
Involvement of Microglia in the Pathophysiology of Intracranial Aneurysms and Vascular Malformations—A Short Overview

 , ,
, ,
Abstract
:1. Background
2. Vasculogenesis and Angiogenesis as Viewed by Microglia
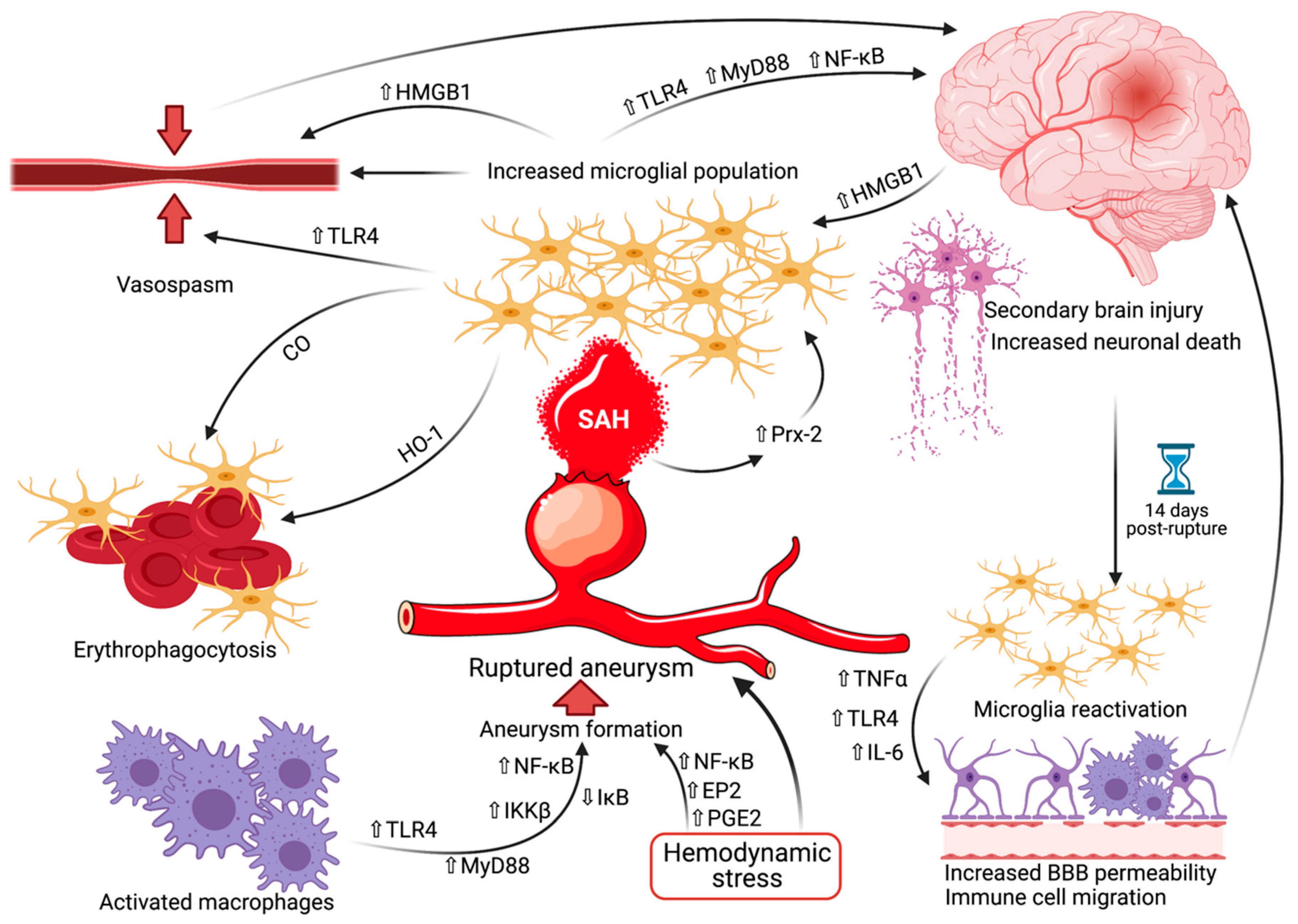
3. Birth by Fire—Are Microglia Responsible for Intracranial Aneurysm Development?
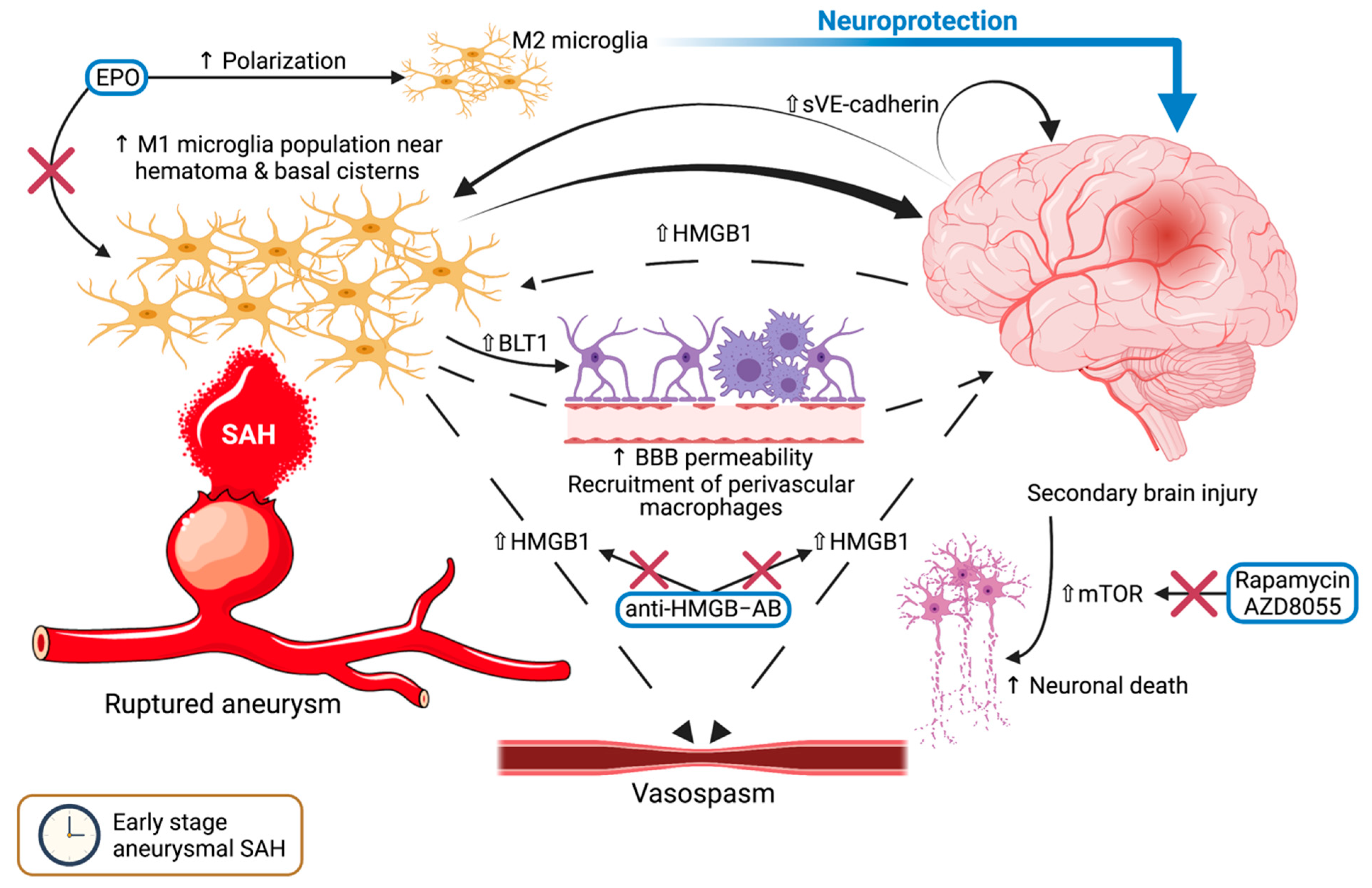
4. Microglia in the Early Stage of SAH—The Arrival of the Storm
4.1. Cerebral Inflammation after SAH
4.2. Autophagy and Neuronal Cell Death
4.3. HMGB1: A Harbinger of Vasospasm?
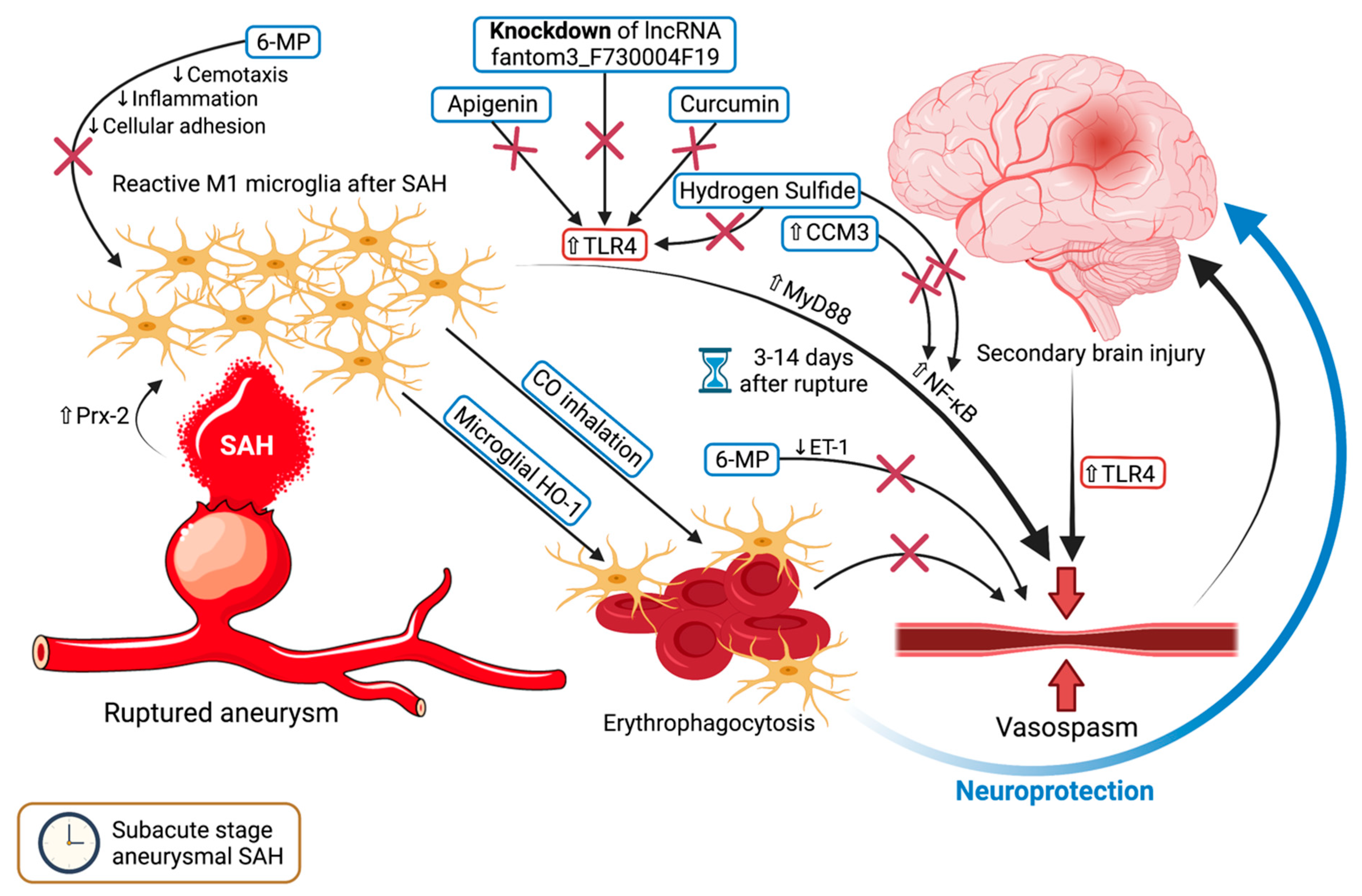
5. Microglia and Vasospasm—Rubbing Salt on the Wound
5.1. For Whom the Bell Tolls—TLR4 Upregulation Triggers Vasospasm
5.2. Damage Control—6-MP and Microglial HO-1
6. Microglia in the Delayed Stage of SAH—Far from Over
7. The Future of Microglia in SAH
8. Microglia in Arteriovenous Malformations and Cavernomas—An Ongoing Enigma
8.1. AVMs—The Evidence on Hand
8.2. CCMs—The Story so Far
9. Limitations and Future Directions
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haase, A.; Schob, S.; Quäschling, U.; Hoffmann, K.-T.; Meixensberger, J.; Nestler, U. Epidemiologic and anatomic aspects comparing incidental and ruptured intracranial aneurysms: A single centre experience. J. Clin. Neurosci. 2020, 81, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Juvela, S.; Poussa, K.; Lehto, H.; Porras, M. Natural History of unruptured Intracranial aneurysms: A long-term follow-up study. Stroke 2013, 44, 2414–2421. [Google Scholar] [CrossRef] [Green Version]
- Flemming, K.D.; Lanzino, G. Management of Unruptured Intracranial Aneurysms and Cerebrovascular Malformations. Contin. Lifelong Learn. Neurol. 2017, 23, 181–210. [Google Scholar] [CrossRef]
- Hua, X.; Gray, A.; Wolstenholme, J.; Clarke, P.; Molyneux, A.J.; Kerr, R.S.C.; Clarke, A.; Sneade, M.; Rivero-Arias, O. Survival, Dependency, and Health-Related Quality of Life in Patients with Ruptured Intracranial Aneurysm: 10-Year Follow-up of the United Kingdom Cohort of the International Subarachnoid Aneurysm Trial. Neurosurgery 2020, 88, 252–260. [Google Scholar] [CrossRef]
- Jeong, H.W.; Seo, J.H.; Kim, S.T.; Jung, C.K.; Suh, S.-I. Clinical Practice Guideline for the Management of Intracranial Aneurysms. Neurointervention 2014, 9, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saqr, K.M.; Tupin, S.; Rashad, S.; Endo, T.; Niizuma, K.; Tominaga, T.; Ohta, M. Physiologic blood flow is turbulent. Sci. Rep. 2020, 10, 15492. [Google Scholar] [CrossRef]
- Aoki, T.; Yamamoto, K.; Fukuda, M.; Shimogonya, Y.; Fukuda, S.; Narumiya, S. Sustained expression of MCP-1 by low wall shear stress loading concomitant with turbulent flow on endothelial cells of intracranial aneurysm. Acta Neuropathol. Commun. 2016, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerramilli, S.K.; Kokula, P.; Gupta, S.K.; Radotra, B.D.; Aggarwal, A.; Aggarwal, D.; Chatterjee, D. Connective Tissue Abnormalities in Patients with Ruptured Intracranial Aneurysms and No Known Systemic Connective Tissue Disorder. World Neurosurg. 2020, 141, e829–e835. [Google Scholar] [CrossRef] [PubMed]
- Heinz, R.; Brandenburg, S.; Nieminen-Kelhä, M.; Kremenetskaia, I.; Boehm-Sturm, P.; Vajkoczy, P.; Schneider, U.C. Microglia as target for anti-inflammatory approaches to prevent secondary brain injury after subarachnoid hemorrhage (SAH). J. Neuroinflamm. 2021, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.K.C.; Chen, J. Microglia accumulation and activation after subarachnoid hemorrhage. Neural Regen. Res. 2021, 16, 1531–1532. [Google Scholar] [CrossRef]
