
Satellitome Analysis of Rhodnius prolixus, One of the Main Chagas Disease Vector Species

 , , and
, , and
Abstract
:1. Introduction
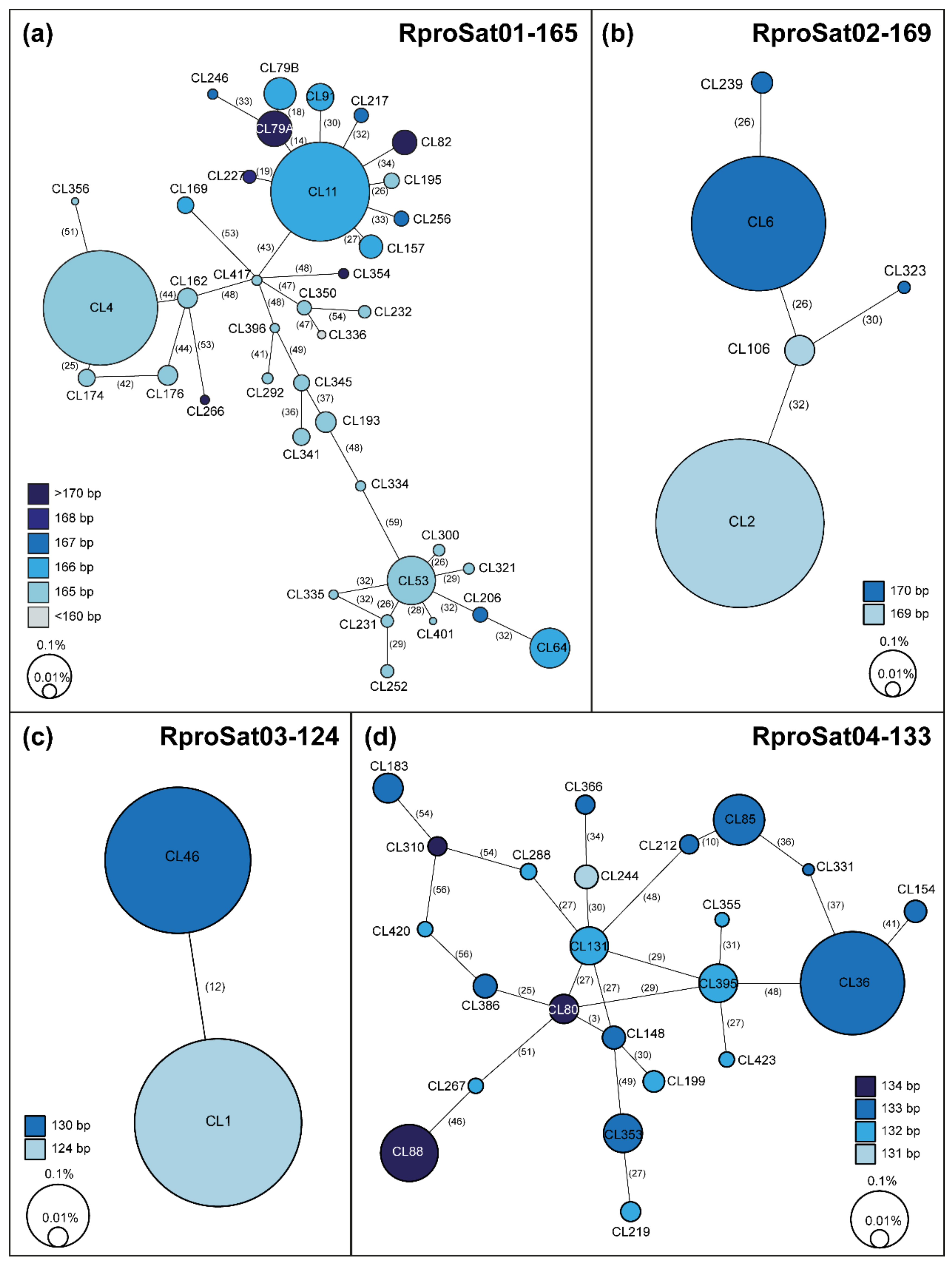
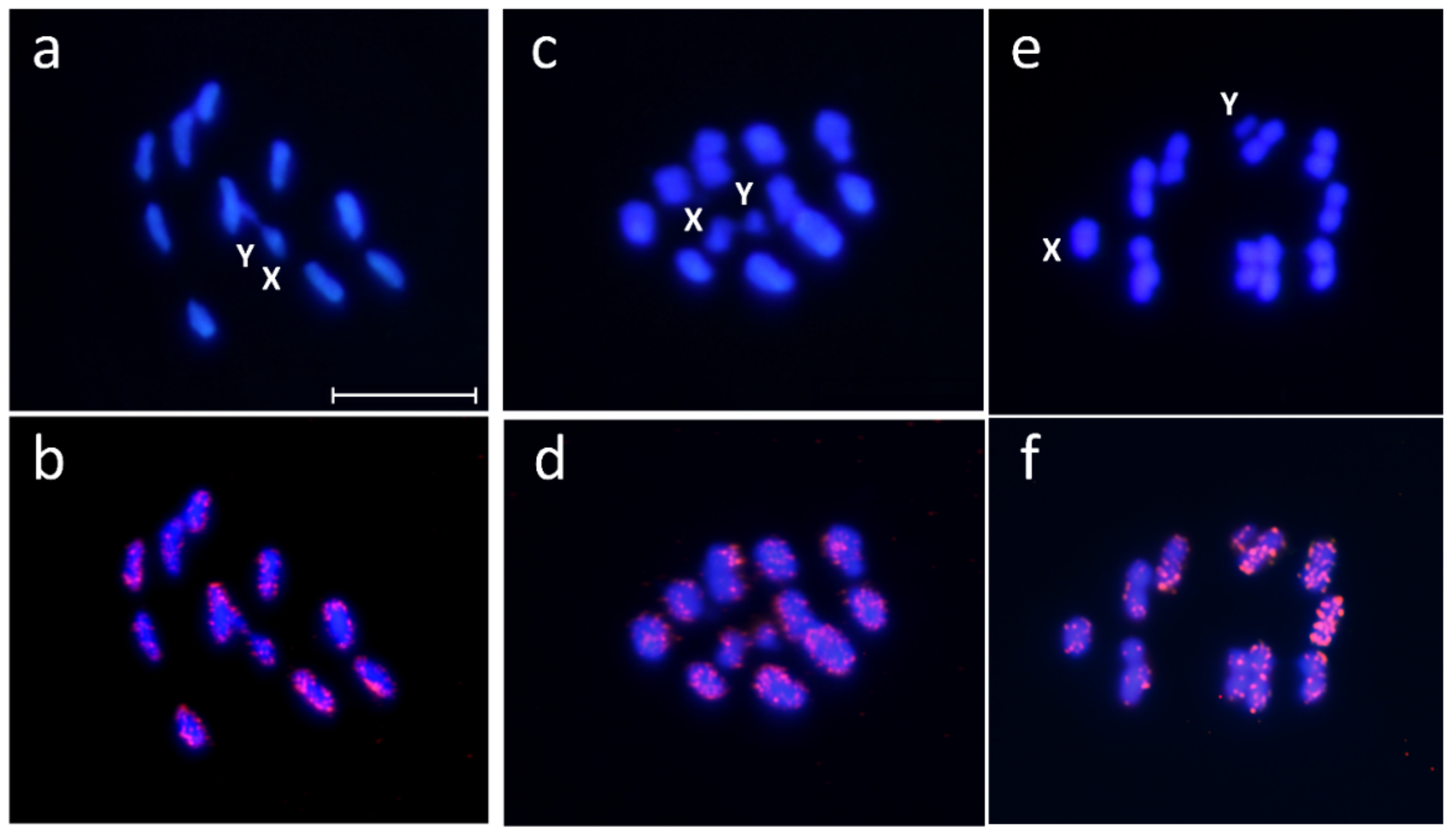
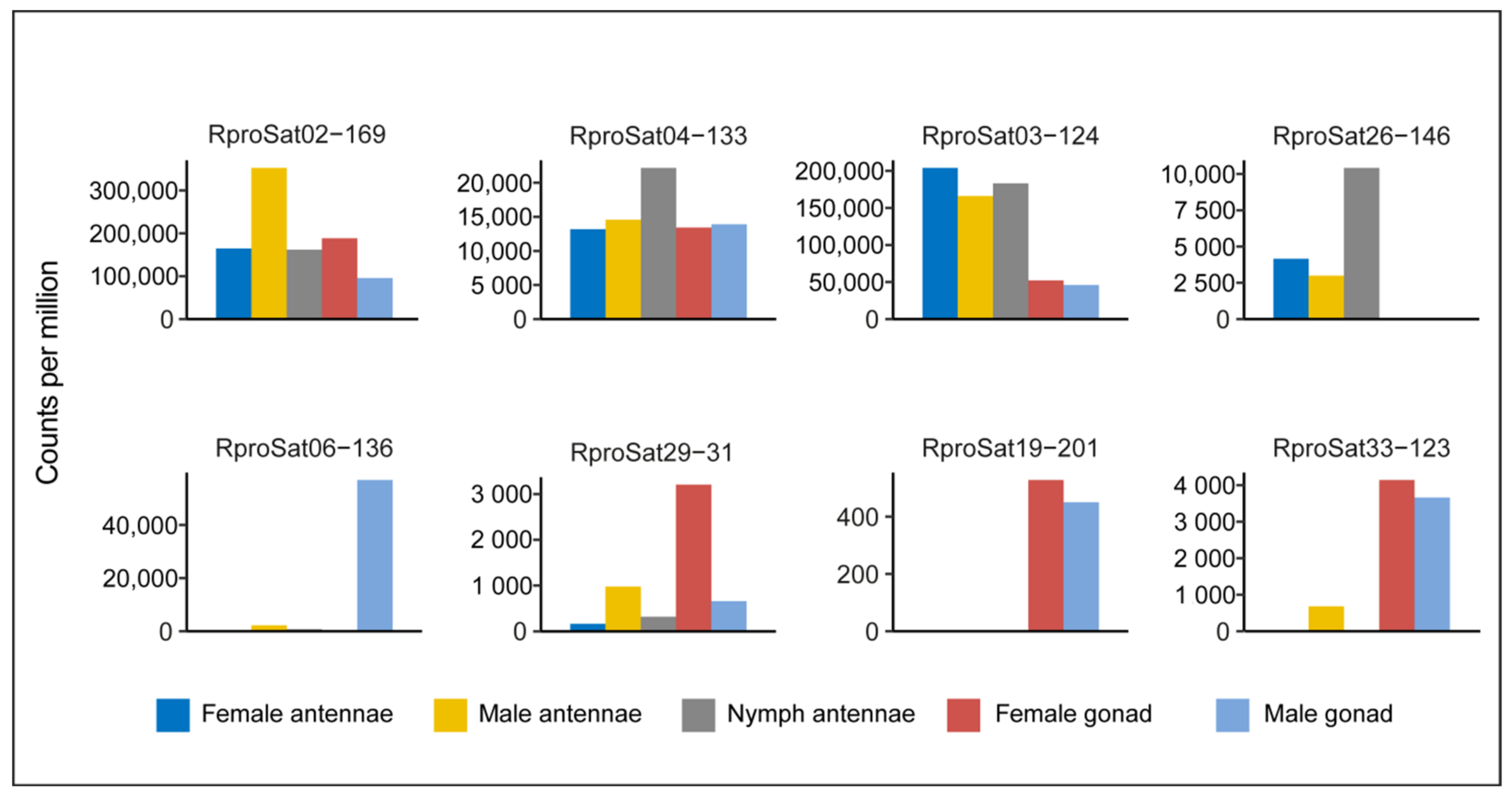
2. Results and Discussion
3. Materials and Methods
3.1. Samples, DNA Extraction and Chromosome Preparation
3.2. DNA Sequencing and Graph-Based Clustering of Sequencing Reads
3.3. Transcription of Satellite DNA
3.4. Cytogenetic Mapping
3.5. Rhodnius Prolixus Genome Assembly satDNA Families Searches
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mesquita, R.D.; Vionette-Amaral, R.J.; Lowenberger, C.; Rivera-Pomar, R.; Monteiro, F.A.; Minx, P.; Spieth, J.; Carvalho, A.B.; Panzera, F.; Lawson, D.; et al. Genome of Rhodnius prolixus, an insect vector of Chagas disease, reveals unique adaptations to hematophagy and parasite infection. Proc. Natl. Acad. Sci. USA 2015, 112, 14936–14941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzera, F.; Pérez, R.; Panzera, Y.; Ferrandis, I.; Ferreiro, M.J.; Calleros, L. Cytogenetics and genome evolution in the subfamily Triatominae (Hemiptera, Reduviidae). Cytogenet. Genome Res. 2010, 128, 77–87. [Google Scholar] [CrossRef]
- Maumus, F.; Quesneville, H. Ancestral repeats have shaped epigenome and genome composition for millions of years in Arabidopsis thaliana. Nat. Commun. 2014, 5, 4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, M.R.J.; Goubert, C.; Monteiro, F.A.; Vieira, C.; Carareto, C.M.A. Homology-free detection of transposable elements unveils their dynamics in three ecologically distinct Rhodnius species. Genes 2020, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Goubert, C.; Modolo, L.; Vieira, C.; ValienteMoro, C.; Mavingui, P.; Boulesteix, M. De novo assembly and annotation of the Asian tiger mosquito (Aedes albopictus) repeatome with dnaPipeTE from raw genomic reads and comparative analysis with the yellow fever mosquito (Aedes aegypti). Genome Biol. Evol. 2015, 7, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, F.A.; Weirauch, C.; Felix, M.; Lazoski, C.; Abad-Franch, F. Chapter five—Evolution, systematics, and biogeography of the Triatominae, vectors of Chagas disease. In Advances in Parasitology; Rollinson, D., Stothard, J.R., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 99, pp. 265–344. [Google Scholar] [CrossRef]
