
Microtubule Acetylation Controls MDA-MB-231 Breast Cancer Cell Invasion through the Modulation of Endoplasmic Reticulum Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
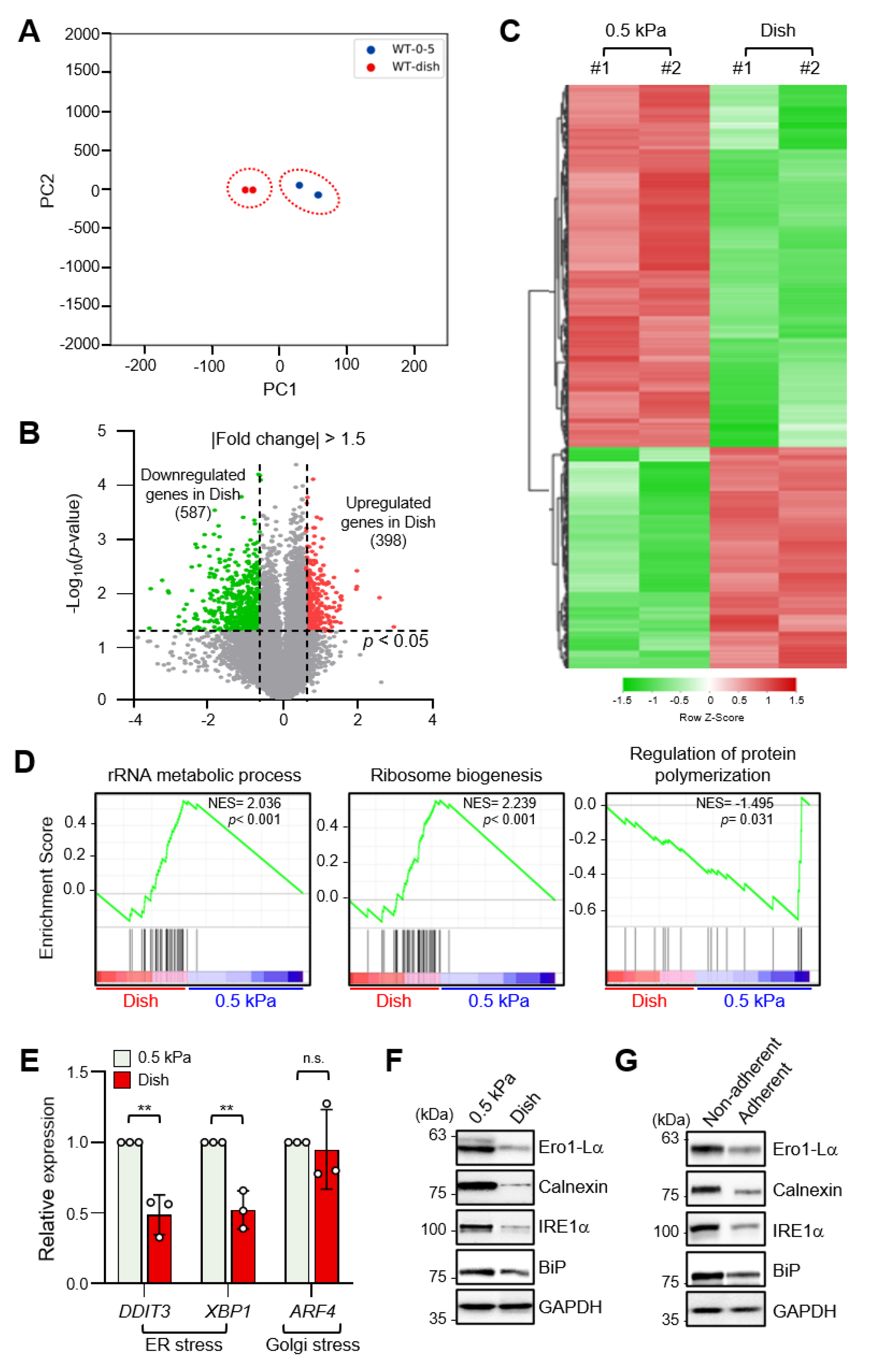
2.1. Increased ECM Stiffness Inhibits ER Stress
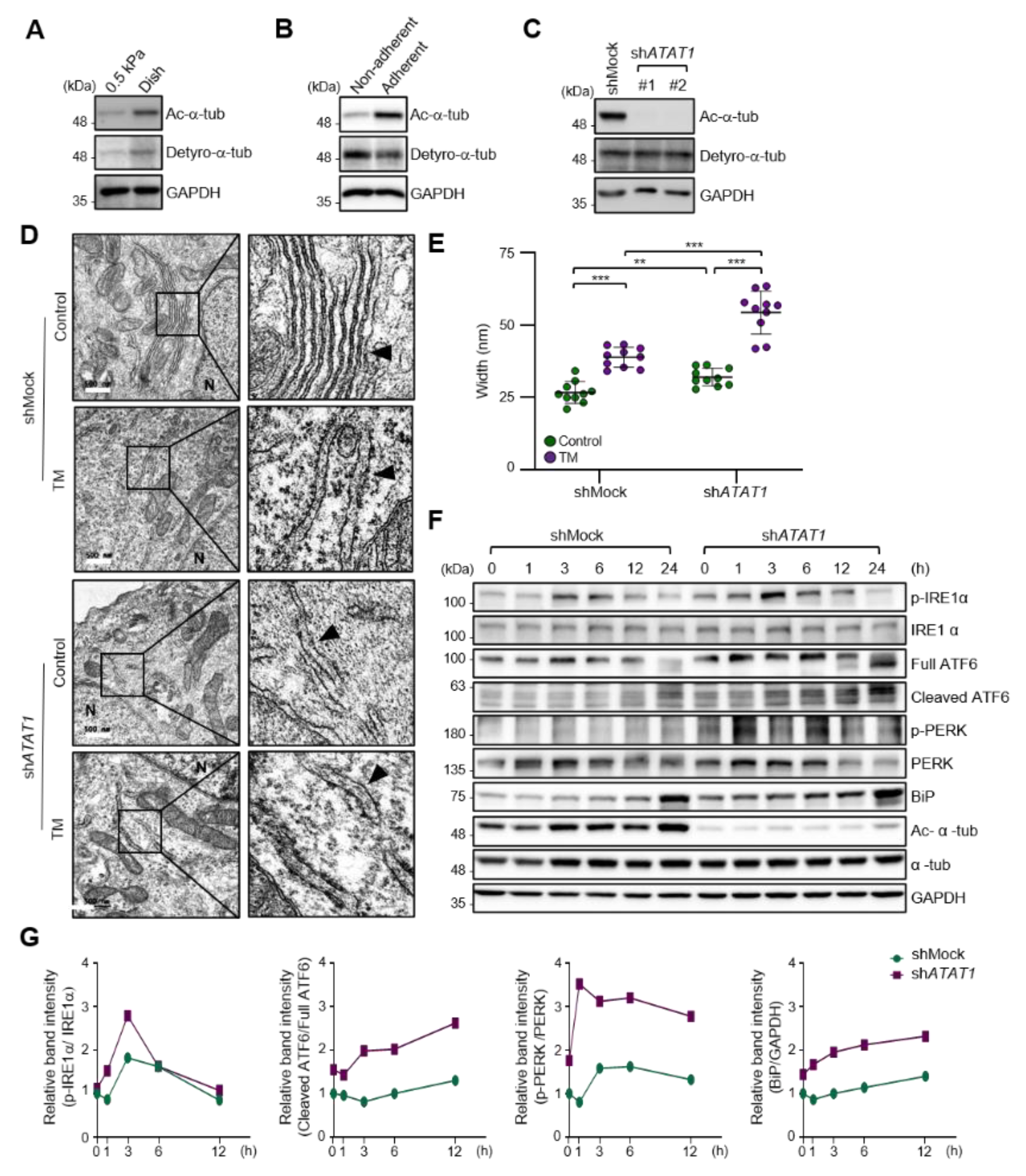
2.2. Microtubule Acetylation Is Required for the Regulation of UPR Signaling
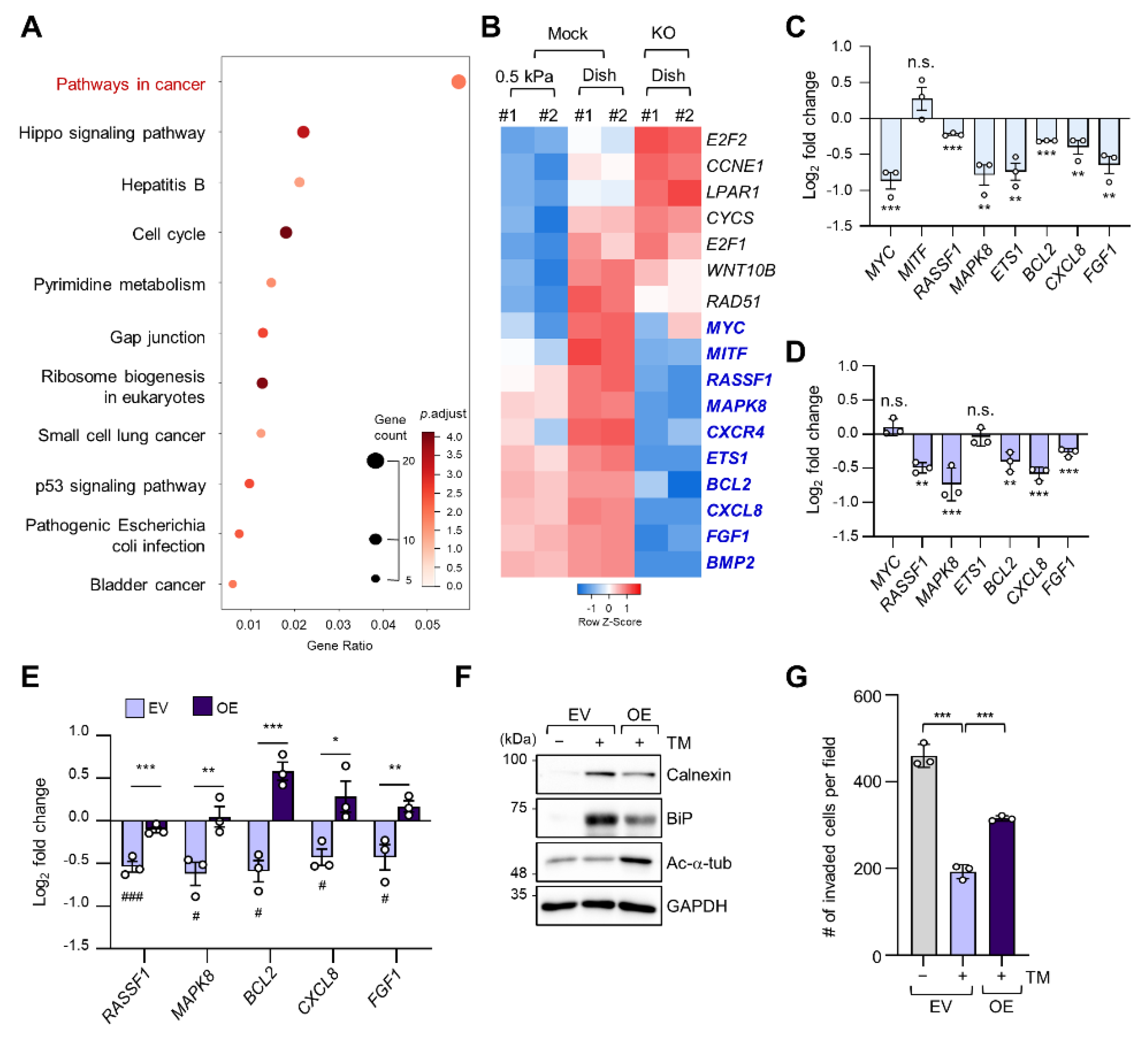
2.3. Regulation of Cancer Pathway-Related Gene Expression by ER Stress in a Stiff Matrix Is Dependent on Microtubule Acetylation
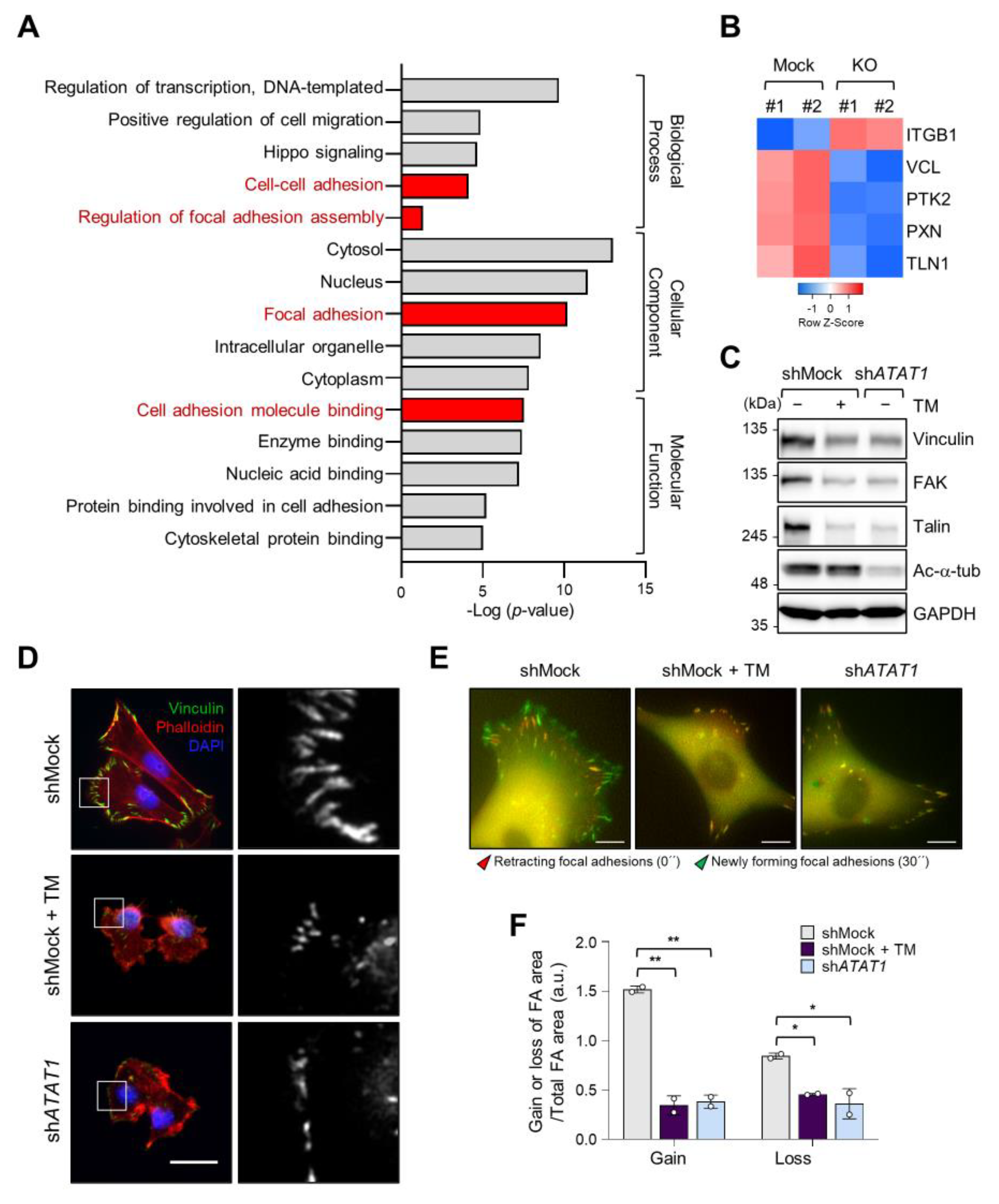
2.4. Microtubule Acetylation and ER Stress Regulate Focal Adhesion Formation
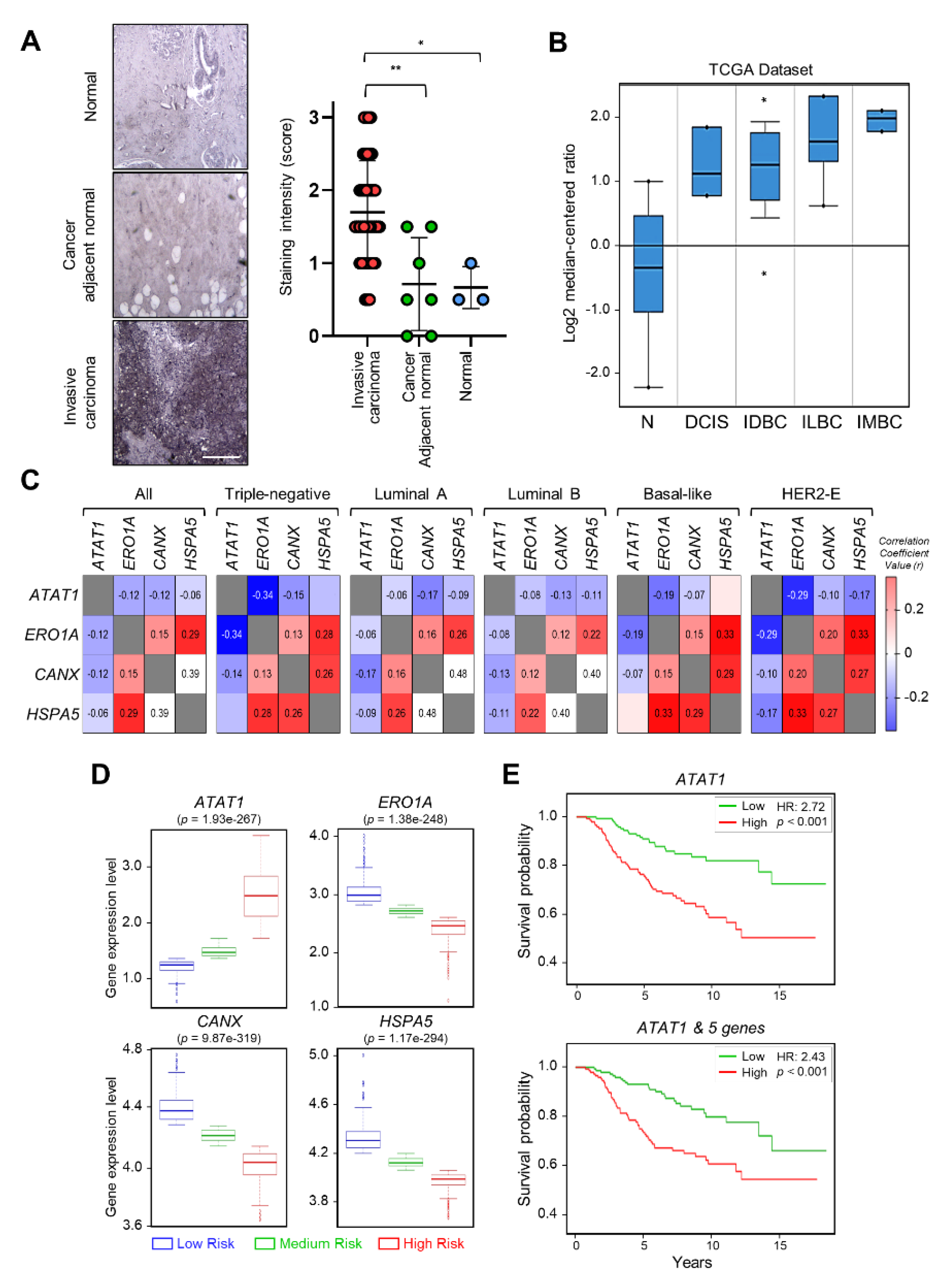
2.5. Expression of ATAT1 and ER Stress Markers Is Negatively Correlated in Breast Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Preparation of PAGs
4.3. Antibodies and Reagents
4.4. Western Blotting
4.5. RT-qPCR
4.6. RNA-seq and Data Analysis
4.7. Single-Cell Tracking
4.8. Invasion Assay
4.9. Immunocytochemistry
4.10. Plasmid Construction
4.11. Generation of ATAT1-Knockout Cell Lines Using the CRISPR/Cas9 System
4.12. Establishment of ATAT1 Knockdown and Overexpression Cell Lines
4.13. Transmission Electron Microscopy
4.14. Immunohistochemistry
4.15. Focal Adhesion Assembly
