
Virus–Host Interaction Gets Curiouser and Curiouser. PART II: Functional Transcriptomics of the E. coli DksA-Deficient Cell upon Phage P1vir Infection


Abstract
:1. Introduction
2. Results and Discussion
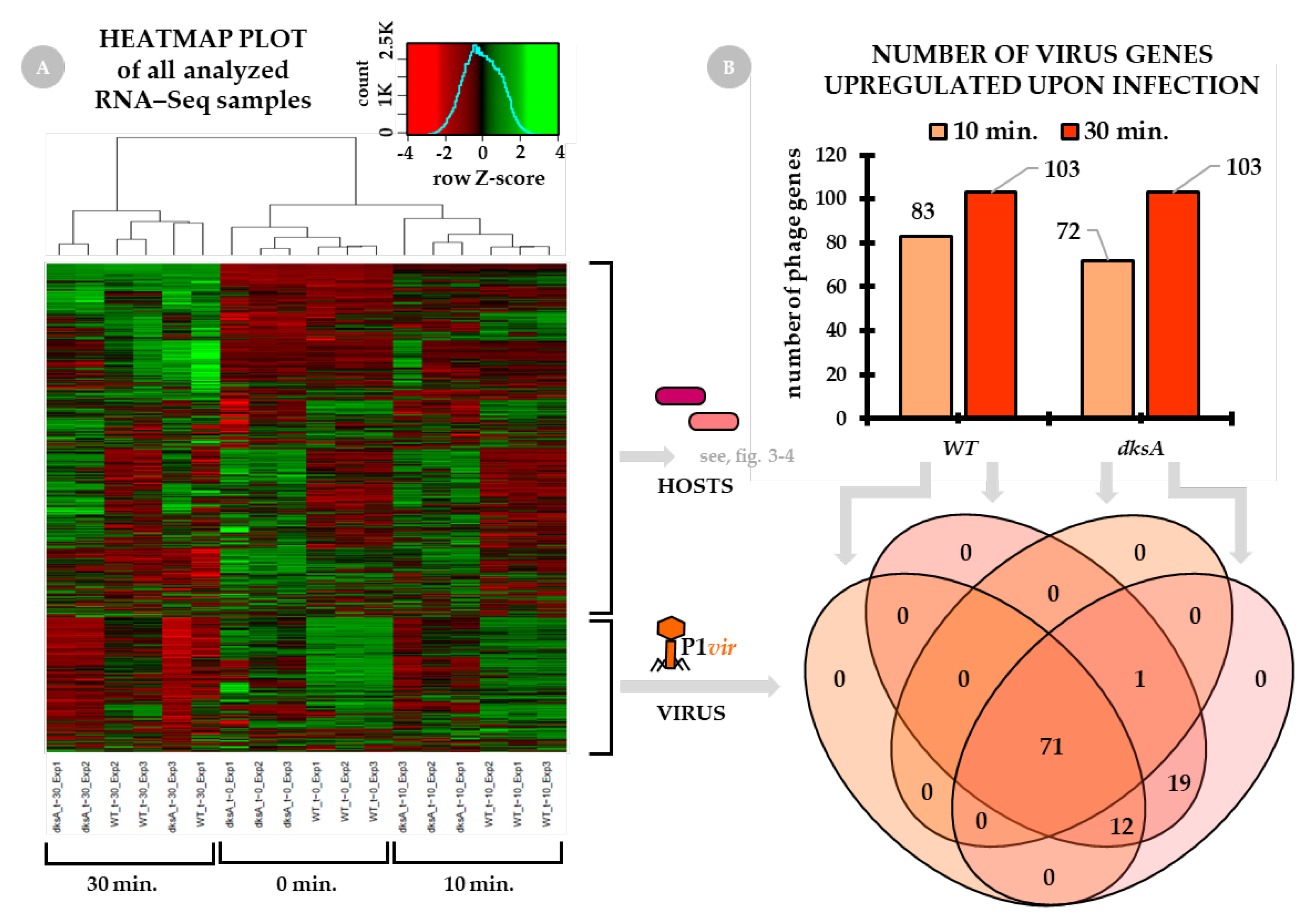
2.1. General Overview of the Virus–Host Transcriptome
2.2. The Virus Transcriptome Upon Infection
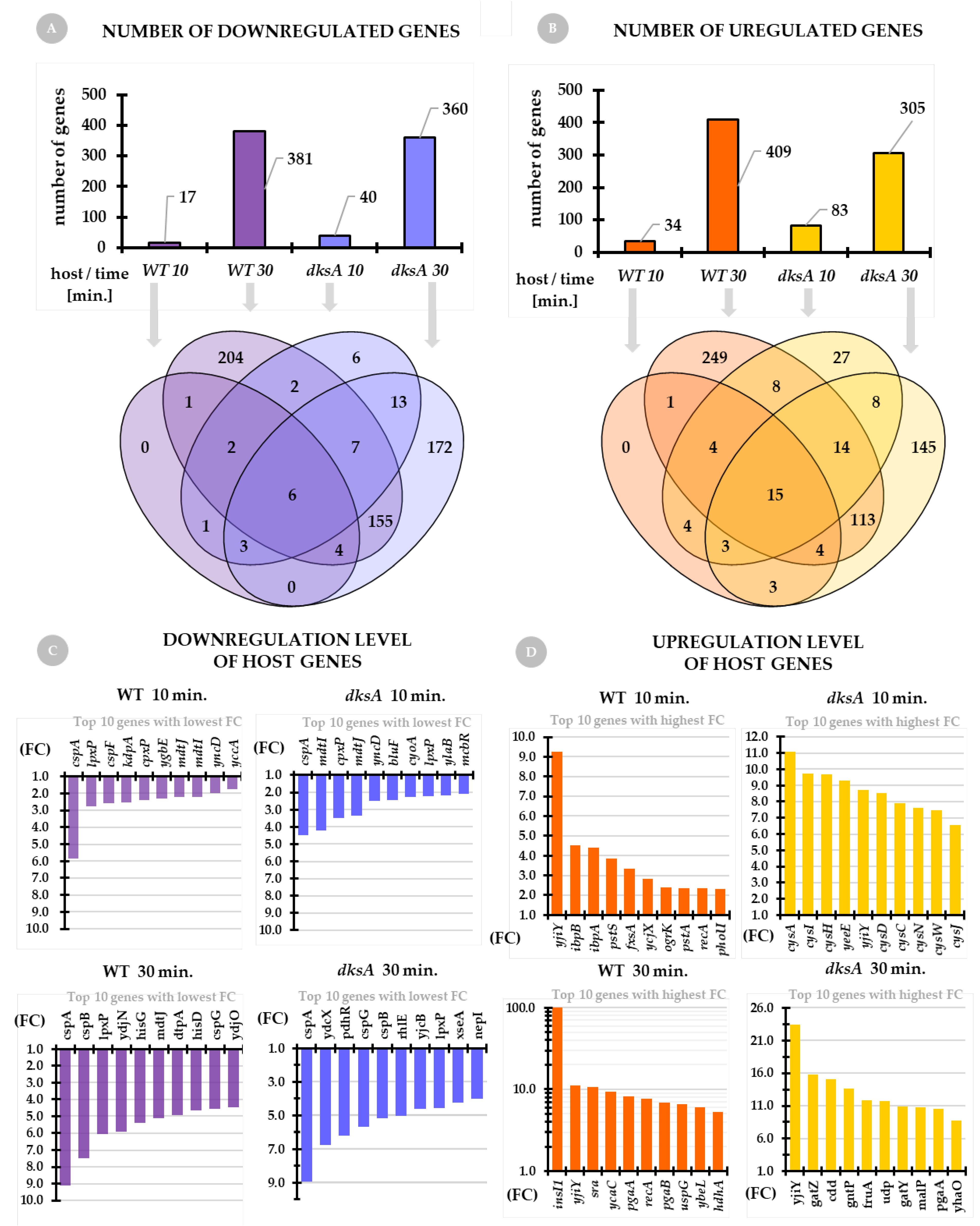
2.3. The Host Transcriptome Upon Infection
3. Materials and Methods
3.1. Bacterial Strains and Phages
3.2. Total RNA Isolation
3.3. RNA-Seq Analysis
3.4. High Throughput Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ↓↑ Reg. | WT | dksA | Gene | Function |
|---|---|---|---|---|
| ↓ | ++ | ++ | cpxP | periplasmic adaptor protein |
| ↓ | ++ | ++ | cspA | RNA chaperone and antiterminator, cold-inducible |
| ↓ | ++ | ++ | cspB | Qin prophage; cold shock protein |
| ↓ | ++ | ++ | cspG | cold shock protein homolog |
| ↓ | ++ | ++ | lpxP | palmitoleoyl-acyl carrier protein |
| ↓ | ++ | ++ | mdtI | multidrug efflux system transporter |
| ↓ | ++ | ++ | mdtJ | multidrug efflux system transporter |
| ↓ | ++ | ++ | nepI | putative transporter |
| ↓ | ++ | ++ | yncD | putative iron outer membrane transporter |
| ↓ | ++ | + | cspF | Qin prophage, cold shock protein |
| ↓ | ++ | + | dtpA | dipeptide / tripeptide permease A |
| ↓ | ++ | − | hisD | bifunctional histidinal/histidinol dehydrogenase |
| ↓ | ++ | − | hisG | ATP phosphoribosyltransfer-ase |
| ↓ | ++ | − | kdpA | potassium translocating ATPase |
| ↓ | ++ | − | yccA | modu-lator of FtsH protease, inner membrane protein |
| ↓ | ++ | − | ydjN | putative transporter |
| ↓ | ++ | − | ydjO | uncharacterized protein |
| ↓ | ++ | − | ygbE | DUF3561 family inner membrane protein |
| ↓ | + | ++ | bluF | anti-repressor for YcgE |
| ↓ | + | ++ | mcbR | biofilm gene transcriptional regulator |
| ↓ | + | ++ | ylaB | put. membrane cyclic-diGMP phosphodiesterase |
| ↓ | − | ++ | cyoA | cyto-chrome o ubiquinol oxidase subunit II |
| ↓ | − | ++ | pdhR | pyruvate dehydrogenase complex repressor |
| ↓ | − | ++ | rhlE | ATP-dependent RNA helicase |
| ↓ | − | ++ | xseA | exonuclease VII |
| ↓ | − | ++ | ydcX | DUF2566 family protein |
| ↓ | − | ++ | yjcB | putative inner membrane protein |
| ↑ | − | ++ | cdd | cytidine/deoxycytidine deaminase |
| ↑ | ++ | + | fxsA | suppressor of F exclusion of phage T7 |
| ↑ | ++ | + | ibpA | heat shock chaperone |
| ↑ | ++ | + | ibpB | heat shock chaperone |
