
V600EBRAF Inhibition Induces Cytoprotective Autophagy through AMPK in Thyroid Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
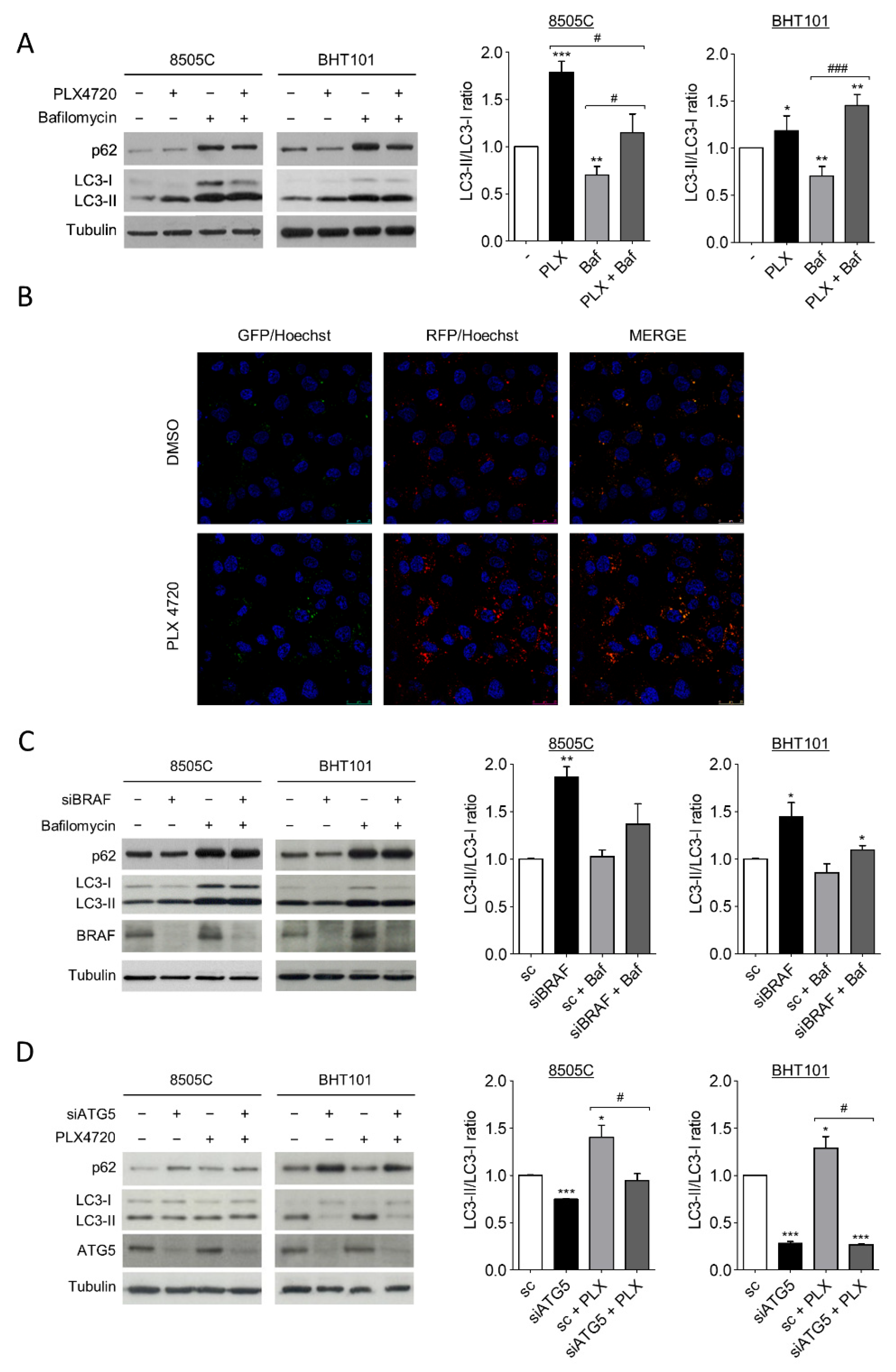
2.1. V600EBRAF Inhibition Induces Autophagy in Thyroid Cancer Cells
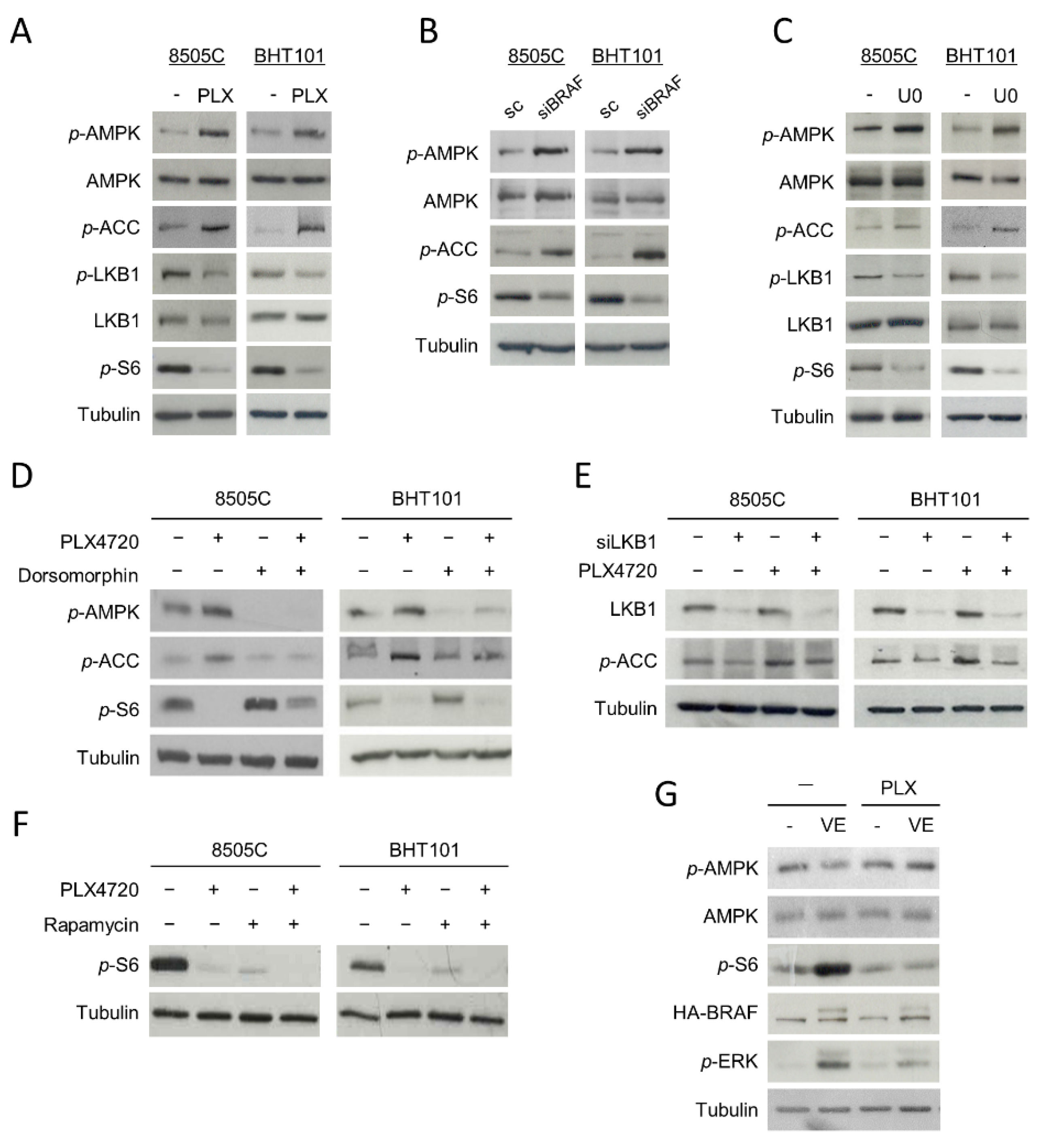
2.2. Inhibition of V600EBRAF Activates the AMPK Pathway in Thyroid Cancer Cells
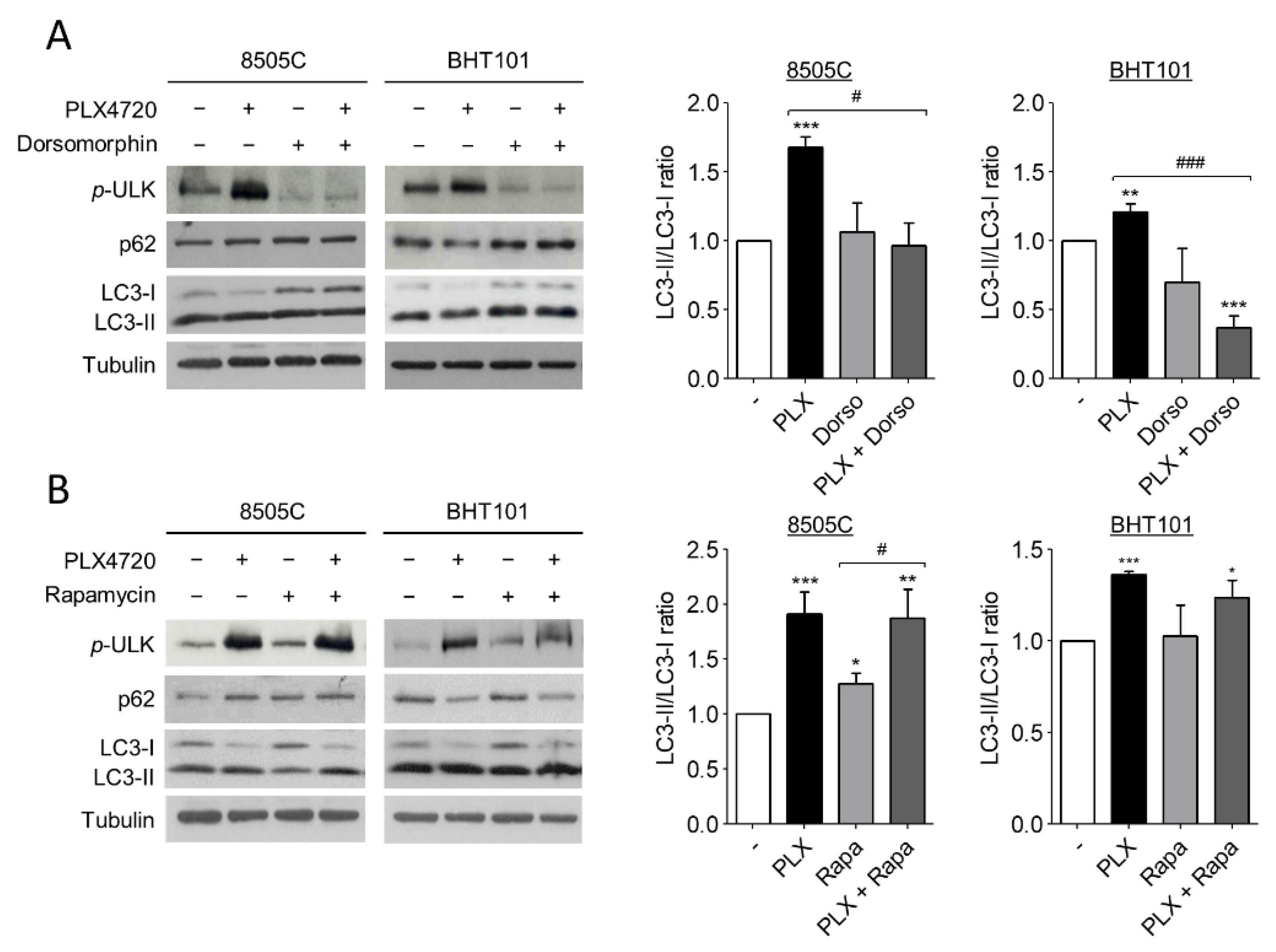
2.3. V600EBRAF Inhibition Induces Autophagy through AMPK-ULK1 Activation in Thyroid Cancer Cells
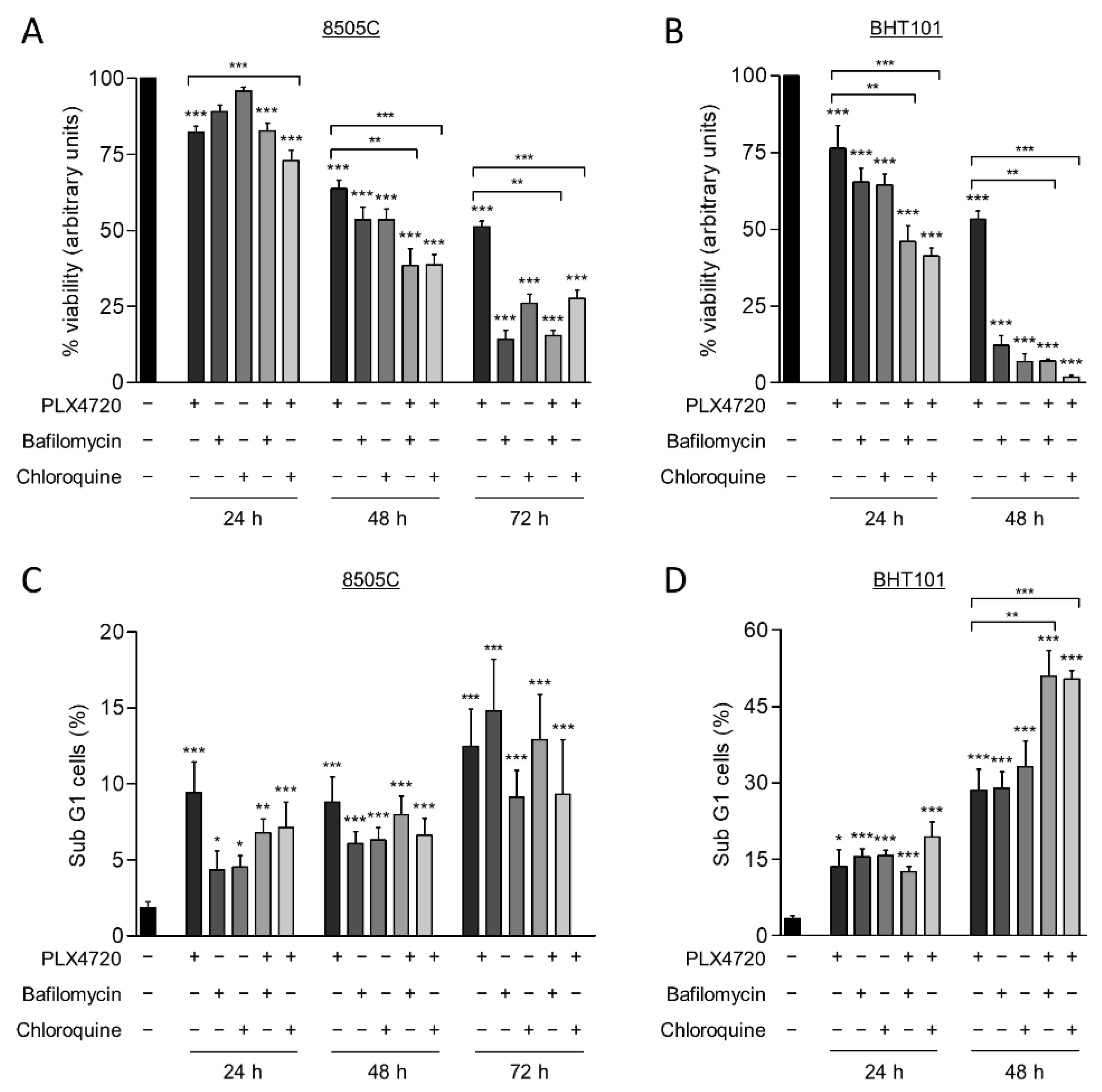
2.4. Autophagy Blockage Decreases Cell Viability and Sensitizes Thyroid Cancer Cells to V600EBRAF Inhibition
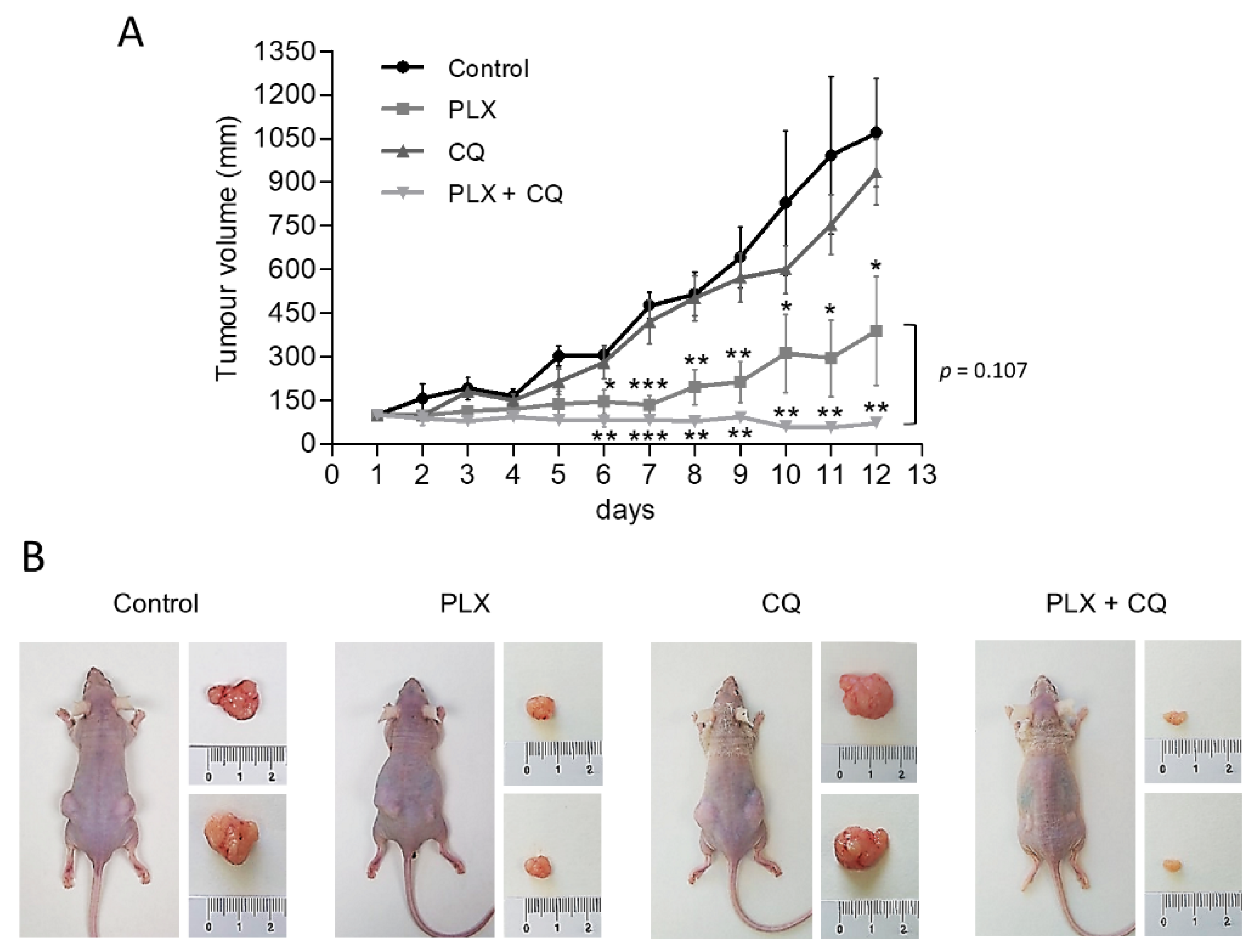
2.5. Targeting Autophagy Enhances Antitumor Effect of PLX4720 In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines, In Vitro Treatments, Plasmids, siRNA and Transfections
4.2. Cell Lysis and Western Blot Analysis
4.3. Immunofluorescence Assays
4.4. Cell Viability Assays
4.5. Quantification of Sub-G1 DNA Content by Flow Cytometry
4.6. Xenograft Models
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, Y.; Li, H.; Wang, M.; Li, N.; Tian, T.; Wu, Y.; Xu, P.; Yang, S.; Zhai, Z.; Zhou, L.; et al. Global Burden of Thyroid Cancer From 1990 to 2017. JAMA Netw. Open 2020, 3, e208759. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, R.Y.; Kloppel, G.; Rosai, J. WHO classification of tumours of endocrine organs. In WHO Classification of Tumours, 4th ed.; IARC Publications: Lyon, France, 2017; Volume 10. [Google Scholar] [CrossRef]