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Boettiger, D. A novel mode for integrin-mediated signaling: Tethering is required for phosphorylation of FAK Y397. Mol. Biol. Cell 2003, 14, 4306–4315. [Google Scholar] [CrossRef] [Green Version]
- Lawson, C.D.; Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014, 5, e27958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Tiffany-Castiglioni, E.; Koh, H.C.; Son, I.H. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxicol. Lett. 2009, 191, 203–210. [Google Scholar] [CrossRef]
- Gadde, S.; Heald, R. Mechanisms and molecules of the mitotic spindle. Curr. Biol. 2004, 14, R797–R805. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.L.; Gelfand, V.I. Membrane trafficking, organelle transport, and the cytoskeleton. Curr. Opin. Cell Biol. 2000, 12, 57–62. [Google Scholar] [CrossRef]
- Boggs, A.E.; Vitolo, M.I.; Whipple, R.A.; Charpentier, M.S.; Goloubeva, O.G.; Ioffe, O.B.; Tuttle, K.C.; Slovic, J.; Lu, Y.; Mills, G.B.; et al. alpha-Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res. 2015, 75, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.; You, E.; Ko, P.; Jeong, J.; Keum, S.; Rhee, S. Genetic disruption of tubulin acetyltransferase, alphaTAT1, inhibits proliferation and invasion of colon cancer cells through decreases in Wnt1/beta-catenin signaling. Biochem. Biophys. Res. Commun. 