- Fernández-Medina, R.D.; Granzotto, A.; Ribeiro, J.M.; Carareto, C.M.A. Transposition burst of mariner-like elements in the sequenced genome of Rhodnius prolixus. Insect Biochem. Mol. Biol. 2016, 69, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. Satellite DNA: An evolving topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: Old ideas, new approaches. Curr. Opin. Genet. Dev. 2018, 48, 70–78. [Google Scholar] [CrossRef]
- Ruíz-Ruano, F.J.; Castillo-Martínez, J.; Cabrero, J.; Gómez, R.; Camacho, J.P.M.; López-León, M.D. High-throughput analysis of satellite DNA in the grasshopper Pyrgomorpha conica reveals abundance of homologous and heterologous higher-order repeats. Chromosoma 2018, 127, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Mora, P.; Vela, J.; Palomeque, T.; Sánchez, A.; Panzera, F.; Lorite, P. Comparative Analysis of repetitive DNA between the main vectors of Chagas disease: Triatoma infestans and Rhodnius prolixus. Int. J. Mol. Sci. 2018, 19, 1277. [Google Scholar] [CrossRef] [Green Version]
- Bardella, V.B.; da Rosa, J.A.; Vanzela, A.L.L. Origin and distribution of AT-rich repetitive DNA families in Triatoma infestans (Heteroptera). Infect. Genet. Evol. 2014, 23, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pita, S.; Panzera, F.; Mora, P.; Vela, J.; Cuadrado, A.; Sánchez, A.; Palomeque, T.; Lorite, P. Comparative repeatome analysis on Triatoma infestans Andean and Non-Andean lineages, main vector of Chagas disease. PLoS ONE 2017, 12, e0181635. [Google Scholar] [CrossRef] [Green Version]
- Ruíz-Ruano, F.J.; López-León, M.D.; Cabrero, J.; Camacho, J.P.M. High-throughput analysis of the satellitome illuminates satellite DNA evolution. Sci. Rep. 2016, 6, 28333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardella, V.B.; Milani, D.; Cabral-de-Mello, D.C. Analysis of Holhymenia histrio genome provides insight into the satDNA evolution in an insect with holocentric chromosomes. Chromosome Res. 2020, 28, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Ávila Robledillo, L.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef]
- Ruíz-Ruano, F.J.; Cabrero, J.; López-León, M.D.; Camacho, J.P.M. Satellite DNA content illuminates the ancestry of a supernumerary (B) chromosome. Chromosoma 2017, 126, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Gimenez, O.M.; Bardella, V.B.; Lemos, B.; Cabral-de-Mello, D.C. Satellite DNAs are conserved and differentially transcribed among Gryllus cricket species. DNA Res. 2018, 25, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Silva, B.S.M.L.; Heringer, P.; Dias, G.B.; Svartman, M.; Kuhn, G.C.S. De novo identification of satellite DNAs in the sequenced genomes of Drosophila virilis and D. americana using the RepeatExplorer and TAREAN pipelines. PLoS ONE 2019, 14, e0223466. [Google Scholar] [CrossRef] [Green Version]