- Soares, P.; Sobrinho-Simões, M. Cancer: Small papillary thyroid cancers-is BRAF of prognostic value? Nat. Rev. Endocrinol. 2011, 7, 9–10. [Google Scholar] [CrossRef]
- Sherman, S.I.; Angelos, P.; Ball, D.W.; Byrd, D.; Clark, O.H.; Daniels, G.H.; Dilawari, R.A.; Ehya, H.; Farrar, W.B.; Gagel, R.F.; et al. National Comprehensive Cancer Network Thyroid Carcinoma Panel. Thyroid carcinoma. J. Natl. Compr. Cancer Netw. 2010, 8, 1228–1274. [Google Scholar] [CrossRef] [Green Version]
- Kazaure, H.S.; Roman, S.A.; Sosa, J.A. Aggressive variants of papillary thyroid cancer: Incidence, characteristics and predictors of survival among 43,738 patients. Ann. Surg. Oncol. 2012, 19, 1874–1880. [Google Scholar] [CrossRef]
- Nagaiah, G.; Hossain, A.; Mooney, C.J.; Parmentier, J.; Remick, S.C. Anaplastic thyroid cancer: A review of epidemiology, pathogenesis, and treatment. J. Oncol. 2011, 2011. [Google Scholar] [CrossRef]
- Tiedje, V.; Stuschke, M.; Weber, F.; Dralle, H.; Moss, L.; Führer, D. Anaplastic thyroid carcinoma: Review of treatment protocols. Endocr. Relat. Cancer. 2018, 25, R153–R161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, M. BRAF mutation in papillary thyroid cancer: Pathogenic role, molecular bases, and clinical implications. Endocr. Rev. 2007, 28, 742–762. [Google Scholar] [CrossRef] [PubMed]
- Riesco-Eizaguirre, G.; Rodriguez, I.; De la Vieja, A.; Costamagna, E.; Carrasco, N.; Nistal, M.; Santisteban, P. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 2009, 69, 8317–8325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauf, J.A.; Ma, X.; Smith, E.P.; Zhang, L.; Mitsutake, N.; Liao, X.H.; Refetoff, S.; Nikiforov, Y.E.; Fagin, J.A. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res. 2005, 65, 4238–4245. [Google Scholar] [CrossRef] [Green Version]