| ↑ | ++ | − | insI1 | IS30 transposase |
| ↑ | ++ | − | ogrK | orphan Ogr protein, positive regulator of P2 growth |
| ↑ | ++ | ++ | pgaA | biofilm adhesin polysaccharide PGA secretin |
| ↑ | ++ | + | pgaB | outer membrane export lipoprotein |
| ↑ | ++ | + | phoU | negative regulator of PhoR/PhoB two-component regulator |
| ↑ | ++ | + | pstA | phosphate ABC transporter permease |
| ↑ | ++ | + | pstS | phosphate ABC transporter periplasmic binding protein |
| ↑ | ++ | + | recA | DNA recombination and repair protein |
| ↑ | ++ | − | sra | stationary-phase-induced ribosome-associated protein |
| ↑ | ++ | + | uspG | universal stress protein UP12 |
| ↑ | ++ | + | ybeL | DUF1451 family protein |
| ↑ | ++ | − | ycaC | putative isochorismatase family hydrolase |
| ↑ | ++ | + | ycjX | DUF463 family protein, puatative P-loop NTPase |
| ↑ | ++ | ++ | yjiY | putative transporter |
| ↑ | ++ | + | hdhA | 7-alpha-hydroxysteroid dehydrogenase, NAD-dependent |
| ↑ | − | ++ | cysA | sulfate/thiosulfate transporter subunit |
| ↑ | − | ++ | cysC | adenosine 5’-phosphosulfate kinase |
| ↑ | − | ++ | cysD | sulfate adenylyltransferase, subunit 2 |
| ↑ | − | ++ | cysH | phosphoadenosine phosphosulfate reductase |
| ↑ | − | ++ | cysI | sulfite reductase, beta subunit, NAD(P)-binding |
| ↑ | − | ++ | cysJ | sulfite reductase, alpha subunit, flavoprotein |
| ↑ | − | ++ | cysN | sulfate adenylyltransferase, subunit 1 |
| ↑ | − | ++ | cysW | sulfate/thiosulfate ABC transporter permease |
| ↑ | − | ++ | fruA | fused fructose-specific PTS enzymes |
| ↑ | − | ++ | gatY | D-tagatose 1,6-bisphosphate aldolase 2, catalytic subunit |
| ↑ | − | ++ | gatZ | D-tagatose 1,6-bisphosphate aldolase 2, subunit |
| ↑ | − | ++ | gntP | fructuronate transporter |
| ↑ | − | ++ | malP | maltodextrin phosphorylase |
| ↑ | − | ++ | udp | uridine phosphorylase |
| ↑ | − | ++ | yeeE | UPF0394 family inner membrane protein |
| ↑ | − | ++ | yhaO | putative transporter |
References
- Łobocka, M.B.; Rose, D.J.; Plunkett, G.; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of Bacteriophage P1. J. Bacteriol. 2004, 186, 7032–7068. [Google Scholar] [CrossRef] [Green Version]
- Heinzel, T.; Velleman, M.; Schuster, H. The c1 repressor inactivator protein coi of bacteriophage P1. Cloning and expression of coi and its interference with c1 repressor function. J. Biol. Chem. 1990, 265, 17928–17934. [Google Scholar] [CrossRef]