- Mora, P.; Vela, J.; Ruíz-Ruano, F.J.; Ruiz-Mena, A.; Montiel, E.E.; Palomeque, T.; Lorite, P. Satellitome analysis in the ladybird beetle Hippodamia variegata (Coleoptera, Coccinellidae). Genes 2020, 11, 783. [Google Scholar] [CrossRef]
- Utsunomia, R.; Ruíz-Ruano, F.J.; Silva, D.M.Z.A.; Serrano, É.A.; Rosa, I.F.; Scudeler, P.E.S.; Hashimoto, D.T.; Oliveira, C.; Camacho, J.P.M.; Foresti, F. A Glimpse into the Satellite DNA Library in Characidae Fish (Teleostei, Characiformes). Front. Genet. 2017, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Utsunomia, R.; Silva, D.; Ruíz-Ruano, F.J.; Goes, C.; Melo, S.; Ramos, L.P.; Oliveira, C.; Porto-Foresti, F.; Foresti, F.; Hashimoto, D.T. Satellitome landscape analysis of Megaleporinus macrocephalus (Teleostei, Anostomidae) reveals intense accumulation of satellite sequences on the heteromorphic sex chromosome. Sci. Rep. 2019, 9, 5856. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, R.G.; Calegari, R.M.; Silva, D.M.Z.A.; Ruiz-Ruano, F.J.; Melo, S.; Oliveira, C.; Foresti, F.; Uliano-Silva, M.; Porto-Foresti, F.; Utsunomia, R. A Long-Term Conserved Satellite DNA That Remains Unexpanded in Several Genomes of Characiformes Fish Is Actively Transcribed. Genome Biol. Evol. 2021, 13, evab002. [Google Scholar] [CrossRef]
- Ruíz-Ruano, F.J.; Navarro-Domínguez, B.; Camacho, J.P.M.; Garrido-Ramos, M.A. Characterization of the satellitome in lower vascular plants: The case of the endangered fern Vandenboschia speciosa. Ann. Bot. 2019, 123, 587–599. [Google Scholar] [CrossRef]
- Belyayev, A.; Jandová, M.; Josefiová, J.; Kalendar, R.; Mahelka, V.; Mandák, B.; Krak, K. The major satellite DNA families of the diploid Chenopodium album aggregate species: Arguments for and against the “library hypothesis”. PLoS ONE 2020, 15, e0241206. [Google Scholar] [CrossRef]
- Sader, M.; Vaio, M.; Cauz-Santos, L.A.; Dornelas, M.C.; Vieira, M.L.C.; Melo, N.; Pedrosa-Harand, A. Large vs small genomes in Passiflora: The influence of the mobilome and the satellitome. Planta 2021, 253, 86. [Google Scholar] [CrossRef]
