
Yes-Associated Protein (Yap) Is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation

 and
and 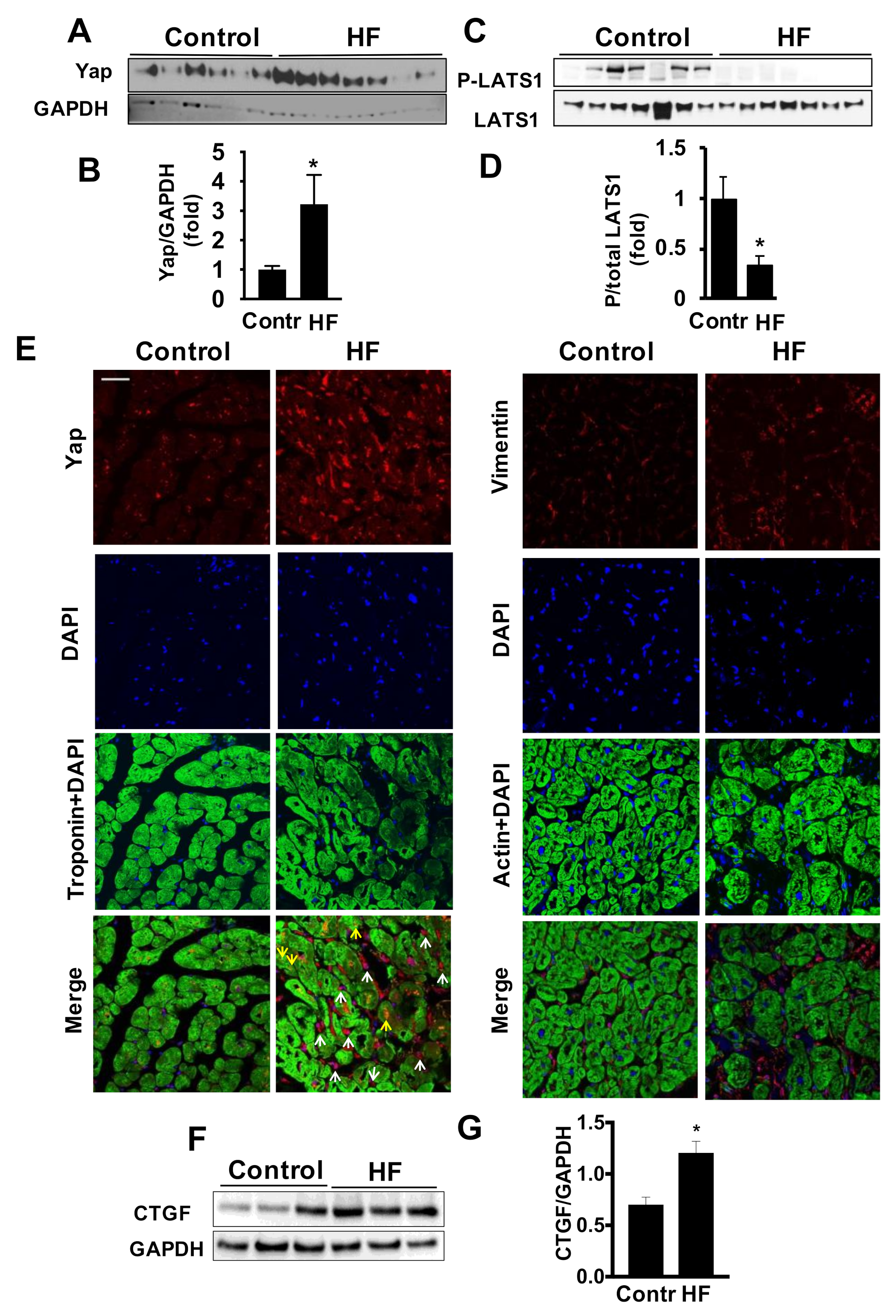{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cardiac Accumulation of Yap in LV of Patients with HF
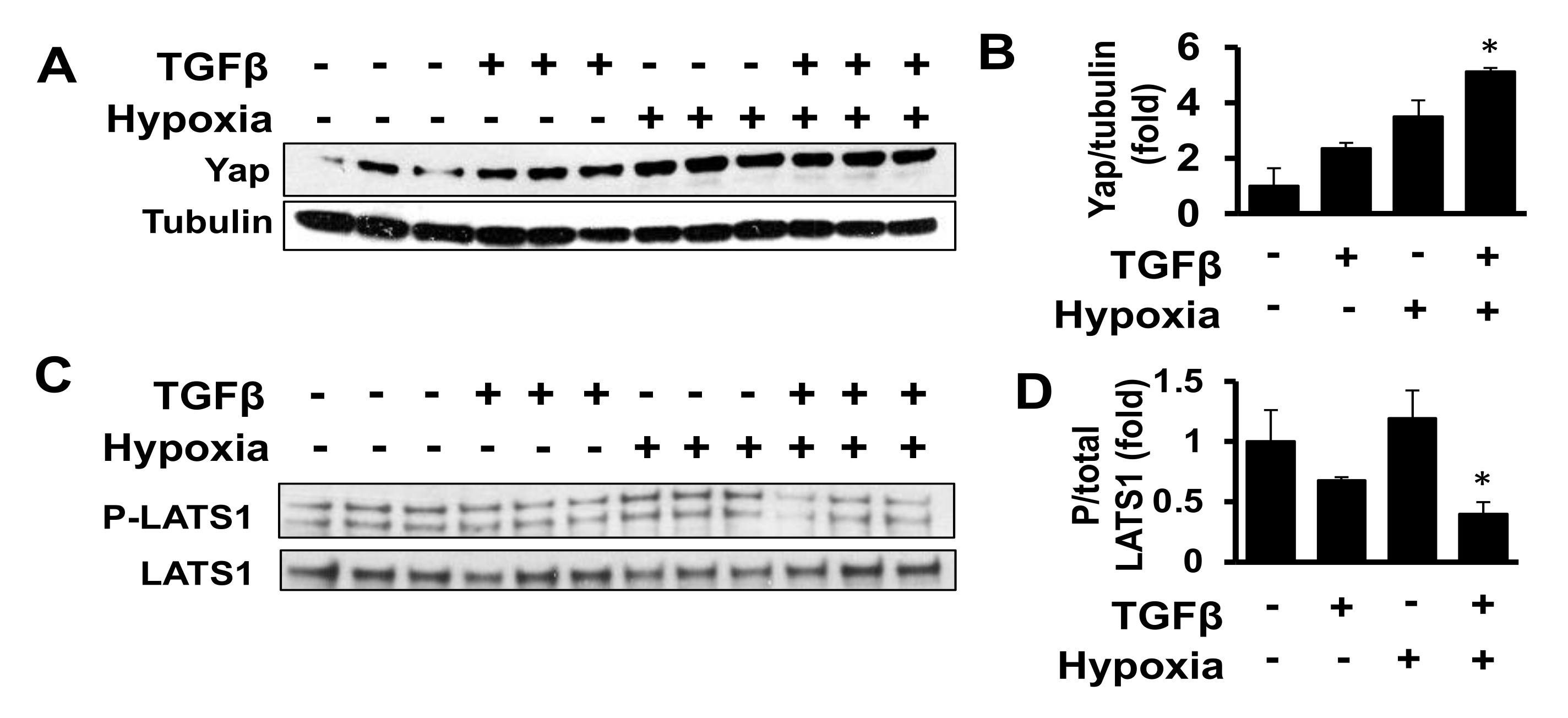
2.2. Combined Exposure to TGF-β and Hypoxia Promotes Yap Accumulation in Human Adult Cardiac Fibroblasts
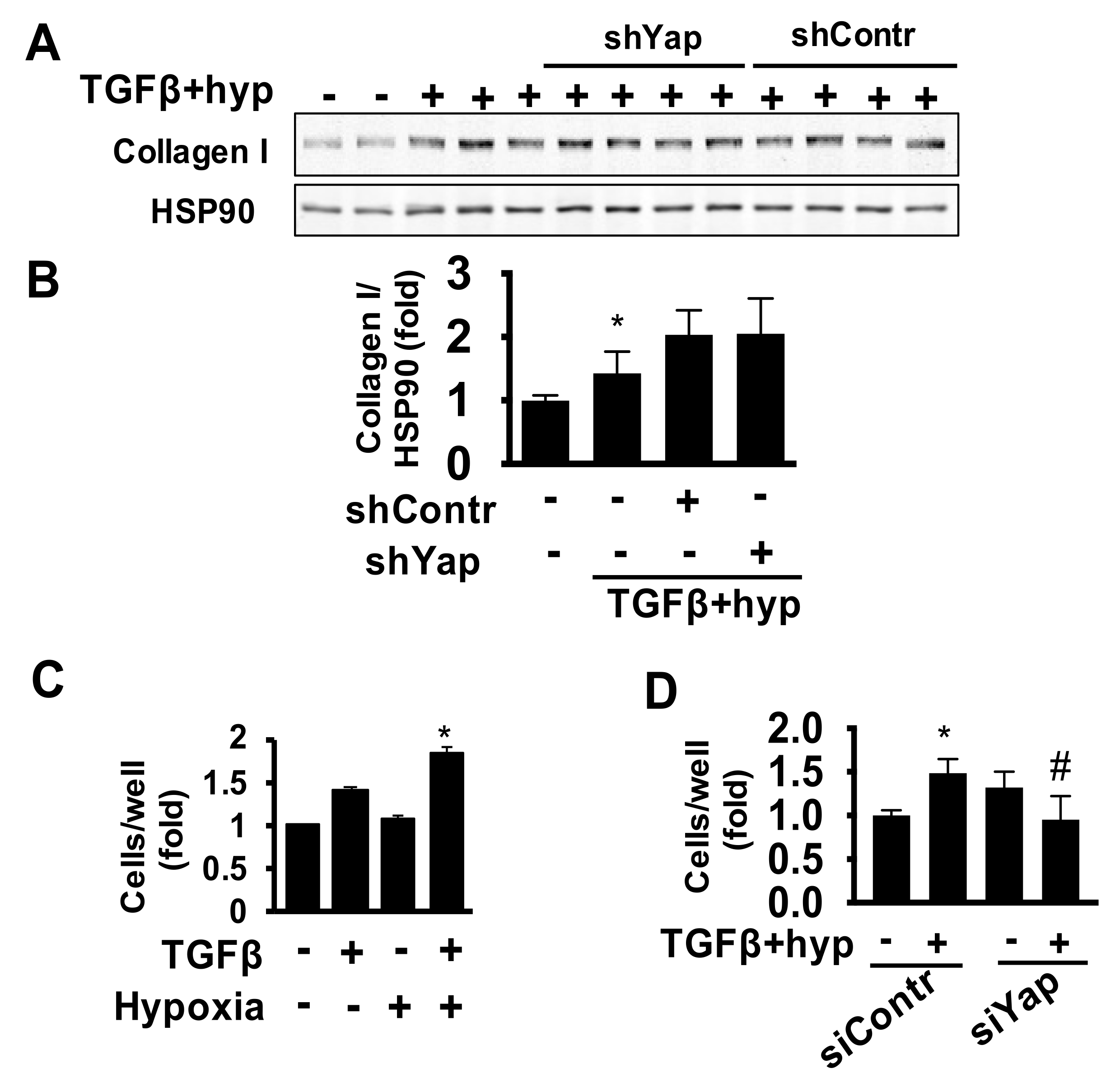
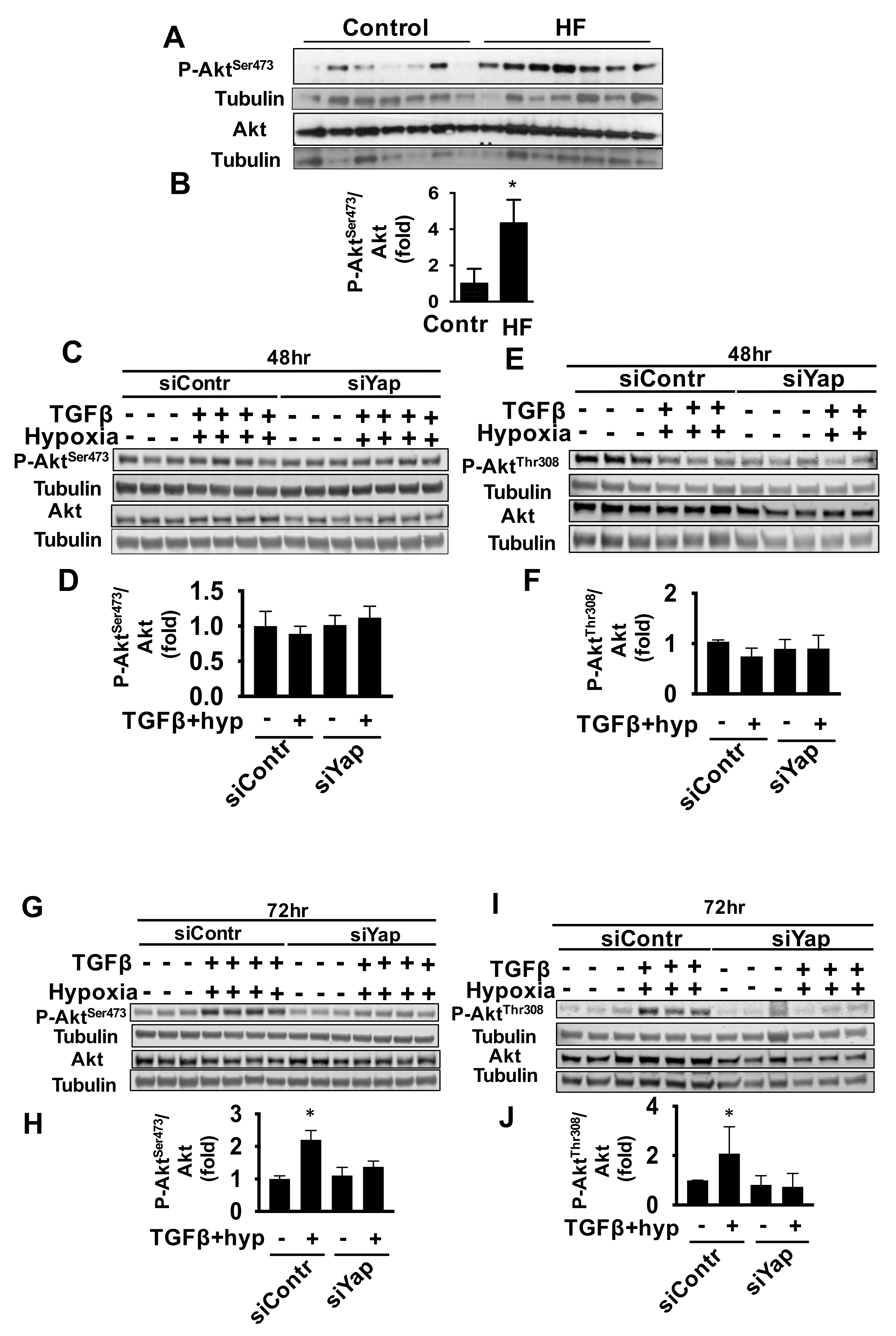
2.3. Yap Is Required for TGFβ/Hypoxia-Induced Cardiac Fibroblast Proliferation and Akt Phosphorylation, But Not Collagen I Production
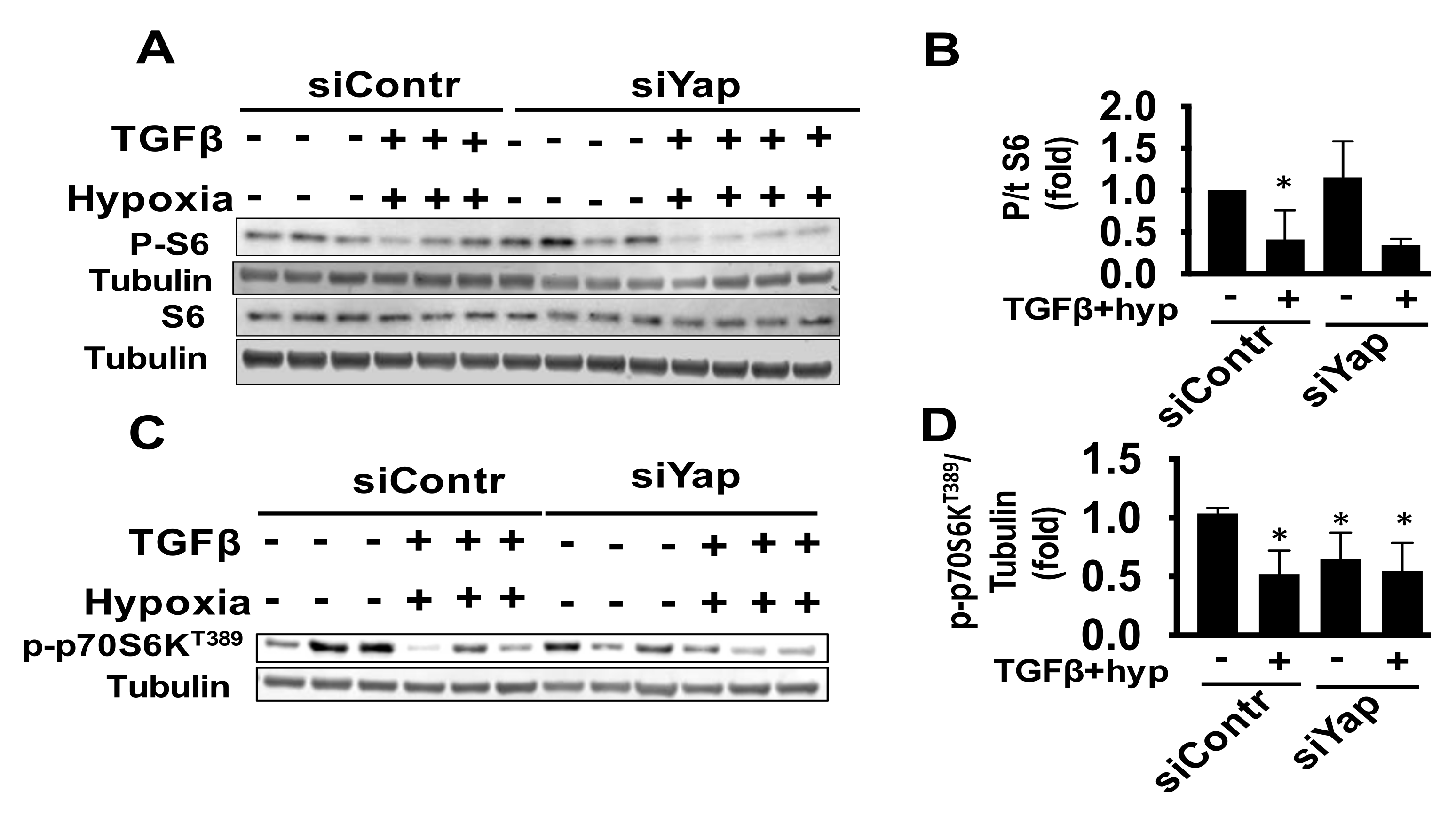
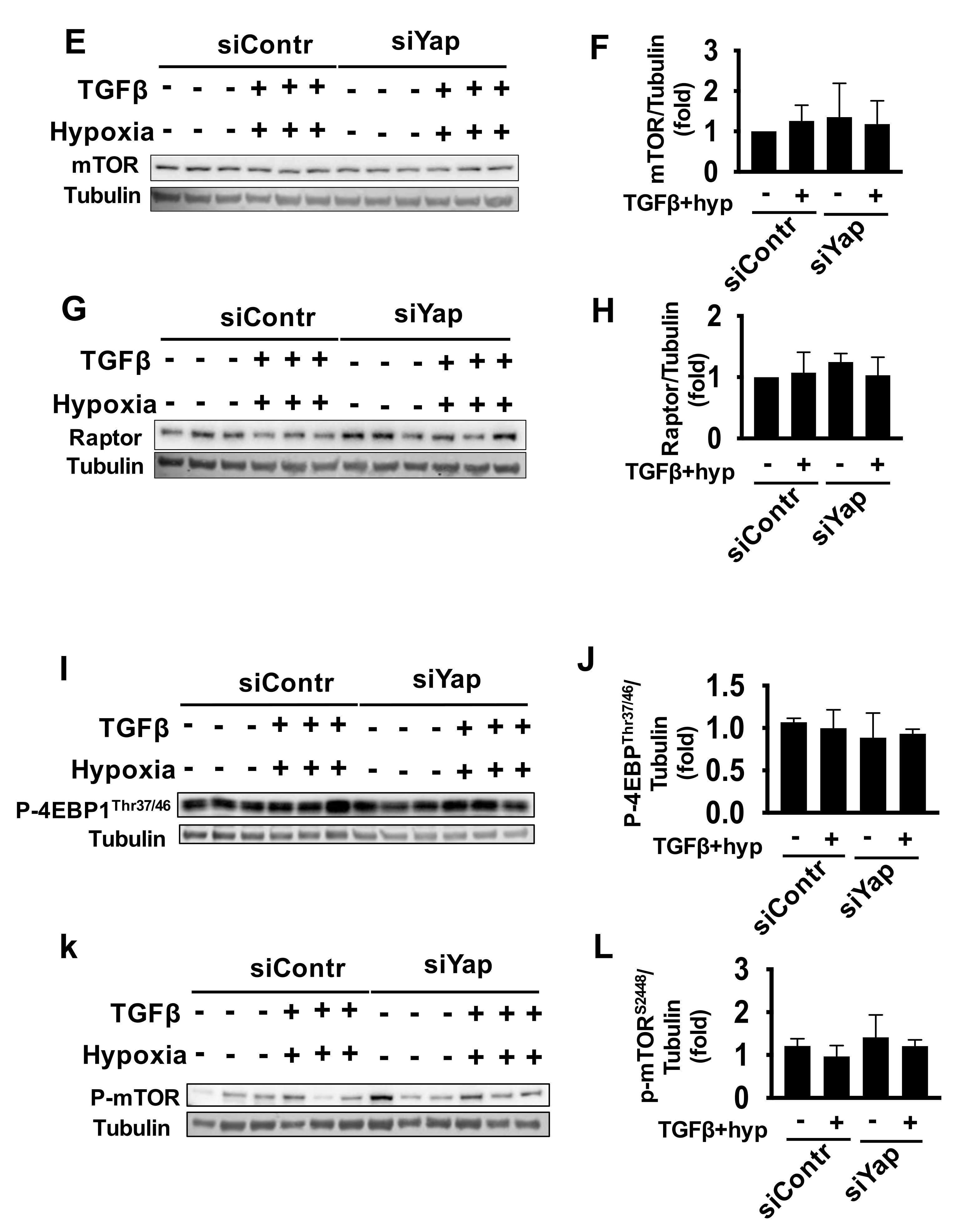
2.4. Combined Exposure of Human Adult Cardiac Fibroblasts to TGF-β and Hypoxia Reduces S6 Activation Independent of Yap
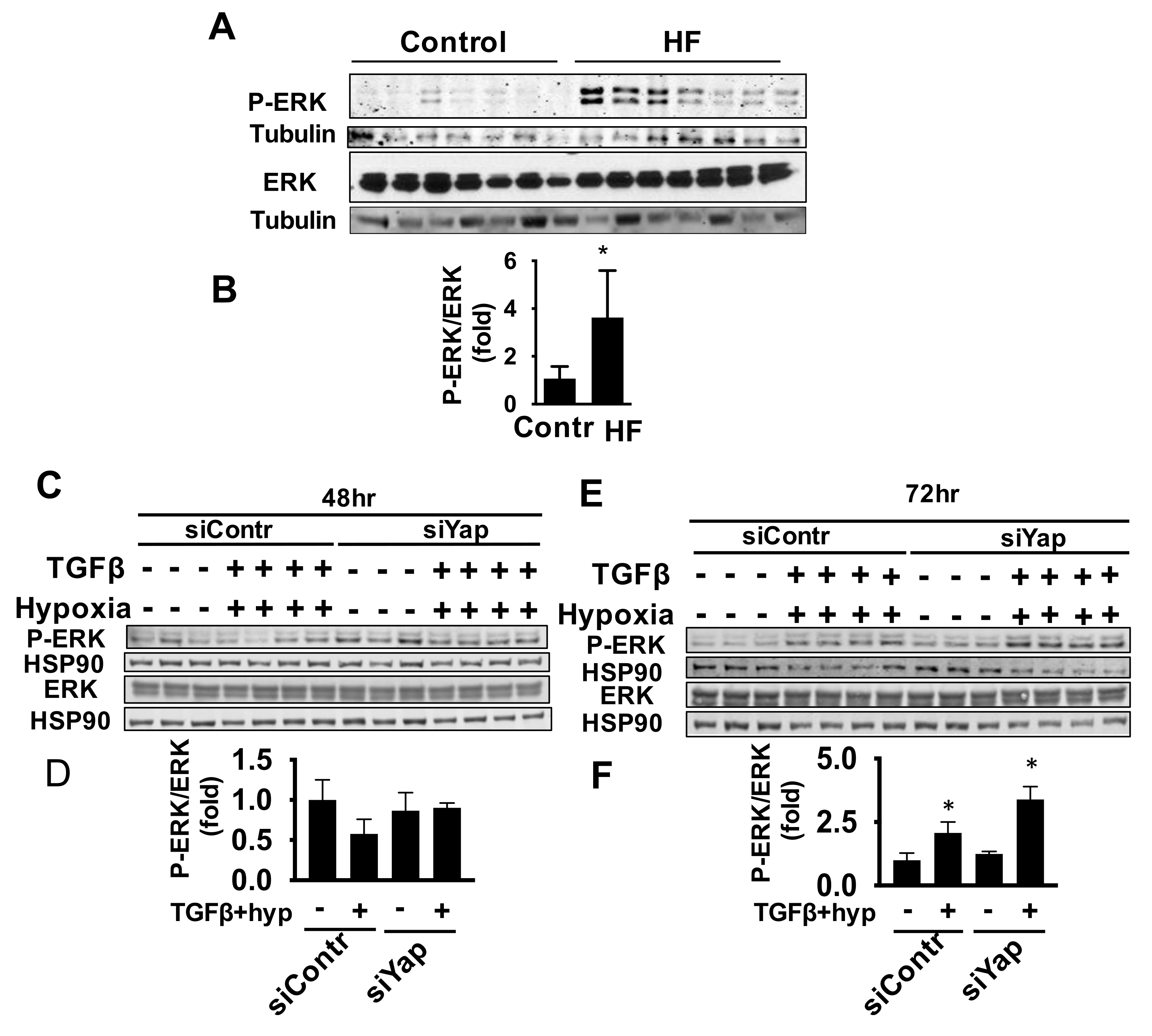
2.5. Yap Is Not Required for TGFβ/Hypoxia-Induced Cardiac Fibroblast ERK Activation
3. Discussion
4. Materials and Methods
4.1. Human Tissue Samples
4.2. Immunofluorescence
4.3. Immunoblot
4.4. Primary Cell Culture of Human Cardiac Fibroblasts
4.5. Cell Growth Assay