- Ruigrok, Y.M. Management of Unruptured Cerebral Aneurysms and Arteriovenous Malformations. Contin. Lifelong Learn. Neurol. 2020, 26, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Abecassis, I.J.; Xu, D.S.; Batjer, H.H.; Bendok, B.R. Natural history of brain arteriovenous malformations: A systematic review. Neurosurg. Focus 2014, 37, E7. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.; Gross, B.A.; Nakaji, P. Vascular Malformations (Arteriovenous Malformations and Dural Arteriovenous Fistulas). In Principles of Neurological Surgery, 4th ed.; Ellenbogen, R.G., Sekhar, L.N., Kitchen, N.D., Eds.; Elsevier: Philadelphia, PA, USA, 2018; Volume 20, pp. 313–324. [Google Scholar]
- Choudhri, O.; Chen, R.P.; Bulsara, K. Cavernous Malformations of the Brain and Spinal Cord. In Principles of Neurological Surgery; Ellenbogen, R.G., Sekhar, L.N., Kitchen, N.D., Eds.; Elsevier Inc.: Philadelphia, PA, USA, 2018; pp. 325–333.e2. [Google Scholar]
- Bertalanffy, H.; Florian, I.A.; Timiș, T.L. Cavernous Malformations of the Pineal Region: Overview, Management, and Controversies. In Pineal Region Lesions; Florian, I.S., Ed.; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Nasi, M.; Pinti, M.; Ferraro, D. Microglia activation: A role for mitochondrial DNA? Neural Regen. Res. 2021, 16, 2393–2394. [Google Scholar] [CrossRef]
- Zhang, J.; Boska, M.; Zheng, Y.; Liu, J.; Fox, H.S.; Xiong, H. Minocycline attenuation of rat corpus callosum abnormality mediated by low-dose lipopolysaccharide-induced microglia activation. J. Neuroinflamm. 2021, 18, 100. [Google Scholar] [CrossRef]
- Chen, Y.; Hong, T.; Chen, F.; Sun, Y.; Wang, Y.; Cui, L. Interplay Between Microglia and Alzheimer’s Disease—Focus on the Most Relevant Risks: APOE Genotype, Sex and Age. Front. Aging Neurosci. 2021, 13, 631827. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Cardona, A. The myeloid cells of the central nervous system parenchyma. Nature 2010, 468, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Wirenfeldt, M.; Babcock, A.A.; Vinters, H.V. Microglia-insights into immune system structure, function, and reactivity in the central nervous system. Histol. Histopathol. 2011, 26, 10. [Google Scholar]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, T.; Betsholtz, C. Correction: The importance of microglia in the development of the vasculature in the central nervous system. Vasc. Cell 2013, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilken, H.M.; Adams, R.H. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 2010, 22, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Rymo, S.F.; Gerhardt, H.; Sand, F.W.; Lang, R.; Uv, A.; Betsholtz, C. A Two-way communication between microglial cells and angiogenic sprouts regulates angiogenesis in aortic ring cultures. PLoS ONE 2011, 6, e15846. [Google Scholar] [CrossRef]
- Pont-Lezica, L.; Béchade, C.; Belarif-Cantaut, Y.; Pascual, O.; Bessis, A. Physiological roles of microglia during development. J. Neurochem. 2011, 119, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Dudiki, T.; Meller, J.; Mahajan, G.; Liu, H.; Zhevlakova, I.; Stefl, S.; Witherow, C.; Podrez, E.; Kothapalli, C.R.; Byzova, T.V. Microglia control vascular architecture via a TGFβ1 dependent paracrine mechanism linked to tissue mechanics. Nat. Commun. 2020, 11, 986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowicki, K.W.; Hosaka, K.; Walch, F.J.; Scott, E.W.; Hoh, B.L. M1 macrophages are required for murine cerebral aneurysm formation. J. NeuroInterventional Surg. 2018, 10, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Hasan, D.; Chalouhi, N.; Jabbour, P.; Hashimoto, T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: Preliminary results. J. Neuroinflamm. 2012, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Yamashiro, S.; Uchikawa, H.; Yoshikawa, M.; Kuriwaki, K.; Hitoshi, Y.; Yoshida, A.; Komohara, Y.; Mukasa, A. Histological analysis of infiltrating macrophages in the cerebral aneurysm walls. J. Clin. Neurosci. 2019, 67, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M. Erratum: Toll-like receptor 4 expression during cerebral aneurysm formation. J. Neurosurg. 