- Baquero, P.; Sánchez-Hernández, I.; Jiménez-Mora, E.; Orgaz, J.L.; Jiménez, B.; Chiloeches, A. (V600E)BRAF promotes invasiveness of thyroid cancer cells by decreasing E-cadherin expression through a Snail-dependent mechanism. Cancer Lett. 2013, 335, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baquero, P.; Jiménez-Mora, E.; Santos, A.; Lasa, M.; Chiloeches, A. TGFβ induces epithelial-mesenchymal transition of thyroid cancer cells by both the BRAF/MEK/ERK and Src/FAK pathways. Mol. Carcinog. 2016, 55, 1639–1654. [Google Scholar] [CrossRef]
- Nucera, C.; Nehs, M.A.; Nagarkatti, S.S.; Sadow, P.M.; Mekel, M.; Fischer, A.H.; Lin, P.S.; Bollag, G.E.; Lawler, J.; Hodin, R.A.; et al. Targeting BRAFV600E with PLX4720 displays potent antimigratory and anti-invasive activity in preclinical models of human thyroid cancer. Oncologist 2011, 16, 296–309. [Google Scholar] [CrossRef]
- Xing, M.; Alzahrani, A.S.; Carson, K.A.; Shong, Y.K.; Kim, T.Y.; Viola, D.; Elisei, R.; Bendlová, B.; Yip, L.; Mian, C.; et al. Association between BRAFV600E mutation and recurrence of papillary thyroid cancer. J. Clin. Oncol. 2015, 33, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Laorden, B.; Viros, A.; Girotti, M.R.; Pedersen, M.; Saturno, G.; Zambon, A.; Niculescu-Duvaz, D.; Turajlic, S.; Hayes, A.; Gore, M.; et al. BRAF Inhibitors Induce Metastasis in RAS Mutant or Inhibitor-Resistant Melanoma Cells by Reactivating MEK and ERK Signaling. Sci. Signal. 2014, 7, ra30. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofir Dovrat, T.; Sokol, E.; Frampton, G.; Shachar, E.; Pelles, S.; Geva, R.; Wolf, I. Unusually long-term responses to vemurafenib in BRAF V600E mutated colon and thyroid cancers followed by the development of rare RAS activating mutations. Cancer Biol. Ther. 2018, 19, 871–874. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Dadu, R.; Iyer, P.; Wanland, K.B.; Busaidy, N.L.; Ying, A.; Gule-Monroe, M.; Wang, J.R.; Zafereo, M.; Hofmann, M.C. Acquired Secondary RAS Mutation in BRAF V600E-Mutated Thyroid Cancer Patients Treated with BRAF Inhibitors. Thyroid 2020, 30, 1288–1296. [Google Scholar] [CrossRef]
- Roelli, M.A.; Ruffieux-Daidié, D.; Stooss, A.; ElMokh, O.; Phillips, W.A.; Dettmer, M.S.; Charles, R.P. PIK3CAH1047R-induced paradoxical ERK activation results in resistance to BRAFV600E specific inhibitors in BRAFV600E PIK3CAH1047R double mutant thyroid tumors. Oncotarget 2017, 8, 103207–103222. [Google Scholar] [CrossRef] [Green Version]
- Crispo, F.; Notarangelo, T.; Pietrafesa, M.; Lettini, G.; Storto, G.; Sgambato, A.; Maddalena, F.; Landriscina, M. BRAF Inhibitors in Thyroid Cancer: Clinical Impact, Mechanisms of Resistance and Future Perspectives. Cancers 2019, 11, 1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef] [Green Version]
- Byeon, H.K.; Na, H.J.; Yang, Y.J.; Kwon, H.J.; Chang, J.W.; Ban, M.J.; Kim, W.S.; Shin, D.Y.; Lee, E.J.; Koh, Y.W.; et al. c-Met-mediated reactivation of PI3K/AKT signaling contributes to insensitivity of BRAF(V600E) mutant thyroid cancer to BRAF inhibition. Mol. Carcinog. 