- Heitkam, T.; Schulte, L.; Weber, B.; Liedtke, S.; Breitenbach, S.; Kögler, A.; Morgenstern, K.; Brückner, M.; Tröber, U.; Wolf, H.; et al. Comparative repeat profiling of two closely related conifers (Larix decidua and Larix kaempferi) reveals high genome similarity with only few fast-evolving satellite DNAs. bioRxiv 2021. [Google Scholar] [CrossRef]
- Thakur, J.; Packiaraj, J.; Henikoff, S. Sequence, chromatin and evolution of satellite DNA. Int. J. Mol. Sci. 2021, 22, 4309. [Google Scholar] [CrossRef]
- Cabral-de-Mello, D.C.; Zrzavá, M.; Kubíčková, S.; Rendón, P.; Marec, F. The role of satellite DNAs in genome architecture and sex chromosome evolution in Crambidae moths. Front. Genet. 2021, 12, 661417. [Google Scholar] [CrossRef]
- Perea-Resa, C.; Blower, M.D. Satellite transcripts locally promote centromere formation. Dev. Cell 2017, 42, 201–202. [Google Scholar] [CrossRef]
- Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-Carvalho, M.; Chaves, R. Decoding the role of satellite DNA in genome architecture and plasticity-an evolutionary and clinical affair. Genes 2020, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Halbach, R.; Miesen, P.; Joosten, J.; Taşköprü, J.; Rondeel, I.; Pennings, B.; Vogels, C.B.F.; Merkling, S.H.; Koenraadt, C.J.; Lambrechts, L.; et al. A satellite repeat-derived piRNA controls embryonic development of Aedes. Nature 2020, 580, 274–277. [Google Scholar] [CrossRef]
- Mills, W.K.; Lee, Y.C.G.; Kochendoerfer, A.M.; Dunleavy, E.M.; Karpen, G.H. RNA from a simple-tandem repeat is required for sperm maturation and male fertility in Drosophila melanogaster. Elife 2019, 8, 48940. [Google Scholar] [CrossRef] [PubMed]
- Pezer, Z.; Ugarković, D. Satellite DNA-associated siRNAs as mediators of heat shock response in insects. RNA Biol. 2012, 9, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Palomeque, T.; Lorite, P. Satellite DNA in insects: A review. Heredity 2008, 100, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Dalíková, M.; Zrzavá, M.; Kubícková, S.; Marec, F. W-enriched satellite sequence in the Indian meal moth, Plodia interpunctella (Lepidoptera, Pyralidae). Chromosome Res. 2017, 25, 241–252. [Google Scholar] [CrossRef]
- Shatskikh, A.S.; Kotov, A.A.; Adashev, V.E.; Bazylev, S.S.; Olenina, L.V. Functional significance of satellite DNAs: Insights from Drosophila. Front. Cell Dev. Biol. 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- De Souza, R.C.M.; Gorla, D.E.; Chame, M.; Jaramillo, N.; Monroy, C.; Diotaiuti, L. Chagas disease in the context of the 2030 agenda: Global warming and vectors. Mem. Inst. Oswaldo Cruz 2021, 116, e200479. [Google Scholar] [CrossRef]
