsson, M.; Erlandsson, A. Alpha-synuclein oligomer-selective antibodies reduce intracellular accumulation and mitochondrial impairment in alpha-synuclein exposed astrocytes. J. Neuroinflamm. 2017, 14, 241. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinsmaa, Y.; Sullivan, P.; Gross, D.; Cooney, A.; Sharabi, Y.; Goldstein, D.S. Divalent metal ions enhance DOPAL-induced oligomerization of alpha-synuclein. Neurosci. Lett. 2014, 569, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinsmaa, Y.; Isonaka, R.; Sharabi, Y.; Goldstein, D.S. 3,4-Dihydroxyphenylacetaldehyde Is More Efficient than Dopamine in Oligomerizing and Quinonizing alpha-Synuclein. J. Pharmacol. Exp. Ther. 2020, 372, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Berti, G.; Ferrari, E.; Tessari, I.; Zanetti, M.; Lunelli, L.; Greggio, E.; Bisaglia, M.; Veronesi, M.; Girotto, S.; et al. DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep. 2017, 7, 40699. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Yacoub, A.; Kunz, A.; Aranki, B.; Serobyan, G.; Cohn, W.; Whitelegge, J.P.; Watson, J.B. Enhanced mitochondrial inhibition by 3,4-dihydroxyphenyl-acetaldehyde (DOPAL)-oligomerized alpha-synuclein. J. Neurosci. Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.G.; Florang, V.R.; Schamp, J.H.; Buettner, G.R.; Doorn, J.A. Antioxidant-mediated modulation of protein reactivity for 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Chem. Res. Toxicol. 2016, 29, 1098–1107. [Google Scholar] [CrossRef]
- Werner-Allen, J.W.; DuMond, J.F.; Levine, R.L.; Bax, A. Toxic dopamine metabolite DOPAL forms an unexpected dicatechol pyrrole adduct with lysines of alpha-synuclein. Angew. Chem. Int. Ed. Engl. 2016, 55, 7374–7378. [Google Scholar] [CrossRef] [Green Version]
- Werner-Allen, J.W.; Monti, S.; DuMond, J.F.; Levine, R.L.; Bax, A. Isoindole linkages provide a pathway for DOPAL-mediated cross-linking of alpha-synuclein. Biochemistry 2018, 57, 1462–1474. [Google Scholar] [CrossRef]
- Werner-Allen, J.W.; Levine, R.L.; Bax, A. Superoxide is the critical driver of DOPAL autoxidation, lysyl adduct formation, and crosslinking of alpha-synuclein. Biochem. Biophys. Res. Commun. 2017, 487, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.B.; Hsu, F.F.; Lakshmi, V.M.; Gillespie, K.N.; Burke, W.J. Aldehyde adducts inhibit 3,4-dihydroxyphenylacetaldehyde-induced alpha-synuclein aggregation and toxicity: Implication for Parkinson neuroprotective therapy. Eur. J. Pharmacol. 2019, 845, 65–73. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotoxic. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef]
- Segura-Aguilar, J. On the role of endogenous neurotoxins and neuroprotection in Parkinson’s disease. Neural Regen. Res. 2017, 12, 897–901. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Mishizen, A.J.; Giasson, B.I.; Lynch, D.R.; Thomas, S.A.; Nakashima, A.; Nagatsu, T.; Ota, A.; Ischiropoulos, H. Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J. Neurosci. 2006, 26, 10068–10078. [Google Scholar] [CrossRef] [PubMed]
- Cagle, B.S.; Crawford, R.A.; Doorn, J.A. Biogenic aldehyde-mediated mechanisms of toxicity in neurodegenerative disease. Curr. Opin. Toxicol. 2019, 13, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.N.; Florang, V.R.; Eckert, L.L.; Doorn, J.A. Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem. Res. Toxicol. 2009, 22, 1256–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Aneman, A.; Friberg, P.; Hooper, D.; Fandriks, L.; Lonroth, H.; Hunyady, B.; Mezey, E. Substantial production of dopamine in the human gastrointestinal tract. J. Clin. Endocrinol. Metab. 1997, 82, 3864–3871. [Google Scholar] [CrossRef]