2016, 55, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Knauf, J.A.; Luckett, K.A.; Chen, K.Y.; Voza, F.; Socci, N.D.; Ghossein, R.; Fagin, J.A. Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J. Clin. Investig. 2018, 128, 4086–4097. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Levin, R.S.; Gordan, J.D.; Oses-Prieto, J.A.; Webber, J.T.; Salt, M.; Hann, B.; Burlingame, A.L.; McCormick, F.; Bandyopadhyay, S.; et al. Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell. Rep. 2014, 8, 1037–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Rybstein, M.D.; Pedro, J.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef]
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, E.; Das, G.; Bergmann, A.; Baehrecke, E.H. Autophagy regulates tissue overgrowth in a context-dependent manner. Oncogene 2015, 34, 3369–3376. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer. 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebecca, V.W.; Amaravadi, R.K. Emerging strategies to effectively target autophagy in cancer. Oncogene 2016, 35, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, J.; Qin, H.; Xu, J. The Role of Autophagy in the Resistance to BRAF Inhibition in BRAF-Mutated Melanoma. Target Oncol. 2018, 13, 437–446. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Goodall, M.L.; Wang, T.; Martin, K.R.; Kortus, M.G.; Kauffman, A.L.; Trent, J.M.; Gately, S.; MacKeigan, J.P. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy 2014, 10, 1120–1136. [Google Scholar] [CrossRef] [Green Version]