- Pita, S.; Panzera, F.; Sánchez, A.; Panzera, Y.; Palomeque, T.; Lorite, P. Distribution and evolution of repeated sequences in genomes of Triatominae (Hemiptera-Reduviidae) inferred from genomic in situ hybridization. PLoS ONE 2014, 9, e114298. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Lorite, P.; Vela, J.; Mora, P.; Palomeque, T.; Thi, K.P.; Panzera, F. Holocentric chromosome evolution in kissing bugs (Hemiptera-Reduviidae-Triatominae): Diversification of repeated sequences. Parasit. Vectors 2017, 10, 410. [Google Scholar] [CrossRef]
- Fry, K.; Salser, W. Nucleotide sequences of HS-alpha satellite DNA from kangaroo rat Dipodomys ordii and characterization of similar sequences in other rodents. Cell 1977, 12, 1069–1084. [Google Scholar] [CrossRef]
- Mestrović, N.; Plohl, M.; Mravinac, B.; Ugarković, D. Evolution of satellite DNAs from the genus Palorus—Experimental evidence for the “library” hypothesis. Mol. Biol. Evol. 1998, 15, 1062–1068. [Google Scholar] [CrossRef]
- De Paula, A.S.; Barreto, C.; Telmo, M.C.M.; Diotaiuti, L.; Galvão, C. Historical biogeography and the evolution of hematophagy in Rhodniini (Heteroptera: Reduviidae: Triatominae). Front. Ecol. Evol. 2021, 9, 660151. [Google Scholar] [CrossRef]
- Bachmann, L.; Sperlich, D. Gradual evolution of a specific satellite DNA family in Drosophila ambigua, D. tristis, and D. obscura. Mol. Biol. Evol. 1993, 10, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Palacios-Gimenez, O.M.; Milani, D.; Song, H.; Marti, D.A.; López-León, M.D.; Ruíz-Ruano, F.J.; Camacho, J.P.M.; Cabral-de-Mello, D.C. Eight million years of satellite DNA evolution in grasshoppers of the genus Schistocerca illuminate the ins and outs of the library hypothesis. Genome Biol. Evol. 2020, 12, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Lorite, P.; Muñoz-López, M.; Carrillo, J.; Sanllorente, O.; Vela, J.; Mora, P.; Tinaut, A.; Torres, M.I.; Palomeque, T. Concerted evolution, a slow process for ant satellite DNA: Study of the satellite DNA in the Aphaenogaster genus (Hymenoptera, Formicidae). Org. Divers. Evol. 2017, 17, 595–606. [Google Scholar] [CrossRef]