4.6. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pagidipati, N.J.; Gaziano, T.A. Estimating deaths from cardiovascular disease: A review of global methodologies of mortality measurement. Circulation 2013, 127, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Norton, G.R.; Woodiwiss, A.J.; Gaasch, W.H.; Mela, T.; Chung, E.S.; Aurigemma, G.P.; Meyer, T.E. Heart failure in pressure overload hypertrophy. The relative roles of ventricular remodeling and myocardial dysfunction. J. Am. Coll. Cardiol. 2002, 39, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Csibi, A.; Blenis, J. Hippo-YAP and mTOR pathways collaborate to regulate organ size. Nat. Cell Biol. 2012, 14, 1244–1245. [Google Scholar] [CrossRef]
- Chiang, J.; Martinez-Agosto, J.A. Effects of mTOR Inhibitors on Components of the Salvador-Warts-Hippo Pathway. Cells 2012, 1, 886–904. [Google Scholar] [CrossRef]
- Kudryashova, T.V.; Goncharov, D.A.; Pena, A.; Kelly, N.; Vanderpool, R.; Baust, J.; Kobir, A.; Shufesky, W.; Mora, A.L.; Morelli, A.E.; et al. HIPPO-Integrin-linked Kinase Cross-Talk Controls Self-Sustaining Proliferation and Survival in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; von Gise, A.; Zhou, P.; Gu, F.; Ma, Q.; Jiang, J.; Yau, A.L.; Buck, J.N.; Gouin, K.A.; van Gorp, P.R.; et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [Green Version]