2017, 482, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Lee, J.; Go, H.; Lee, C.G.; Kim, S.; Kim, H.S.; Cho, H.; Choi, K.S.; Ha, G.H.; Lee, C.W. Oncogenic microtubule hyperacetylation through BEX4-mediated sirtuin 2 inhibition. Cell Death Dis. 2016, 7, e2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.T.; Chang, Y.J.; Yang, L.X.; Wei, P.L.; Liu, T.Z.; Liu, J.J. A novel microtubule-disrupting agent induces endoplasmic reticular stress-mediated cell death in human hepatocellular carcinoma cells. PLoS ONE 2015, 10, e0136340. [Google Scholar] [CrossRef] [Green Version]
- Tilghman, R.W.; Blais, E.M.; Cowan, C.R.; Sherman, N.E.; Grigera, P.R.; Jeffery, E.D.; Fox, J.W.; Blackman, B.R.; Tschumperlin, D.J.; Papin, J.A.; et al. Matrix rigidity regulates cancer cell growth by modulating cellular metabolism and protein synthesis. PLoS ONE 2012, 7, e37231. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, P.; Kim, D.; You, E.; Jung, J.; Oh, S.; Kim, J.; Lee, K.H.; Rhee, S. Extracellular matrix rigidity-dependent sphingosine-1-phosphate secretion regulates metastatic cancer cell invasion and adhesion. Sci. Rep. 2016, 6, 21564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, E.; Ko, P.; Jeong, J.; Keum, S.; Kim, J.-W.; Seo, Y.