- Dreiseikelmann, B.; Velleman, M.; Schuster, H. The c1 repressor of bacteriophage P1. Isolation and characterization of the repressor protein. J. Biol. Chem. 1988, 263, 1391–1397. [Google Scholar] [CrossRef]
- Citron, M.; Schuster, H. The c4 repressors of bacteriophages P1 and P7 are antisense RNAs. Cell 1990, 62, 591–598. [Google Scholar] [CrossRef]
- Riedel, H.D.; Heinrich, J.; Schuster, H. Cloning, expression, and characterization of the icd gene in the immI operon of bacteriophage P1. J. Bacteriol. 1993, 175, 2833–2838. [Google Scholar] [CrossRef] [Green Version]
- Lehnherr, H. Bacteriophage P1. In The Bacteriophages; Calendar, R., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 350–379. ISBN 0-19-514850-9. [Google Scholar]
- Lehnherr, H.; Guidolin, A.; Arber, W. Bacteriophage P1 gene 10 encodes a trans-activating factor required for late gene expression. J. Bacteriol. 1991, 173, 6438–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, D.E.; Landy, A. The role of host RNA polymerase in P1 phage development. Virology 1973, 51, 521–524. [Google Scholar] [CrossRef]
- Hansen, A.-M.; Lehnherr, H.; Wang, X.; Mobley, V.; Jin, D.J. Escherichia coli SspA is a transcription activator for bacteriophage P1 late genes. Mol. Microbiol. 2003, 48, 1621–1631. [Google Scholar] [CrossRef]
- Kang, P.J.; Craig, E.A. Identification and characterization of a new Escherichia coli gene that is a dosage-dependent suppressor of a dnaK deletion mutation. J. Bacteriol. 1990, 172, 2055–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.J.; Barker, M.M.; Ross, W.; Schneider, D.A.; Webb, C.; Foster, J.W.; Gourse, R.L. DksA: A critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 2004, 118, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.J.; Berkmen, M.B.; Gourse, R.L. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc. Natl. Acad. Sci. USA 2005, 102, 7823–7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnusson, L.U.; Gummesson, B.; Joksimović, P.; Farewell, A.; Nyström, T. Identical, Independent, and Opposing Roles of ppGpp and DksA in Escherichia coli. J. Bacteriol. 2007, 189, 5193–5202. [Google Scholar] [CrossRef] [Green Version]
- Aberg, A.; Fernández-Vázquez, J.; Cabrer-Panes, J.D.; Sánchez, A.; Balsalobre, C. Similar and Divergent Effects of ppGpp and DksA Deficiencies on Transcription in Escherichia coli. J. Bacteriol. 2009, 191, 3226–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyzen, R.; Kochanowska, M.; Wegrzyn, G.; Szalewska-Palasz, A. Transcription from bacteriophage pR promoter is regulated independently and antagonistically by DksA and ppGpp. Nucleic Acids Res. 2009, 37, 6655–6664. [Google Scholar] [CrossRef] [Green Version]