- Wackerhage, H.; Del Re, D.P.; Judson, R.N.; Sudol, M.; Sadoshima, J. The Hippo signal transduction network in skeletal and cardiac muscle. Sci. Signal 2014, 7, re4. [Google Scholar] [CrossRef]
- Flinn, M.A.; Jeffery, B.E.; O’Meara, C.C.; Link, B.A. Yap is required for scar formation but not myocyte proliferation during heart regeneration in zebrafish. Cardiovasc. Res. 2019, 115, 570–577. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Eghbali, M.; Czaja, M.J.; Zeydel, M.; Weiner, F.R.; Zern, M.A.; Seifter, S.; Blumenfeld, O.O. Collagen chain mRNAs in isolated heart cells from young and adult rats. J. Mol. Cell. Cardiol. 1988, 20, 267–276. [Google Scholar] [CrossRef]
- Song, J.; Zhu, Y.; Li, J.; Liu, J.; Gao, Y.; Ha, T.; Que, L.; Liu, L.; Zhu, G.; Chen, Q.; et al. Pellino1-mediated TGF-beta1 synthesis contributes to mechanical stress induced cardiac fibroblast activation. J. Mol. Cell. Cardiol. 2015, 79, 145–156. [Google Scholar] [CrossRef]
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Investig. 2010, 120, 254–265. [Google Scholar] [CrossRef] [Green Version]