-J.; Song, W.K.; Rhee, S. Dynein-mediated nuclear translocation of yes-associated protein through microtubule acetylation controls fibroblast activation. Cell. Mol. Life Sci. 2020, 77, 4143–4161. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Emon, B.; Bauer, J.; Jain, Y.; Jung, B.; Saif, T. Biophysics of tumor microenvironment and cancer metastasis—A mini review. Comput. Struct. Biotechnol. J. 2018, 16, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Kasetti, R.B.; Maddineni, P.; Millar, J.C.; Clark, A.F.; Zode, G.S. Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci. Rep. 2017, 7, 14951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Ke, W.; Wang, K.; Li, G.; Ma, L.; Lu, S.; Xiang, Q.; Liao, Z.; Luo, R.; Song, Y.; et al. Mechanosensitive ion channel Piezo1 activated by matrix stiffness regulates oxidative stress-induced senescence and apoptosis in human intervertebral disc degeneration. Oxid. Med. Cell. Longev. 2021, 2021, 8884922. [Google Scholar]
- Zhou, D.W.; Lee, T.T.; Weng, S.; Fu, J.; García, A.J. Effects of substrate stiffness and actomyosin contractility on coupling between force transmission and vinculin–paxillin recruitment at single focal adhesions. Mol. Biol. Cell 2017, 28, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, A.F.; Eng, C.H.; Schlaepfer, D.D.; Marcantonio, E.E.; Gundersen, G.G. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science 2004, 303, 836–839. [Google Scholar] [CrossRef]