- Łyżeń, R.; Maitra, A.; Milewska, K.; Kochanowska-Łyżeń, M.; Hernandez, V.J.; Szalewska-Pałasz, A. The dual role of DksA protein in the regulation of Escherichia coli pArgX promoter. Nucleic Acids Res. 2016, 44, gkw912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, L.M.D.; Johansson, L.U.M.; Skärfstad, E.; Shingler, V. σ54-Promoter Discrimination and Regulation by ppGpp and DksA. J. Biol. Chem. 2009, 284, 828–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perederina, A.; Svetlov, V.; Vassylyeva, M.N.; Tahirov, T.H.; Yokoyama, S.; Artsimovitch, I.; Vassylyev, D.G. Regulation through the Secondary Channel—Structural Framework for ppGpp-DksA Synergism during Transcription. Cell 2004, 118, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Gourse, R.L.; Chen, A.Y.; Gopalkrishnan, S.; Sanchez-Vazquez, P.; Myers, A.; Ross, W. Transcriptional Responses to ppGpp and DksA. Annu. Rev. Microbiol. 2018, 72, 163–184. [Google Scholar] [CrossRef]
- Fernández-Coll, L.; Potrykus, K.; Cashel, M. Puzzling conformational changes affecting proteins binding to the RNA polymerase. Proc. Natl. Acad. Sci. USA 2018, 115, 12550–12552. [Google Scholar] [CrossRef] [Green Version]
- Myka, K.K.; Gottesman, M.E. DksA and DNA double-strand break repair. Curr. Genet. 2019, 65, 1297–1300. [Google Scholar] [CrossRef]
- Jude, F.; Kóhler, T.; Branny, P.; Perron, K.; Mayer, M.P.; Comte, R.; van Delden, C. Posttranscriptional Control of Quorum-Sensing-Dependent Virulence Genes by DksA in Pseudomonas aeruginosa. J. Bacteriol. 2003, 185, 3558–3566. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.; Gentry, D.; Elliott, T.; Cashel, M. DksA Affects ppGpp Induction of RpoS at a Translational Level. J. Bacteriol. 2002, 184, 4455–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.B.; Yoon, S.S. Transcriptome analysis reveals that the RNA polymerase–binding protein DksA1 has pleiotropic functions in Pseudomonas aeruginosa. J. Biol. Chem. 2020, 295, 3851–3864. [Google Scholar] [CrossRef]