- Abad, J.P.; Carmena, M.; Baars, S.; Saunders, R.D.; Glover, D.M.; Ludeña, P.; Sentis, C.; Tyler-Smith, C.; Villasante, A. Dodeca satellite: A conserved G+C- rich satellite from the centromeric heterochromatin of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1992, 89, 4663–4667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šatović, E.; Plohl, M. Distribution of DTHS3 satellite DNA across 12 bivalve species. J. Genet. 2018, 97, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Panzera, F.; Mora, P.; Vela, J.; Palomeque, T.; Lorite, P. The presence of the ancestral insect telomeric motif in kissing bugs (Triatominae) rules out the hypothesis of its loss in evolutionarily advanced Heteroptera (Cimicomorpha). Comp. Cytogenet. 2016, 10, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Panzera, F.; Dujardin, J.P.; Nicolini, P.; Caraccio, M.N.; Rose, V.; Tellez, T.; Bermúdez, H.; Bargues, M.D.; Mas Coma, S.; O’Connor, J.E.; et al. Genomic changes of Chagas disease vector, South America. Emerg. Infect. Dis. 2004, 10, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Panzera, F.; Ferreiro, M.J.; Pita, S.; Calleros, L.; Pérez, R.; Basmadjián, Y.; Guevara, Y.; Breniére, S.F.; Panzera, Y. Evolutionary and dispersal history of Triatoma infestans, main vector of Chagas disease, by chromosomal markers. Infect. Genet. Evol. 2014, 27, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lorite, P.; Carrillo, J.A.; Aguilar, J.A.; Palomeque, T. Isolation and characterization of two families of satellite DNA with repetitive units of 135 bp and 2.5 kb in the ant Monomorium subopacum (Hymenoptera, Formicidae). Cytogen. Genome Res. 2004, 105, 83–92. [Google Scholar] [CrossRef]
- Wang, S.; Lorenzen, M.D.; Beeman, R.W.; Brown, S.J. Analysis of repetitive DNA distribution patterns in the Tribolium castaneum genome. Genome Biol. 2008, 9, R61. [Google Scholar] [CrossRef] [Green Version]
- Brito, R.N.; Souza, R.C.M.; Abad-Franch, F. Dehydration-stress resistance in two sister, cryptic Rhodnius species—Rhodnius prolixus and Rhodnius robustus genotype I (Hemiptera: Reduviidae). J. Med. Entomol. 2019, 56, 1019–1026. [Google Scholar] [CrossRef]
- Ferreira, D.; Escudeiro, A.; Adega, F.; Chaves, R. DNA Methylation patterns of a satellite non-coding sequence—FA-SAT in cancer cells: Its expression cannot be explained solely by DNA methylation. Front. Genet. 2019, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Pezer, Z.; Ugarković, D. Transcription of pericentromeric heterochromatin in beetles—Satellite DNAs as active regulatory elements. Cytogenet. Genome Res. 2009, 124, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Feliciello, I.; Akrap, I.; Ugarković, D. Satellite DNA modulates gene expression in the beetle Tribolium castaneum after heat stress. PLoS Genet. 2015, 11, e1005466. [Google Scholar] [CrossRef] [Green Version]