2013, 119, 825–827. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, K.; Ikedo, T.; Kamio, Y.; Furukawa, H.; Lawton, M.T.; Hashimoto, T. TLR4 (Toll-Like Receptor 4) Mediates the Development of Intracranial Aneurysm Rupture. Hypertension 2020, 75, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Häkkinen, S.-K.; Frösen, J.; Tulamo, R.; Fraunberg, M.V.U.Z.; Wong, G.; Tromp, G.; Niemelä, M.; Hernesniemi, J.; Jääskeläinen, J.E.; et al. Upregulated signaling pathways in ruptured human saccular intracranial aneurysm wall: An emerging regulative role of toll-like receptor signaling and nuclear factor-κB, hypoxia-Inducible factor-1A, and ETS transcription factors. Neurosurgery 2011, 68, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Frȍsen, J.; Fukuda, M.; Bando, K.; Shioi, G.; Tsuji, K.; Ollikainen, E.; Nozaki, K.; Laakkonen, J.; Narumiya, S. Prostaglandin E2–EP2–NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal. 2017, 10, eaah6037. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Nishimura, M.; Matsuoka, T.; Yamamoto, K.; Furuyashiki, T.; Kataoka, H.; Kitaoka, S.; Ishibashi, R.; Ishibazawa, A.; Miyamoto, S.; et al. PGE2-EP2signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-κB. Br. J. Pharmacol. 2011, 163, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Furukawa, H.; Wada, K.; Korai, M.; Wei, Y.; Tada, Y.; Kuwabara, A.; Shikata, F.; Kitazato, K.T.; Nagahiro, S.; et al. Protective Role of Peroxisome Proliferator–Activated Receptor-γ in the Development of Intracranial Aneurysm Rupture. Stroke 2015, 46, 1664–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, D.; Starke, R.M.; Gu, H.; Wilson, K.; Chu, Y.; Chalouhi, N.; Heistad, D.D.; Faraci, F.M.; Sigmund, C.D. Smooth Muscle Peroxisome Proliferator–Activated Receptor γ Plays a Critical Role in Formation and Rupture of Cerebral Aneurysms in Mice In Vivo. Hypertension 2015, 66, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikedo, T.; Minami, M.; Kataoka, H.; Hayashi, K.; Nagata, M.; Fujikawa, R.; Higuchi, S.; Yasui, M.; Aoki, T.; Fukuda, M.; et al. Dipeptidyl Peptidase-4 Inhibitor Anagliptin Prevents Intracranial Aneurysm Growth by Suppressing Macrophage Infiltration and Activation. J. Am. Heart Assoc. 2017, 6, e004777. [Google Scholar] [CrossRef]
- Shinjo, T.; Nakatsu, Y.; Iwashita, M.; Sano, T.; Sakoda, H.; Ishihara, H.; Kushiyama, A.; Fujishiro, M.; Fukushima, T.; Tsuchiya, Y.; et al. DPP-IV inhibitor anagliptin exerts anti-inflammatory effects on macrophages, adipocytes, and mouse livers by suppressing NF-κB activation. Am. J. Physiol. Metab. 2015, 309, E214–E223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashijima, Y.; Tanaka, T.; Yamaguchi, J.; Tanaka, S.; Nangaku, M. Anti-inflammatory role of DPP-4 inhibitors in a nondiabetic model of glomerular injury. Am. J. Physiol. Physiol. 2015, 308, F878–F887. [Google Scholar] [CrossRef] [Green Version]
- Takahara, Y.; Tokunou, T.; Ichiki, T. Suppression of Abdominal Aortic Aneurysm Formation in Mice by Teneligliptin, a Dipeptidyl Peptidase-4 Inhibitor. J. Atheroscler. Thromb. 2018, 25, 698–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provencio, J.J.; Altay, T.; Smithason, S.; Moore, S.K.; Ransohoff, R.M. Depletion of Ly6G/C+ cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J. Neuroimmunol. 2011, 232, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Coulibaly, A.; Provencio, J. Aneurysmal Subarachnoid Hemorrhage: An Overview of Inflammation-Induced Cellular Changes. Neurotherapeutics 2020, 17, 436–445. [Google Scholar] [CrossRef]
- Sun, Q.; Wu, W.; Hu, Y.-C.; Li, H.; Zhang, D.; Li, S.; Li, W.-D.; Ma, B.; Zhu, J.-H.; Zhou, M.-L.; et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2014, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Luo, C.; Yu, S.; Gao, J.; Liu, C.; Wei, Z.; Zhang, Z.; Wei, L.; Yi, B. Erythropoietin ameliorates early brain injury after subarachnoid haemorrhage by modulating microglia polarization via the EPOR/JAK2-STAT3 pathway. Exp. Cell Res. 2017, 361, 342–352. [Google Scholar] [CrossRef]