- Goulielmaki, M.; Koustas, E.; Moysidou, E.; Vlassi, M.; Sasazuki, T.; Shirasawa, S.; Zografos, G.; Oikonomou, E.; Pintzas, A. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget 2016, 7, 9188–9221. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-I.; Whang, E.E.; Abramson, M.A.; Jiang, X.; Price, B.D.; Donner, D.B.; Moore, F.D.; Ruan, D.T., Jr. Autophagy: A new target for advanced papillary thyroid cancer therapy. Surgery 2009, 146, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-I.; Whang, E.E.; Donner, D.B.; Du, J.; Lorch, J.; He, F.; Jiang, X.; Price, B.D.; Moore, F.D., Jr.; Ruan, D.T. Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through Met inhibition in papillary thyroid cancer. Mol. Cancer Res. 2010, 8, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.M.; Jang, H.W.; Sohn, S.Y.; Kim, N.K.; Joung, J.Y.; Cho, Y.Y.; Kim, S.W.; Chung, J.H. Role of autophagy in the resistance to tumour necrosis factor-related apoptosis-inducing ligand-induced apoptosis in papillary and anaplastic thyroid cancer cells. Endocrine 2014, 45, 256–262. [Google Scholar] [CrossRef]
- Wang, W.; Kang, H.; Zhao, Y.; Min, I.; Wyrwas, B.; Moore, M.; Teng, L.; Zarnegar, R.; Jiang, X.; Fahey, T.J., 3rd. Targeting autophagy sensitizes BRAF-mutant thyroid cancer to vemurafenib. J. Clin. Endocrinol. Metab. 2017, 102, 634–643. [Google Scholar] [CrossRef]
- Sueda, T.; Sakai, D.; Kawamoto, K.; Konno, M.; Nishida, N.; Koseki, J.; Colvin, H.; Takahashi, H.; Haraguchi, N.; Nishimura, J.; et al. BRAF V600E inhibition stimulates AMP-activated protein kinase-mediated autophagy in colorectal cancer cells. Sci. Rep. 2016, 6, 18949. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Long, M.; Li, X.; Li, L.; Dodson, M.; Zhang, D.D.; Zheng, H. Multifunctional p62 Effects underlie diverse metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 818–830. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Zheng, B.; Jeong, J.H.; Asara, J.M.; Yuan, Y.; Granter, S.R.; Chin, L.; Cantley, L.C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell. 2009, 33, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk: Implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yin, L.; Song, G.; Han, X.; Yin, Z.; Luo, D. LKB1 loss cooperating with BRAF(V600E) promotes melanoma cell invasion and migration by up-regulation MMP-2 via PI3K/Akt/mTOR pathway. Oncotarget 2017, 8, 113847–113857. [Google Scholar] [CrossRef] [Green Version]
- Mack, H.I.D.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef] [Green Version]
- Sanduja, S.; Feng, Y.; Mathis, R.A.; Sokol, E.S.; Reinhardt, F.; Halaban, R.; Gupta, P.B. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene 2016, 35, 5295–5303. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of Braf(V600E)-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddodi, N.; Huang, W.; Havighurst, T.; Kim, K.; Longley, B.J.; Setaluri, V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J. Invest. Dermatol. 2010, 130, 1657–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cheng, X.; Gao, Y.; Zheng, J.; Xu, Q.; Sun, Y.; Guan, H.; Yu, H.; Sun, Z. Apigenin induces autophagic cell death in human papillary thyroid carcinoma BCPAP cells. Food Funct. 2015, 6, 3464–3472. [Google Scholar] [CrossRef]