- Mora, P.; Vela, J.; Ruiz-Mena, A.; Palomeque, T.; Lorite, P. Characterization and transcriptional analysis of a subtelomeric satellite DNA family in the ladybird beetle Henosepilachna argus (Coleoptera: Coccinellidae). Eur. J. Entomol. 2017, 114, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Rouleux-Bonnin, F.; Renault, S.; Bigot, Y.; Periquet, G. Transcription of four satellite DNA subfamilies in Diprion pini (Hymenoptera, Symphyta, Diprionidae). Eur. J. Biochem. 1996, 238, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Renault, S.; Rouleux-Bonnin, F.; Periquet, G.; Bigot, Y. Satellite DNA transcription in Diadromus pulchellus (Hymenoptera). Insect Biochem. Mol. Biol. 1999, 29, 103–111. [Google Scholar] [CrossRef]
- Lorite, P.; Renault, S.; Rouleux-Bonnin, F.; Bigot, S.; Periquet, G.; Palomeque, T. Genomic organization and transcription of satellite DNA in the ant Aphaenogaster subterranea (Hymenoptera, Formicidae). Genome 2002, 45, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Věchtová, P.; Dalíková, M.; Sýkorová, M.; Žurovcová, M.; Füssy, Z.; Zrzavá, M. CpSAT-1, a transcribed satellite sequence from the codling moth, Cydia pomonella. Genetica 2016, 144, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.G.; Svartman, M.; Kuhn, G.C.S. Dissecting the Satellite DNA landscape in three cactophilic Drosophila sequenced genomes. G3-Genes Genom. Genet. 2017, 7, 2831–2843. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.S.; Meller, V.H. Satellite repeats identify X chromatin for dosage compensation in Drosophila melanogaster males. Curr. Biol. 2017, 27, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. PopART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 3 May 2021).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2. WIREs Comp. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Palomeque, T.; Muñoz-López, M.; Carrillo, J.A.; Lorite, P. Characterization and evolutionary dynamics of a complex family of satellite DNA in the leaf beetle Chrysolina carnifex (Coleoptera, Chrysomelidae). Chromosome Res. 2005, 13, 795–807. [Google Scholar] [CrossRef]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [Green Version]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. gplots: Various R Programming Tools for Plotting Data. R Package Version 3.1.1. 2020. Available online: https://CRAN.R-project.org/package=gplots (accessed on 3 May 2021).