- Li, Y.; Wu, P.; Dai, J.; Zhang, T.; Bihl, J.; Wang, C.; Liu, Y.; Shi, H. Inhibition of mTOR Alleviates Early Brain Injury After Subarachnoid Hemorrhage Via Relieving Excessive Mitochondrial Fission. Cell. Mol. Neurobiol. 2019, 40, 629–642. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Wang, Z.; Li, H.; Shen, H.; Xu, X.; Jia, G.; Chen, G. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J. Neurol. Sci. 2016, 367, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Takase, H.; Chou, S.H.-Y.; Hamanaka, G.; Ohtomo, R.; Islam, M.R.; Lee, J.W.; Hsu, L.; Mathew, J.; Reyes-Bricio, E.; Hayakawa, K.; et al. Soluble vascular endothelial-cadherin in CSF after subarachnoid hemorrhage. Neurology 2020, 94, e1281–e1293. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.-N.; Zhuang, Z.; Wu, L.-Y.; Liu, J.-P.; Chen, Q.; Zhang, X.-S.; Zhou, M.-L.; Zhang, Z.-H.; Li, W.; Wang, X.-L.; et al. Expression and cell distribution of leukotriene B4 receptor 1 in the rat brain cortex after experimental subarachnoid hemorrhage. Brain Res. 2016, 1652, 127–134. [Google Scholar] [CrossRef]
- Saiwai, H.; Ohkawa, Y.; Yamada, H.; Kumamaru, H.; Harada, A.; Okano, H.; Yokomizo, T.; Iwamoto, Y.; Okada, S. The LTB4-BLT1 Axis Mediates Neutrophil Infiltration and Secondary Injury in Experimental Spinal Cord Injury. Am. J. Pathol. 2010, 176, 2352–2366. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Koide, M.; Dumont, T.M.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Subarachnoid Hemorrhage Induces Gliosis and Increased Expression of the Pro-inflammatory Cytokine High Mobility Group Box 1 Protein. Transl. Stroke Res. 2010, 2, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Tsuruta, R.; Kaneko, T.; Yamashita, S.; Fujita, M.; Kasaoka, S.; Hashiguchi, T.; Suzuki, M.; Maruyama, I.; Maekawa, T. High-Mobility Group Box 1 Protein in CSF of Patients with Subarachnoid Hemorrhage. Neurocrit. Care 2009, 11, 362–368. [Google Scholar] [CrossRef]
- King, M.D.; Laird, M.D.; Ramesh, S.S.; Youssef, P.; Shakir, B.; Vender, J.R.; Alleyne, C.H.; Dhandapani, K.M. Elucidating novel mechanisms of brain injury following subarachnoid hemorrhage: An emerging role for neuroproteomics. Neurosurg. Focus 2010, 28, E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokół, B.; Woźniak, A.; Jankowski, R.; Jurga, S.; Wąsik, N.; Shahid, H.; Grzeskowiak, B. HMGB1 Level in Cerebrospinal Fluid as a Marker of Treatment Outcome in Patients with Acute Hydrocephalus Following Aneurysmal Subarachnoid Hemorrhage. J. Stroke Cerebrovasc. Dis. 2015, 24, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.A. Elucidating the novel biomarker and therapeutic potentials of High-mobility group box 1 in Subarachnoid hemorrhage: A review. AIMS Neurosci. 2019, 6, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Mao, H.Y.; Lv, J.; Lu, X.J. Expression of high-mobility group box-1 (HMGB1) in the basilar artery after experimental subarachnoid hemorrhage. J. Clin. Neurosci. 2016, 27, 161–165. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Liang, L.; Wu, Y.; Zhong, J.; Sun, X. Anti-high mobility group box-1 antibody attenuated vascular smooth muscle cell phenotypic switching and vascular remodelling after subarachnoid haemorrhage in rats. Neurosci. Lett. 2019, 708, 134338. [Google Scholar] [CrossRef] [PubMed]
- Roa, J.A.; Sarkar, D.; Zanaty, M.; Ishii, D.; Lu, Y.; Karandikar, N.J.; Hasan, D.M.; Ortega, S.B.; Samaniego, E.A. Preliminary results in the analysis of the immune response after aneurysmal subarachnoid hemorrhage. Sci. Rep. 2020, 10, 11809. [Google Scholar] [CrossRef]