- Tsoutsman, T.; Wang, X.; Garchow, K.; Riser, B.; Twigg, S.; Semsarian, C. CCN2 plays a key role in extracellular matrix gene expression in severe hypertrophic cardiomyopathy and heart failure. J. Mol. Cell. Cardiol. 2013, 62, 164–178. [Google Scholar] [CrossRef]
- Dorn, L.E.; Petrosino, J.M.; Wright, P.; Accornero, F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell. Cardiol. 2018, 121, 205–211. [Google Scholar] [CrossRef]
- Hara, S.; Aoki, S.; Nagata, M.; Shirasuna, K.; Noguchi, T.; Iwata, H. Xanthan gum and locust bean gum substrate improves bovine embryo development. Reprod. Domest. Anim. 2020. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Hou, N.; Wen, Y.; Yuan, X.; Xu, H.; Wang, X.; Li, F.; Ye, B. Activation of Yap1/Taz signaling in ischemic heart disease and dilated cardiomyopathy. Exp. Mol. Pathol. 2017, 103, 267–275. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Sharov, V.G.; Goldstein, S. Cell death, tissue hypoxia and the progression of heart failure. Heart Fail. Rev. 2000, 5, 131–138. [Google Scholar] [CrossRef]
- Gao, Y.; Chu, M.; Hong, J.; Shang, J.; Xu, D. Hypoxia induces cardiac fibroblast proliferation and phenotypic switch: A role for caveolae and caveolin-1/PTEN mediated pathway. J. Thorac. Dis. 2014, 6, 1458–1468. [Google Scholar] [CrossRef]