- Boyle, W.K.; Groshong, A.M.; Drecktrah, D.; Boylan, J.A.; Gherardini, F.C.; Blevins, J.S.; Samuels, D.S.; Bourret, T.J. DksA Controls the Response of the Lyme Disease Spirochete Borrelia burgdorferi to Starvation. J. Bacteriol. 2019, 201, e00582-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holley, C.L.; Zhang, X.; Fortney, K.R.; Ellinger, S.; Johnson, P.; Baker, B.; Liu, Y.; Janowicz, D.M.; Katz, B.P.; Munson, R.S.; et al. DksA and (p)ppGpp Have Unique and Overlapping Contributions to Haemophilus ducreyi Pathogenesis in Humans. Infect. Immun. 2015, 83, 3281–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage–host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef] [Green Version]
- Pilát, Z.; Jonáš, A.; Pilátová, J.; Klementová, T.; Bernatová, S.; Šiler, M.; Maňka, T.; Kizovský, M.; Růžička, F.; Pantůček, R.; et al. Analysis of Bacteriophage–Host Interaction by Raman Tweezers. Anal. Chem. 2020, 92, 12304–12311. [Google Scholar] [CrossRef]
- Cech, G.M.; Kloska, A.; Krause, K.; Potrykus, K.; Cashel, M.; Szalewska-Pałasz, A. Virus–Host Interaction Gets Curiouser and Curiouser. PART I: Phage P1vir Enhanced Development in an E. coli DksA-Deficient Cell. Int. J. Mol. Sci. 2021, 22, 5890. [Google Scholar] [CrossRef]
- Scott, J.R. Immunity and repression in bacteriophages P1 and P7. Curr. Top. Microbiol. Immunol. 1980, 90, 49–65. [Google Scholar] [CrossRef]
- Hansen, E.B. Structure and regulation of the lytic replicon of phage P1. J. Mol. Biol. 1989, 207, 135–149. [Google Scholar] [CrossRef]
- Heisig, A.; Riedel, H.-D.; Dobrinski, B.; Lurz, R.; Schuster, H. Organization of the immunity region immI of bacteriophage P1 and synthesis of the P1 antirepressor. J. Mol. Biol. 1989, 209, 525–538. [Google Scholar] [CrossRef]
- Riedel, H.-D.; Heinrich, J.; Heisig, A.; Choli, T.; Schuster, H. The antirepressor of phage P1 Isolation and interaction with the C1 repressor of P1 and P7. FEBS Lett. 1993, 334, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.R.; Kropf, M.; Mendelson, L. Clear plaque mutants of phage P7. Virology 1977, 76, 39–46. [Google Scholar] [CrossRef]
- Scott, J.R.; Kropf, M.M. Location of new clear plaque genes on the P1 map. Virology 1977, 82, 362–368. [Google Scholar] [CrossRef]
- Walker, J.T.; Walker, D.H. Coliphage P1 morphogenesis: Analysis of mutants by electron microscopy. J. Virol. 1983, 45, 1118–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, S.; Hiestand-Nauer, R.; Sandmeier, H.; Lehnherr, H.; Arber, W. Accessory Genes in thedarAOperon of Bacteriophage P1 Affect Antirestriction Function, Generalized Transduction, Head Morphogenesis, and Host Cell Lysis. Virology 1998, 251, 49–58. [Google Scholar] [CrossRef]