- Alaraj, A.; Charbel, F.T.; Amin-Hanjani, S. Peri-operative measures for treatment and prevention of cerebral vasospasm following subarachnoid hemorrhage. Neurol. Res. 2009, 31, 651–659. [Google Scholar] [CrossRef]
- Maruhashi, T.; Higashi, Y. An overview of pharmacotherapy for cerebral vasospasm and delayed cerebral ischemia after subarachnoid hemorrhage. Expert Opin. Pharmacother. 2021, 13, 1–14. [Google Scholar] [CrossRef]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 868. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, X.-S.; Zhang, Z.-H.; Zhou, X.-M.; Gao, Y.-Y.; Liu, G.-J.; Wang, H.; Wu, L.-Y.; Li, W.; Hang, C.-H. Peroxiredoxin 2 activates microglia by interacting with Toll-like receptor 4 after subarachnoid hemorrhage. J. Neuroinflamm. 2018, 15, 87. [Google Scholar] [CrossRef]
- Sun, Q.; Dai, Y.; Zhang, X.; Hu, Y.-C.; Zhang, D.; Li, W.; Zhang, X.-S.; Zhu, J.-H.; Zhou, M.-L.; Hang, C.-H. Expression and cell distribution of myeloid differentiation primary response protein 88 in the cerebral cortex following experimental subarachnoid hemorrhage in rats: A pilot study. Brain Res. 2013, 1520, 134–144. [Google Scholar] [CrossRef]
- Peng, W.; Wu, X.; Feng, D.; Zhang, Y.; Chen, X.; Ma, C.; Shen, H.; Li, X.; Li, H.; Zhang, J.; et al. Cerebral cavernous malformation 3 relieves subarachnoid hemorrhage-induced neuroinflammation in rats through inhibiting NF-kB signaling pathway. Brain Res. Bull. 2020, 160, 74–84. [Google Scholar] [CrossRef]
- Gao, Y.; Zhuang, Z.; Lu, Y.; Tao, T.; Zhou, Y.; Liu, G.; Wang, H.; Zhang, D.; Wu, L.; Dai, H.; et al. Curcumin Mitigates Neuro-Inflammation by Modulating Microglia Polarization Through Inhibiting TLR4 Axis Signaling Pathway Following Experimental Subarachnoid Hemorrhage. Front. Neurosci. 2019, 13, 1223. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Li, L.; Shen, S.; Ma, Y.; Yin, X.; Liu, Z.; Yuan, C.; Wang, Y.; Zhang, J. Hydrogen Sulfide Reduces Cognitive Impairment in Rats After Subarachnoid Hemorrhage by Ameliorating Neuroinflammation Mediated by the TLR4/NF-κB Pathway in Microglia. Front. Cell. Neurosci. 2020, 14, 210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Su, J.; Guo, B.; Wang, K.; Li, X.; Liang, G. Apigenin protects blood–brain barrier and ameliorates early brain injury by inhibiting TLR4-mediated inflammatory pathway in subarachnoid hemorrhage rats. Int. Immunopharmacol. 2015, 28, 79–87. [Google Scholar] [CrossRef]
- Peng, J.; Wu, Y.; Tian, X.; Pang, J.; Kuai, L.; Cao, F.; Qin, X.; Zhong, J.; Li, X.; Li, Y.; et al. High-Throughput Sequencing and Co-Expression Network Analysis of lncRNAs and mRNAs in Early Brain Injury Following Experimental Subarachnoid Haemorrhage. Sci. Rep. 2017, 7, 46577. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Z.; Wu, S.-C.; Kwan, A.-L.; Lin, C.-L.; Hwang, S.-L. 6-Mercaptopurine reverses experimental vasospasm and alleviates the production of endothelins in NO-independent mechanism—A laboratory study. Acta Neurochir. 2010, 153, 939–949. [Google Scholar] [CrossRef]
- Chang, C.-Z.; Lin, C.-L.; Kassel, N.F.; Kwan, A.-L.; Howng, S.-L. 6-Mercaptopurine attenuates adhesive molecules in experimental vasospasm. Acta Neurochir. 2010, 152, 861–867. [Google Scholar] [CrossRef]
- Schallner, N.; Pandit, R.; Leblanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, S.; Selzner, L.; Weber, J.; Schallner, N. Carbon monoxide controls microglial erythrophagocytosis by regulating CD36 surface expression to reduce the severity of hemorrhagic injury. Glia 2020. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.H.; Chen, R.; Selim, M.H.; Hanafy, K.A. Heme oxygenase-1-mediated neuroprotection in subarachnoid hemorrhage via intracerebroventricular deferoxamine. J. Neuroinflamm. 2016, 13, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, U.C.; Davids, A.-M.; Brandenburg, S.; Müller, A.; Elke, A.; Magrini, S.; Atangana, E.; Turkowski, K.; Finger, T.; Gutenberg, A.; et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015, 130, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Kooijman, E.; Nijboer, C.H.; Van Velthoven, C.T.J.; Mol, W.; Dijkhuizen, R.M.; Kesecioğlu, J.; Heijnen, C.J. Long-Term Functional Consequences and Ongoing Cerebral Inflammation after Subarachnoid Hemorrhage in the Rat. PLoS ONE 2014, 9, e90584. [Google Scholar] [CrossRef]