- Gao, Q.; Guo, M.; Zeng, W.; Wang, Y.; Yang, L.; Pang, X.; Li, H.; Suo, Y.; Jiang, X.; Yu, C. Matrix metalloproteinase 9 secreted by hypoxia cardiac fibroblasts triggers cardiac stem cell migration in vitro. Stem Cells Int. 2015, 2015, 836390. [Google Scholar] [CrossRef] [Green Version]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef] [Green Version]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef] [Green Version]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [Green Version]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Zhu, Y.Z. Role of transforming growth factor-beta in the progression of heart failure. Cell Mol. Life Sci. 2006, 63, 2584–2596. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xu, Y.; Li, X.; Guo, Y.; Liu, G. Inhibition of Rho-kinase ameliorates myocardial remodeling and fibrosis in pressure overload and myocardial infarction: Role of TGF-beta1-TAK1. Toxicol. Lett. 2012, 211, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.B.; Liu, W.J.; Che, H.; Liu, J.; Sun, H.Y.; Li, G.R. Adenosine-5’-triphosphate up-regulates proliferation of human cardiac fibroblasts. Br. J. Pharmacol. 2012, 166, 1140–1150. [Google Scholar] [CrossRef] [Green Version]
- Diez, C.; Nestler, M.; Friedrich, U.; Vieth, M.; Stolte, M.; Hu, K.; Hoppe, J.; Simm, A. Down-regulation of Akt/PKB in senescent cardiac fibroblasts impairs PDGF-induced cell proliferation. Cardiovasc. Res. 2001, 49, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Hart, J.R.; Vogt, P.K. Phosphorylation of AKT: A Mutational Analysis. Oncotarget 2011, 2, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef]
- Yang, W.; Wu, Z.; Yang, K.; Han, Y.; Chen, Y.; Zhao, W.; Huang, F.; Jin, Y.; Jin, W. BMI1 promotes cardiac fibrosis in ischemia-induced heart failure via the PTEN-PI3K/Akt-mTOR signaling pathway. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H61–H69. [Google Scholar] [CrossRef] [Green Version]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [Green Version]
- Boluyt, M.O.; Li, Z.B.; Loyd, A.M.; Scalia, A.F.; Cirrincione, G.M.; Jackson, R.R. The mTOR/p70S6KSignal Transduction Pathway Plays a Role in Cardiac Hypertrophy and Influences Expression of Myosin Heavy Chain Genes in vivo. Cardiovasc. Drugs Ther. 2004, 18, 257–267. [Google Scholar] [CrossRef]