- Guidolin, A.; Zingg, J.-M.; Arber, W. Organization of the bacteriophage P1 tail-fibre operon. Gene 1989, 76, 239–243. [Google Scholar] [CrossRef]
- Yarmolinsky, M.B.; Sternberg, N. Bacteriophage P1. In The Bacteriophages; Calendar, R., Ed.; Springer US: Boston, MA, USA, 1988; pp. 291–438. ISBN 978-1-4684-5426-0. [Google Scholar]
- Tanabe, H.; Goldstein, J.; Yang, M.; Inouye, M. Identification of the promoter region of the Escherichia coli major cold shock gene, cspA. J. Bacteriol. 1992, 174, 3867–3873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandi, A. Massive presence of the Escherichia coli ‘major cold-shock protein’ CspA under non-stress conditions. EMBO J. 1999, 18, 1653–1659. [Google Scholar] [CrossRef]
- Ivancic, T.; Jamnik, P.; Stopar, D. Cold shock CspA and CspB protein production during periodic temperature cycling in Escherichia coli. BMC Res. Notes 2013, 6, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.G.; VanBogelen, R.A.; Neidhardt, F.C. Induction of proteins in response to low temperature in Escherichia coli. J. Bacteriol. 1987, 169, 2092–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.; Pollitt, N.S.; Inouye, M. Major cold shock protein of Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Brandi, A.; Giangrossi, M.; Giuliodori, A.M.; Falconi, M. An Interplay among FIS, H-NS, and Guanosine Tetraphosphate Modulates Transcription of the Escherichia coli cspA Gene under Physiological Growth Conditions. Front. Mol. Biosci. 2016, 3, 19. [Google Scholar] [CrossRef]
- Jiang, W.; Fang, L.; Inouye, M. The role of the 5’-end untranslated region of the mRNA for CspA, the major cold-shock protein of Escherichia coli, in cold-shock adaptation. J. Bacteriol. 1996, 178, 4919–4925. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Jones, P.; Inouye, M. Chloramphenicol induces the transcription of the major cold shock gene of Escherichia coli, cspA. J. Bacteriol. 1993, 175, 5824–5828. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, J.-P.; Inouye, M. CspA, CspB, and CspG, Major Cold Shock Proteins of Escherichia coli, Are Induced at Low Temperature under Conditions That Completely Block Protein Synthesis. J. Bacteriol. 1999, 181, 1827–1830. [Google Scholar] [CrossRef] [Green Version]
- Kristoficova, I.; Vilhena, C.; Behr, S.; Jung, K. BtsT, a Novel and Specific Pyruvate/H+ Symporter in Escherichia coli. J. Bacteriol. 2017, 200. [Google Scholar] [CrossRef] [Green Version]