- Sun, J.; He, C.; Yan, Q.X.; Wang, H.D.; Li, K.X.; Sun, X.; Feng, Y.; Zha, R.R.; Cui, C.P.; Xiong, X.; et al. Parkinson-like early autonomic dysfunction induced by vagal application of DOPAL in rats. CNS Neurosci. Ther. 2021, 27, 540–551. [Google Scholar] [CrossRef]
- Berg, D.; Borghammer, P.; Fereshtehnejad, S.M.; Heinzel, S.; Horsager, J.; Schaeffer, E.; Postuma, R.B. Prodromal Parkinson disease subtypes—Key to understanding heterogeneity. Nat. Rev. Neurol. 2021. [Google Scholar] [CrossRef]
- Kang, S.S.; Ahn, E.H.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Sandoval, I.M.; Dhakal, S.; Iuvone, P.M.; Cao, X.; Ye, K. alpha-Synuclein stimulation of monoamine oxidase-B and legumain protease mediates the pathology of Parkinson’s disease. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Jia, C.; Cheng, C.; Li, T.; Chen, X.; Yang, Y.; Liu, X.; Li, S.; Le, W. alpha-Synuclein Up-regulates Monoamine Oxidase A Expression and Activity via Trans-Acting Transcription Factor 1. Front. Aging Neurosci. 2021, 13, 653379. [Google Scholar] [CrossRef]
- Kang, S.S.; Zhang, Z.; Liu, X.; Manfredsson, F.P.; Benskey, M.J.; Cao, X.; Xu, J.; Sun, Y.E.; Ye, K. TrkB neurotrophic activities are blocked by alpha-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.S.; Liu, X.; Ahn, E.H.; Xiang, J.; Manfredsson, F.P.; Yang, X.; Luo, H.R.; Liles, L.C.; Weinshenker, D.; Ye, K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Investig. 2020, 130, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Dina, O.A.; Khasar, S.G.; Alessandri-Haber, N.; Bogen, O.; Chen, X.; Green, P.G.; Reichling, D.B.; Messing, R.O.; Levine, J.D. Neurotoxic catecholamine metabolite in nociceptors contributes to painful peripheral neuropathy. Eur. J. Neurosci. 2008, 28, 1180–1190. [Google Scholar] [CrossRef] [Green Version]
- Greengard, P. The neurobiology of slow synaptic transmission. Science 2001, 294, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Vizi, E.S. Non-synaptic intercellular communication: Presynaptic inhibition. Acta Biol. Acad. Sci. Hung. 1982, 33, 331–351. [Google Scholar]
- Van der Velden, L.; Vinck, M.; Werkman, T.R.; Wadman, W.J. Tuning of Neuronal Interactions in the Lateral Ventral Tegmental Area by Dopamine Sensitivity. Neuroscience 2017, 366, 62–69. [Google Scholar] [CrossRef]
- Goldstein, D.S. Biomarkers, mechanisms, and potential prevention of catecholamine neuron loss in Parkinson disease. Adv. Pharmacol. 2013, 68, 235–272. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Leaky catecholamine stores: Undue waste or a stress response coping mechanism? Ann. N. Y. Acad. Sci. 2004, 1018, 224–230. [Google Scholar] [CrossRef]
- Benarroch, E.E. The central autonomic network: Functional organization, dysfunction, and perspective. Mayo Clin. Proc. 1993, 68, 988–1001. [Google Scholar] [CrossRef]
- Goldstein, D.S. The extended autonomic system, dyshomeostasis, and COVID-19. Clin. Auton. Res. 2020, 30, 299–315. [Google Scholar] [CrossRef]
- Valentino, R.J.; Guyenet, P.; Hou, X.H.; Herman, M. Central Network Dynamics Regulating Visceral and Humoral Functions. J. Neurosci. 2017, 37, 10848–10854. [Google Scholar] [CrossRef] [Green Version]
- Carlson, L.A.; Levi, L.; Oro, L. Plasma lipids and urinary excretion of catecholamines in man during experimentally induced emotional stress, and their modification by nicotinic acid. J. Clin. Investig. 1968, 47, 1795–1805. [Google Scholar] [CrossRef]