- Colello, D.; Mathew, S.; Ward, R.; Pumiglia, K.; LaFlamme, S.E. Integrins regulate microtubule nucleating activity of centrosome through mitogen-activated protein kinase/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase (MEK/ERK) signaling. J. Biol. Chem. 2012, 287, 2520–2530. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Kumar, S.; Zaman, N.; Pan, C.C.; Bloodworth, J.C.; Lei, W.; Streicher, J.M.; Hempel, N.; Mythreye, K.; Lee, N.Y. TAK1 activation of alpha-TAT1 and microtubule hyperacetylation control AKT signaling and cell growth. Nat. Commun. 2018, 9, 1696. [Google Scholar] [CrossRef] [Green Version]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhou, Z.; Zhang, L.; Meng, S.; Li, S.; Wang, X. Inhibition of SIRT2 by targeting GSK3beta-mediated phosphorylation alleviates SIRT2 toxicity in SH-SY5Y cells. Front. Cell. Neurosci. 2019, 13, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandewynckel, Y.-P.; Laukens, D.; Bogaerts, E.; Paridaens, A.; Van Den Bussche, A.; Verhelst, X.; Van Steenkiste, C.; Descamps, B.; Vanhove, C.; Libbrecht, L.; et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: A PERK for hepatocellular carcinoma therapy. Hepatol. Int. 2015, 9, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.E.; Baldwin, S.W.; Baldwin, M.L.; Chen, S.