- Yi, H.; Ye, T.; Ge, M.; Yang, M.; Zhang, L.; Jin, S.; Ye, X.; Long, B.; Li, L. Inhibition of autophagy enhances the targeted therapeutic effect of sorafenib in thyroid cancer. Oncol Rep. 2018, 39, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Cheng, X.; Kuang, J.; Chen, L.; Yuen, S.; Shi, M.; Liang, J.; Shen, B.; Jin, Z.; Yan, J.; et al. Apatinib-induced protective autophagy and apoptosis through the AKT-mTOR pathway in anaplastic thyroid cancer. Cell Death Dis. 2018, 9, 1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Liu, W.; Sha, S.; Fan, S.; Yu, Y.; Chen, L.; Dong, M. Adenosine 5’-monophosphate-activated protein kinase-dependent mTOR pathway is involved in flavokawain B-induced autophagy in thyroid cancer cells. Cancer Sci. 2018, 109, 2576–2589. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Mora, E.; Gallego, B.; Díaz-Gago, S.; Lasa, M.; Baquero, P.; Chiloeches, A. V600EBRAF Inhibition Induces Cytoprotective Autophagy through AMPK in Thyroid Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6033. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116033
Jiménez-Mora E, Gallego B, Díaz-Gago S, Lasa M, Baquero P, Chiloeches A. V600EBRAF Inhibition Induces Cytoprotective Autophagy through AMPK in Thyroid Cancer Cells. International Journal of Molecular Sciences. 2021; 22(11):6033. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116033
Chicago/Turabian StyleJiménez-Mora, Eva, Beatriz Gallego, Sergio Díaz-Gago, Marina Lasa, Pablo Baquero, and Antonio Chiloeches. 2021. "V600EBRAF Inhibition Induces Cytoprotective Autophagy through AMPK in Thyroid Cancer Cells" International Journal of Molecular Sciences 22, no. 11: 6033. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116033