- Shanks, N.; Griffiths, J.; Anisman, H. Central catecholamine alterations induced by stressor exposure: Analyses in recombinant inbred strains of mice. Behav. Brain Res. 1994, 63, 25–33. [Google Scholar] [CrossRef]
- Axelrod, J.; Kopin, I.J. The uptake, storage, release and metabolism of noradrenaline in sympathetic nerves. Prog. Brain Res. 1969, 31, 21–32. [Google Scholar]
- Hertting, G.; Axelrod, J. Fate of tritiated noradrenaline at sympathetic nerve endings. Nature 1961, 192, 172–173. [Google Scholar] [CrossRef]
- Pacak, K.; Armando, I.; Fukuhara, K.; Kvetnansky, R.; Palkovits, M.; Kopin, I.J.; Goldstein, D.S. Noradrenergic activation in the paraventricular nucleus during acute and chronic immobilization stress in rats: An in vivo microdialysis study. Brain Res. 1992, 589, 91–96. [Google Scholar] [CrossRef]
- Pacak, K.; Palkovits, M.; Kvetnansky, R.; Fukuhara, K.; Armando, I.; Kopin, I.J.; Goldstein, D.S. Effects of single or repeated immobilization on release of norepinephrine and its metabolites in the central nucleus of the amygdala in conscious rats. Neuroendocrinology 1993, 57, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Kvetnansky, R.; Armando, I.; Weise, V.K.; Holmes, C.; Fukuhara, K.; Deka-Starosta, A.; Kopin, I.J.; Goldstein, D.S. Plasma dopa responses during stress: Dependence on sympathoneural activity and tyrosine hydroxylation. J. Pharmacol. Exp. Ther. 1992, 261, 899–909. [Google Scholar]
- Kvetnansky, R.; Goldstein, D.S.; Weise, V.K.; Holmes, C.; Szemeredi, K.; Bagdy, G.; Kopin, I.J. Effects of handling or immobilization on plasma levels of 3,4-dihydroxyphenylalanine, catecholamines, and metabolites in rats. J. Neurochem. 1992, 58, 2296–2302. [Google Scholar]
- Sugama, S.; Sekiyama, K.; Kodama, T.; Takamatsu, Y.; Takenouchi, T.; Hashimoto, M.; Bruno, C.; Kakinuma, Y. Chronic restraint stress triggers dopaminergic and noradrenergic neurodegeneration: Possible role of chronic stress in the onset of Parkinson’s disease. Brain Behav. Immun. 2016, 51, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Zarow, C.; Lyness, S.A.; Mortimer, J.A.; Chui, H.C. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 2003, 60, 337–341. [Google Scholar] [CrossRef]
- Hemmerle, A.M.; Dickerson, J.W.; Herman, J.P.; Seroogy, K.B. Stress exacerbates experimental Parkinson’s disease. Mol. Psychiatry 2014, 19, 638–640. [Google Scholar] [CrossRef]
- Janakiraman, U.; Manivasagam, T.; Justin Thenmozhi, A.; Dhanalakshmi, C.; Essa, M.M.; Song, B.J.; Guillemin, G.J. Chronic mild stress augments MPTP induced neurotoxicity in a murine model of Parkinson’s disease. Physiol. Behav. 2017, 173, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Forsyth, C.B.; Voigt, R.M.; Engen, P.A.; Patel, J.; Shaikh, M.; Green, S.J.; Naqib, A.; Roy, A.; Kordower, J.H.; et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol. Dis. 2020, 135, 104352. [Google Scholar] [CrossRef] [PubMed]
- Group, P.S. Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP subjects not requiring levodopa. Parkinson Study Group. Ann. Neurol. 1996, 39, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.D. Does selegiline delay progression of Parkinson’s disease? A critical re-evaluation of the DATATOP study. J. Neurol. Neurosurg. Psychiatry 1994, 57, 217–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, G.; Abbruzzese, G.; Marconi, S.; Zappia, M. Selegiline: A reappraisal of its role in Parkinson disease. Clin. Neuropharmacol. 2012, 35, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Rascol, O.; Hauser, R.; Feigin, P.D.; Jankovic, J.; Lang, A.; Langston, W.; Melamed, E.; Poewe, W.; Stocchi, F.; et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1268–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Fuente-Fernandez, R.; Schulzer, M.; Mak, E.; Sossi, V. Trials of neuroprotective therapies for Parkinson’s disease: Problems and limitations. Parkinsonism Relat. Disord. 2010, 16, 365–369. [Google Scholar] [CrossRef]