- Kawabori, M.; Yenari, M.A. The role of the microglia in acute CNS injury. Metab. Brain Dis. 2015, 30, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.-P.; Tian, Y.; Zhang, W.; Tian, T.; Gong, S.-X.; Huang, W.-Q.; Zhou, Q.-Y. Microglia-associated neuroinflammation is a potential therapeutic target for ischemic stroke. Neural Regen. Res. 2021, 16, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Atangana, E.; Schneider, U.C.; Blecharz, K.; Magrini, S.; Wagner, J.; Nieminen-Kelhä, M.; Kremenetskaia, I.; Heppner, F.L.; Engelhardt, B.; Vajkoczy, P. Intravascular Inflammation Triggers Intracerebral Activated Microglia and Contributes to Secondary Brain Injury After Experimental Subarachnoid Hemorrhage (eSAH). Transl. Stroke Res. 2016, 8, 144–156. [Google Scholar] [CrossRef]
- Zhao, X.; Eyo, U.B.; Murugan, M.; Wu, L.-J.; Murguan, M. Microglial interactions with the neurovascular system in physiology and pathology. Dev. Neurobiol. 2018, 78, 604–617. [Google Scholar] [CrossRef]
- Haage, V.; Semtner, M.; Vidal, R.O.; Hernandez, D.P.; Pong, W.W.; Chen, Z.; Hambardzumyan, D.; Magrini, V.; Ly, A.; Walker, J.; et al. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol. Commun. 2019, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeter, C.; Herrmann, A.; Bock, S.; Vogelsang, A.; Eichler, S.; Albrecht, P.; Meuth, S.; Ruck, T. One Brain—All Cells: A Comprehensive Protocol to Isolate All Principal CNS-Resident Cell Types from Brain and Spinal Cord of Adult Healthy and EAE Mice. Cells 2021, 10, 651. [Google Scholar] [CrossRef]
- Ma, L.; Zhu, W.; Zhan, L.; Zhang, R.; Li, Q.; Bao, C.; Weiss, M.; Wang, A.; Su, H. Abstract 76: Transient Depletion of Microglia Reduces the Severity of Brain Arteriovenous Malformation in a Mouse Model. Stroke 2018, 49. [Google Scholar] [CrossRef]
- Zhang, R.; Degos, V.; Kang, S.; Wong, M.; Zhan, L.; Su, H. Abstract 191: Persistent Infiltration of Active Microglia in Brain Arterio-venous Malformation Causes Unresolved Inflammation and Lesion Progression. Stroke 2015, 46, A191. [Google Scholar]
- Chen, Y.; Zhu, W.; Bollen, A.W.; Lawton, M.T.; Barbaro, N.M.; Dowd, C.F.; Hashimoto, T.; Yang, G.-Y.; Young, W.L. Evidence of inflam-matory cell involvement in brain arteriovenous malformations. Neurosurgery 2008, 62, 1340–1350. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Han, Z.; Degos, V.; Shen, F.; Choi, E.-J.; Sun, Z.; Kang, S.; Wong, M.; Zhu, W.; Zhan, L.; et al. Persistent infiltration and pro-inflammatory differentiation of monocytes cause unresolved inflammation in brain arteriovenous malformation. Angiogenesis 2016, 19, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Guo, Y.; Walker, E.J.; Shen, F.; Jun, K.; Oh, S.P.; Degos, V.; Lawton, M.T.; Tihan, T.; Davalos, D.; et al. Reduced mural cell coverage and impaired vessel integrity after angiogenic stimulation in the Alk1-deficient brain. Arter. Thromb. Vasc. Biol. 2013, 33, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sun, Z.; Han, Z.; Jun, K.; Camus, M.; Wankhede, M.; Mao, L.; Arnold, T.; Young, W.L.; Su, H. De Novo Cerebrovascular Malformation in the Adult Mouse After Endothelial Alk1 Deletion and Angiogenic Stimulation. Stroke 2014, 45, 900–902. [Google Scholar] [CrossRef] [Green Version]
- Ferrier, C.H.; Aronica, E.; Leijten, F.S.S.; Spliet, W.G.M.; Boer, K.; Van Rijen, P.C.; Van Huffelen, A.C. Electrocorticography discharge patterns in patients with a cavernous hemangioma and pharmacoresistent epilepsy. J. Neurosurg. 2007, 107, 495–503. [Google Scholar] [CrossRef]