- Beauloye, C.; Bertrand, L.; Krause, U.; Marsin, A.-S.; Dresselaers, T.; Vanstapel, F.; Vanoverschelde, J.-L.; Hue, L. No-Flow Ischemia Inhibits Insulin Signaling in Heart by Decreasing Intracellular pH. Circ. Res. 2001, 88, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Colbert, M.; Krenz, M.; Molkentin, J.D.; Hahn, H.S.; Dorn, G.W., II; Robbins, J. Mediating ERK 1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. J. Clin. Investig. 2007, 117, 2123–2132. [Google Scholar] [CrossRef] [Green Version]
- Purcell, N.H.; Wilkins, B.J.; York, A.; Saba-El-Leil, M.K.; Meloche, S.; Robbins, J.; Molkentin, J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 14074–14079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, B.; Yang, Y.L.; Xu, Z.; Dai, Y.; Liu, S.; Mao, J.H.; Tetsu, O.; Li, H.; Jablons, D.M.; You, L. Inhibition of ERK1/2 down-regulates the Hippo/YAP signaling pathway in human NSCLC cells. Oncotarget 2015, 6, 4357–4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Mao, B.; Luo, W.; Wei, B.; Jiang, W.; Liu, D.; Song, L.; Ji, G.; Yang, Z.; Lai, Y.Q.; et al. The alteration of Hippo/YAP signaling in the development of hypertrophic cardiomyopathy. Basic Res Cardiol 2014, 109, 435. [Google Scholar] [CrossRef]