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes. R Package Version 1.1-2. 2014. Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 3 May 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | No. of RE Clusters | Genome Proportion | Repeat Unit Length (bp) | A + T Percentage | Kimura Divergence (%) |
|---|---|---|---|---|---|
| RproSat01-165 | 38 | 2.13% | 165 | 72.1% | 13.42% |
| RproSat02-169 | 5 | 1.94% | 169 | 69.2% | 9.81% |
| RproSat03-124 | 2 | 1.18% | 124 | 62.1% | 8.81% |
| RproSat04-133 | 23 | 0.861% | 133 | 66.2% | 17.38% |
| RproSat05-208 | 1 | 0.460% | 208 | 53.4% | 15.48% |
| RproSat06-136 1 | 1 | 0.320% | 136 | 62.5% | 12.42% |
| RproSat07-375 | 1 | 0.200% | 375 | 68.5% | 7.77% |
| RproSat08-67 | 1 | 0.194% | 67 | 61.2% | 20.58% |
| RproSat09-499 | 1 | 0.107% | 499 | 71.1% | 12.20% |
| RproSat10-104 | 1 | 0.085% | 104 | 62.5% | 10.10% |
| RproSat11-198 | 1 | 0.073% | 198 | 83.3% | 2.23% |
| RproSat12-41 | 1 | 0.069% | 41 | 58.5% | 10.03% |
| RproSat13-293 1 | - | 0.067% | 293 | 61.1% | 28.28% |
| RproSat14-461 | 1 | 0.062% | 461 | 64.2% | 6.19% |
| RproSat15-161 | 1 | 0.052% | 161 | 60.9% | 8.38% |
| RproSat16-821 | 1 | 0.044% | 821 | 65.3% | 6.83% |
| RproSat17-584 | 1 | 0.027% | 584 | 64.7% | 11.71% |
| RproSat18-122 | 1 | 0.019% | 122 | 63.1% | 25.27% |
| RproSat19-201 | 1 | 0.019% | 201 | 76.1% | 19.52% |
| RproSat20-134 | 1 | 0.017% | 134 | 68.7% | 16.23% |
| RproSat21-167 | 1 | 0.016% | 167 | 71.3% | 16.25% |
| RproSat22-980 | 2 | 0.013% | 980 | 69.4% | 4.75% |
| RproSat23-412 | 1 | 0.010% | 412 | 75.2% | 5.59% |
| RproSat24-673 | 1 | 0.009% | 673 | 70.0% | 2.89% |
| RproSat25-84 1 | - | 0.009% | 84 | 65.5% | 25.75% |
| RproSat26-146 | 1 | 0.009% | 146 | 63.7% | 3.55% |
| RproSat27-187 | 1 | 0.008% | 187 | 27.8% | 13.06% |
| RproSat28-199 | 1 | 0.008% | 199 | 53.8% | 12.72% |
| RproSat29-31 | 1 | 0.006% | 31 | 77.4% | 11.21% |
| RproSat30-201 | 1 | 0.006% | 201 | 61.2% | 3.02% |
| RproSat31-75 | 1 | 0.005% | 75 | 68.0% | 2.64% |
| RproSat32-59 | 1 | 0.004% | 59 | 59.3% | 0.88% |
| RproSat33-123 | 1 | 0.003% | 123 | 78.0% | 0.96% |
| RproSat34-415 | 1 | 0.003% | 415 | 72.8% | 5.15% |
| RproSat35-279 | 1 | 0.003% | 279 | 60.6% | 5.71% |
| RproSat36-40 | 1 | 0.003% | 40 | 70.0% | 4.17% |
| RproSat37-98 1 | - | 0.0005% | 98 | 69.4% | 6.96% |
| Telomeric repeat 1 | - | 0.003% | 5 | 60.0% | 13.9% |
| (GATA)n repeat 1 | - | 0.001% | 4 | 75.0% | 10.1% |
| Total | 8.05% | ||||
| Mean | 235.23 | 65.72% | 10.56% | ||
| SD | 223.13 | 9.10% | 6.91% | ||
| Median | 165 | 65.50% | 10.03% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montiel, E.E.; Panzera, F.; Palomeque, T.; Lorite, P.; Pita, S. Satellitome Analysis of Rhodnius prolixus, One of the Main Chagas Disease Vector Species. Int. J. Mol. Sci. 2021, 22, 6052. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116052
Montiel EE, Panzera F, Palomeque T, Lorite P, Pita S. Satellitome Analysis of Rhodnius prolixus, One of the Main Chagas Disease Vector Species. International Journal of Molecular Sciences. 2021; 22(11):6052. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116052
Chicago/Turabian StyleMontiel, Eugenia E., Francisco Panzera, Teresa Palomeque, Pedro Lorite, and Sebastián Pita. 2021. "Satellitome Analysis of Rhodnius prolixus, One of the Main Chagas Disease Vector Species" International Journal of Molecular Sciences 22, no. 11: 6052. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116052