- Liu, J.; Reeves, C.; Michalak, Z.; Coppola, A.; Diehl, B.; Sisodiya, S.M.; Thom, M. Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol. Commun. 2014, 2, 71. [Google Scholar] [CrossRef] [Green Version]
- Boer, K.; Spliet, W.; van Rijen, P.; Redeker, S.; Troost, D.; Aronica, E. Evidence of activated microglia in focal cortical dysplasia. J. Neuroimmunol. 2006, 173, 188–195. [Google Scholar] [CrossRef]
- Abid, K.A.; Sobowale, O.A.; Parkes, L.M.; Naish, J.; Parker, G.J.; Du Plessis, D.; Brough, D.; Barrington, J.; Allan, S.M.; Hinz, R.; et al. Assessing Inflammation in Acute Intracerebral Hemorrhage with PK11195 PET and Dynamic Contrast-Enhanced MRI. J. Neuroimaging 2017, 28, 158–161. [Google Scholar] [CrossRef]
- Chang, C.-F.; Wan, J.; Li, Q.; Renfroe, S.C.; Heller, N.M.; Wang, J. Alternative activation-skewed microglia/macrophages promote hematoma resolution in experimental intracerebral hemorrhage. Neurobiol. Dis. 2017, 103, 54–69. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Ting, S.-M.; Song, S.; Zhang, J.; Edwards, N.J.; Aronowski, J. Cleaning up after ICH: The role of Nrf2 in modulating microglia function and hematoma clearance. J. Neurochem. 2015, 133, 144–152. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Song, W.; Gonzales, N.; Grotta, J.C.; Aronowski, J. Hematoma resolution as a target for intracerebral hemorrhage treatment: Role for peroxisome proliferator-activated receptor γ in microglia/macrophages. Ann. Neurol. 2007, 61, 352–362. [Google Scholar] [CrossRef]
- Zhao, X.; Grotta, J.; Gonzales, N.; Aronowski, J. Hematoma Resolution as a Therapeutic Target: The Role of Microglia/Macrophages. Stroke 2008, 40, S92–S94. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef]
- Springborg, J.B.; Møller, C.; Gideon, P.; Jørgensen, O.S.; Juhler, M.; Olsen, N.V. Erythropoietin in patients with aneurysmal subarachnoid haemorrhage: A double blind randomised clinical trial. Acta Neurochir. 2007, 149, 1089–1101. [Google Scholar] [CrossRef]
- Tseng, M.-Y.; Hutchinson, P.J.; Richards, H.K.; Czosnyka, M.; Pickard, J.D.; Erber, W.; Brown, S.J.; Kirkpatrick, P.J. Acute systemic erythropoietin therapy to reduce delayed ischemic deficits following aneurysmal subarachnoid hemorrhage: A Phase II randomized, double-blind, placebo-controlled trial. J. Neurosurg. 2009, 111, 171–180. [Google Scholar] [CrossRef]
- Helbok, R.; Shaker, E.; Beer, R.; Chemelli, A.; Sojer, M.; Sohm, F.; Broessner, G.; Lackner, P.; Beck, M.; Zangerle, A.; et al. High dose Erythropoietin increases Brain Tissue Oxygen Tension in Severe Vasospasm after Subarachnoid Hemorrhage. BMC Neurol. 2012, 12, 32. [Google Scholar] [CrossRef] [Green Version]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [Green Version]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [Green Version]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Sidhaye, J.; Knoblich, J.A. Brain Organoids: An Ensemble of Bioassays to Investigate Human Neurodevelopment and Disease. Cell Death Differ. 2021, 28, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S.; et al. Engineering of Human Brain Organoids with a Functional Vascular-Like System. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A. Brain organoids get vascularized. Nat. Biotechnol. 2018, 36, 407–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Author, Year | Pathology | Agent/Drug | Target | Type of Study | Outcome |
|---|---|---|---|---|---|
| Wei et al., 2017 [46] | Aneurysm (acute stage SAH) | EPO | EPOR/JAK2/STAT3 pathway | in vitro (mouse brain SAH model) | Amelioration of brain injury. |
| Springborg et al., 2007 [98] | Aneurysm (subacute stage SAH) | EPO | NS | Double-blind randomized clinical trial | No conclusive evidence of beneficial effects from EPO. |