- Kraxenberger, T.; Fried, L.; Behr, S.; Jung, K. First Insights into the Unexplored Two-Component System YehU/YehT in Escherichia coli. J. Bacteriol. 2012, 194, 4272–4284. [Google Scholar] [CrossRef] [Green Version]
- Behr, S.; Fried, L.; Jung, K. Identification of a Novel Nutrient-Sensing Histidine Kinase/Response Regulator Network in Escherichia coli. J. Bacteriol. 2014, 196, 2023–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, P.; Kouzel, I.U.; Berger, M.; Haarmann, N.; Dobrindt, U.; Koudelka, G.B.; Mellmann, A. Carriage of Shiga toxin phage profoundly affects Escherichia coli gene expression and carbon source utilization. BMC Genomics 2019, 20, 504. [Google Scholar] [CrossRef] [PubMed]
- Stalder, R.; Caspers, P.; Olasz, F.; Arber, W. The N-terminal domain of the insertion sequence 30 transposase interacts specifically with the terminal inverted repeats of the element. J. Biol. Chem. 1990, 265, 3757–3762. [Google Scholar] [CrossRef]
- Olasz, F.; Farkas, T.; Kiss, J.; Arini, A.; Arber, W. Terminal inverted repeats of insertion sequence IS30 serve as targets for transposition. J. Bacteriol. 1997, 179, 7551–7558. [Google Scholar] [CrossRef] [Green Version]
- Sirko, A.; Hryniewicz, M.; Hulanicka, D.; Böck, A. Sulfate and thiosulfate transport in Escherichia coli K-12: Nucleotide sequence and expression of the cysTWAM gene cluster. J. Bacteriol. 1990, 172, 3351–3357. [Google Scholar] [CrossRef] [Green Version]
- Linton, K.J.; Higgins, C.F. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. Microbiol. 2002, 28, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Aquino, P.; Honda, B.; Jaini, S.; Lyubetskaya, A.; Hosur, K.; Chiu, J.G.; Ekladious, I.; Hu, D.; Jin, L.; Sayeg, M.K.; et al. Coordinated regulation of acid resistance in Escherichia coli. BMC Syst. Biol. 2017, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Al Mamun, A.A.M.; Lombardo, M.-J.; Shee, C.; Lisewski, A.M.; Gonzalez, C.; Lin, D.; Nehring, R.B.; Saint-Ruf, C.; Gibson, J.L.; Frisch, R.L.; et al. Identity and Function of a Large Gene Network Underlying Mutagenic Repair of DNA Breaks. Science 2012, 338, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, P.; Abedon, S.T. Bacteriophage (overview). In Encyclopedia of Microbiology; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 322–338. ISBN 9780123739445. [Google Scholar]
- Patterson-West, J.; Arroyo-Mendoza, M.; Hsieh, M.-L.; Harrison, D.; Walker, M.; Knipling, L.; Hinton, D. The Bacteriophage T4 MotB Protein, a DNA-Binding Protein, Improves Phage Fitness. Viruses 2018, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef]