- Lamotte, G.; Holmes, C.; Wu, T.; Goldstein, D.S. Long-term trends in myocardial sympathetic innervation and function in synucleinopathies. Parkinsonism Relat. Disord. 2019, 67, 27–33. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Holmes, C.; Lopez, G.J.; Wu, T.; Sharabi, Y. Cerebrospinal fluid biomarkers of central dopamine deficiency predict Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 50, 108–112. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Sharabi, Y. N-Acetylcysteine prevents the increase in spontaneous oxidation of dopamine during monoamine oxidase inhibition in PC12 cells. Neurochem. Res. 2017, 42, 3289–3295. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Kopin, I.J.; Sharabi, Y. 3,4-Dihydroxyphenylethanol (hydroxytyrosol) mitigates the increase in spontaneous oxidation of dopamine during monoamine oxidase inhibition in PC12 cells. Neurochem. Res. 2016, 41, 2173–2178. [Google Scholar] [CrossRef] [Green Version]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Cai, J.; Wei, X.; Bazzan, A.J.; Zhong, L.; Bowen, B.; et al. N-Acetyl cysteine may support dopamine neurons in Parkinson’s disease: Preliminary clinical and cell line data. PLoS ONE 2016, 11, e0157602. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-Acetyl Cysteine Is Associated With Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Holmes, C.; Lopez, G.J.; Wu, T.; Sharabi, Y. Cardiac sympathetic denervation predicts PD in at-risk individuals. Parkinsonism Relat. Disord. 2018, 52, 90–93. [Google Scholar] [CrossRef]
- Kaufmann, H.; Norcliffe-Kaufmann, L.; Palma, J.A.; Biaggioni, I.; Low, P.A.; Singer, W.; Goldstein, D.S.; Peltier, A.C.; Shibao, C.A.; Gibbons, C.H.; et al. Natural history of pure autonomic failure: A United States prospective cohort. Ann. Neurol. 2017, 81, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Pekker, M.J.; Eisenhofer, G.; Sharabi, Y. Computational modeling reveals multiple abnormalities of myocardial noradrenergic function in Lewy body diseases. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, D.S. “Sick-but-not-dead”: Multiple paths to catecholamine deficiency in Lewy Body diseases. Stress 2020, 1–16. [Google Scholar] [CrossRef]
- Isonaka, R.; Rosenberg, A.Z.; Sullivan, P.; Corrales, A.; Holmes, C.; Sharabi, Y.; Goldstein, D.S. Alpha-Synuclein deposition within sympathetic noradrenergic neurons Is associated with myocardial noradrenergic deficiency in neurogenic orthostatic hypotension. Hypertension 2019, 73, 910–918. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldstein, D.S. The Catecholaldehyde Hypothesis for the Pathogenesis of Catecholaminergic Neurodegeneration: What We Know and What We Do Not Know. Int. J. Mol. Sci. 2021, 22, 5999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115999
Goldstein DS. The Catecholaldehyde Hypothesis for the Pathogenesis of Catecholaminergic Neurodegeneration: What We Know and What We Do Not Know. International Journal of Molecular Sciences. 2021; 22(11):5999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115999
Chicago/Turabian StyleGoldstein, David S. 2021. "The Catecholaldehyde Hypothesis for the Pathogenesis of Catecholaminergic Neurodegeneration: What We Know and What We Do Not Know" International Journal of Molecular Sciences 22, no. 11: 5999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115999