| Tseng et al., 2009 [99] | Aneurysm (subacute stage SAH) | EPO | NS | Double-blind randomized clinical trial | No differences in the incidence of vasospasm and adverse events; Patients receiving EPO had a decreased incidence of severe vasospasm. |
| Helbok et al., 2012 [100] | Aneurysm (vasospasm) | EPO | NS | Non-randomized clinical trial | Brain tissue oxygen tension improved; Brain metabolic parameters remained unchanged. |
| You et al., 2016 [48] | Aneurysm (acute stage SAH) | Rapamycin | mTOR | in vitro (rat brain SAH model) | Microglia polarization toward the M2 phenotype, early brain injury amelioration. |
| AZD8055 | mTOR | in vitro (rat brain SAH model) | Microglia polarization toward the M2 phenotype, early brain injury amelioration. | ||
| Wang et al., 2019 [58] | Aneurysm (acute stage SAH) | Anti-HMGB1–AB | HMGB1 | in vitro and in vivo (rat brain SAH model) | Decreased cerebral vasoconstriction, improved cerebral blood flow, lessened brain edema, and microglial activation. |
| Heinz et al., 2021 [9] | Aneurysm (subacute stage SAH) | Perxidartinib | CSF1R | in vitro (mouse brain SAH model) | Reduced microglial accumulation and activation, diminished neuronal death. |
| Gao et al., 2019 [66] | Aneurysm (vasospasm) | Curcumin | TLR4 | in vitro and in vivo (mouse brain SAH model) | Reduction of cerebral proinflammatory cytokines and edema; Promotion of M2 phenotype polarization; Diminished neurological deficit. |
| Duan et al., 2020 [67] | Aneurysm (vasospasm) | Hydrogen sulfide | TLR4/NF-κB | in vitro and in vivo (rat brain SAH model) | Reduced the cognitive impairment; reduced the expression of TNF-α, TLR4, and NF-κB |
| Zhang et al., 2015 [68] | Aneurysm (vasospasm) | Apigenin | TLR4/NF-κB | in vitro and in vivo (rat brain SAH model) | Reduced neuronal apoptosis, inhibition of BBB disruption, and improved neurological results |
| Peng et al., 2017 [69] | Aneurysm (vasospasm) | Lentivirus vectors: lentivirus-50305 (KD1) and lentivirus-50307 (KD3) | lncRNA fantom3_F730004F19 and TLR4 | in vitro (mouse brain SAH model) | Attenuated microglial inflammation. |
| Chang et al., 2011 [70]; Chang et al., 2010 [71] | Aneurysm (vasospasm) | 6-Mercaptopurine | ET-1 | in vitro (rat brain SAH model) | Inhibition of vasospasm, amelioration of microglial inflammation, and chemotaxis. |
| Schallner et al., 2015 | Aneurysm (vasospasm) | CO inhalation | Microglial HO-1 | in vitro and in vivo (mouse brain SAH model) | Amelioration of neuronal cell death, vasospasm, impaired cognitive function, and clearance of cerebral blood burden. |
| LeBlanc et al., 2016 [74] | Aneurysm (vasospasm) | Deferoxamine | Microglial HO-1 | in vitro and in vivo (mouse brain SAH model) | Amelioration of neurological damage, cognitive outcome, and increased HO-1 expression in microglia, but no effect on vasospasm. |
| Ma et al., 2018 [83] | AVM | CSF1R inhibitor | CSF1R | in vitro (mouse brain AVM model) | Microglia depletion, AVM development inhibition. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timis, T.L.; Florian, I.A.; Susman, S.; Florian, I.S. Involvement of Microglia in the Pathophysiology of Intracranial Aneurysms and Vascular Malformations—A Short Overview. Int. J. Mol. Sci. 2021, 22, 6141. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116141
Timis TL, Florian IA, Susman S, Florian IS. Involvement of Microglia in the Pathophysiology of Intracranial Aneurysms and Vascular Malformations—A Short Overview. International Journal of Molecular Sciences. 2021; 22(11):6141. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116141
Chicago/Turabian StyleTimis, Teodora Larisa, Ioan Alexandru Florian, Sergiu Susman, and Ioan Stefan Florian. 2021. "Involvement of Microglia in the Pathophysiology of Intracranial Aneurysms and Vascular Malformations—A Short Overview" International Journal of Molecular Sciences 22, no. 11: 6141. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116141