- Kloska, A.; Cech, G.M.; Sadowska, M.; Krause, K.; Szalewska-Pałasz, A.; Olszewski, P. Adaptation of the Marine Bacterium Shewanella baltica to Low Temperature Stress. Int. J. Mol. Sci. 2020, 21, 4338. [Google Scholar] [CrossRef]
- Xiao, H.; Kalman, M.; Ikehara, K.; Zemel, S.; Glaser, G.; Cashel, M. Residual guanosine 3’,5’-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. J. Biol. Chem. 1991, 266, 5980–5990. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- FastQC Software. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 February 2018).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2015, 32, btv566. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Ayllon-Benitez, A.; Bourqui, R.; Thébault, P.; Mougin, F. GSAn: An alternative to enrichment analysis for annotating gene sets. NAR Genomics Bioinforma. 2020, 2, lqaa017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Ritchie, M.; Law, C. Glimma: Interactive HTML Graphics. R Package Version 1.2.1. Available online: https://github.com/Shians/Glimma (accessed on 6 February 2018).
- Mazandu, G.K.; Chimusa, E.R.; Mulder, N.J. Gene Ontology semantic similarity tools: Survey on features and challenges for biological knowledge discovery. Brief. Bioinform. 2016, 18, bbw067. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cech, G.M.; Szalewska-Pałasz, A.; Potrykus, K.; Kloska, A. Virus–Host Interaction Gets Curiouser and Curiouser. PART II: Functional Transcriptomics of the E. coli DksA-Deficient Cell upon Phage P1vir Infection. Int. J. Mol. Sci. 2021, 22, 6159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116159
Cech GM, Szalewska-Pałasz A, Potrykus K, Kloska A. Virus–Host Interaction Gets Curiouser and Curiouser. PART II: Functional Transcriptomics of the E. coli DksA-Deficient Cell upon Phage P1vir Infection. International Journal of Molecular Sciences. 2021; 22(11):6159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116159
Chicago/Turabian StyleCech, Grzegorz M., Agnieszka Szalewska-Pałasz, Katarzyna Potrykus, and Anna Kloska. 2021. "Virus–Host Interaction Gets Curiouser and Curiouser. PART II: Functional Transcriptomics of the E. coli DksA-Deficient Cell upon Phage P1vir Infection" International Journal of Molecular Sciences 22, no. 11: 6159. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116159