-T.; Moretz, J.M.; Aragon, R.J.; Li, X.; Strong, D.D.; Mohan, S.; Amaar, Y.G. Ras-association domain family 1C protein promotes breast cancer cell migration and attenuates apoptosis. BMC Cancer 2010, 10, 562. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Wang, S.Q.; Sun, C.R.; Wang, M.; Wang, B.; Tang, J.W. Inhibition of JNK1 expression decreases migration and invasion of mouse hepatocellular carcinoma cell line in vitro. Med. Oncol. 2011, 28, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Um, H.-D. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: A review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 2016, 7, 5193–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, X.; Zhang, Q.; Shi, X.; Xu, G.; Zou, C. Cancer-associated fibroblasts secrete FGF-1 to promote ovarian proliferation, migration, and invasion through the activation of FGF-1/FGFR4 signaling. Tumor Biol. 2017, 39, 101042831771259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubici, C.; Papa, S. JNK signalling in cancer: In need of new, smarter therapeutic targets. Br. J. Pharmacol. 2014, 171, 24–37. [Google Scholar] [CrossRef] [PubMed]
- García-Gutiérrez, L.; McKenna, S.; Kolch, W.; Matallanas, D. RASSF1A tumour suppressor: Target the network for effective cancer therapy. Cancers 2020, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.J.F.; Marchi, S.; Petroni, G.; Kroemer, G.; Galluzzi, L.; Pervaiz, S. Noncanonical cell fate regulation by Bcl-2 proteins. Trends Cell Biol. 2020, 30, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, H.; Qin, C.; Sun, H.; Han, Y. Up-regulation of CXCL8 expression is associated with a poor prognosis and enhances tumor cell malignant behaviors in liver cancer. Biosci. Rep. 2020, 40, BSR20201169. [Google Scholar] [CrossRef]
- Francisco, J.; Byun, J.; Zhang, Y.; Kalloo, O.B.; Mizushima, W.; Oka, S.; Zhai, P.; Sadoshima, J.; Del Re, D.P. The tumor suppressor RASSF1A modulates inflammation and injury in the reperfused murine myocardium. J. Biol. Chem. 2019, 294, 13131–13144. [Google Scholar] [CrossRef]
- Wang, C.; Ke, Y.; Liu, S.; Pan, S.; Liu, Z.; Zhang, H.; Fan, Z.; Zhou, C.; Liu, J.; Wang, F. Ectopic fibroblast growth factor receptor 1 promotes inflammation by promoting nuclear factor-κB signaling in prostate cancer cells. J. Biol. Chem. 2018, 293, 14839–14849. [Google Scholar] [CrossRef] [Green Version]
- Hammouda, M.; Ford, A.; Liu, Y.; Zhang, J. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Kwon, A.; Lee, G.B.; Park, T.; Lee, J.H.; Ko, P.; You, E.; Ahn, J.H.; Eom, S.H.; Rhee, S.; Song, W.K. Potent small-molecule inhibitors targeting acetylated microtubules as anticancer agents against triple-negative breast cancer. Biomedicines 2020, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Osanai, M.; Lee, G.H. The retinoic acid-metabolizing enzyme CYP26A1 upregulates fascin and promotes the malignant behavior of breast carcinoma cells. Oncol. Rep. 2015, 34, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, P.; Choi, J.-H.; Song, S.; Keum, S.; Jeong, J.; Hwang, Y.E.; Kim, J.W.; Rhee, S. Microtubule Acetylation Controls MDA-MB-231 Breast Cancer Cell Invasion through the Modulation of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2021, 22, 6018. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116018
Ko P, Choi J-H, Song S, Keum S, Jeong J, Hwang YE, Kim JW, Rhee S. Microtubule Acetylation Controls MDA-MB-231 Breast Cancer Cell Invasion through the Modulation of Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2021; 22(11):6018. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116018
Chicago/Turabian StyleKo, Panseon, Jee-Hye Choi, Seongeun Song, Seula Keum, Jangho Jeong, Ye Eun Hwang, Jung Woong Kim, and Sangmyung Rhee. 2021. "Microtubule Acetylation Controls MDA-MB-231 Breast Cancer Cell Invasion through the Modulation of Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 22, no. 11: 6018. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116018