- Zhou, J. An emerging role for Hippo-YAP signaling in cardiovascular development. J. Biomed. Res. 2014, 28, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153–169.e6. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, Q.; Dang, K.; Ma, S.; Cotton, J.L.; Yang, S.; Zhu, L.J.; Deng, A.C.; Ip, Y.T.; Johnson, R.L.; et al. YAP/TAZ Activation Drives Uveal Melanoma Initiation and Progression. Cell Rep. 2019, 29, 3200–3211. [Google Scholar] [CrossRef] [Green Version]
- Lian, H.; Ma, Y.; Feng, J.; Dong, W.; Yang, Q.; Lu, D.; Zhang, L. Heparin-binding EGF-like growth factor induces heart interstitial fibrosis via an Akt/mTor/p70s6k pathway. PLoS ONE 2012, 7, e44946. [Google Scholar] [CrossRef] [Green Version]
- Ai, F.; Chen, M.; Yu, B.; Yang, Y.; Xu, G.; Gui, F.; Liu, Z.; Bai, X.; Chen, Z. Berberine regulates proliferation, collagen synthesis and cytokine secretion of cardiac fibroblasts via AMPK-mTOR-p70S6K signaling pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 12509–12516. [Google Scholar]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Sanjani, M.; Shoushtari, A.H.; Quiroz, M.; Baust, J.; Sestito, S.F.; Mosher, M.; Ross, M.; McTiernan, C.F.; St Croix, C.M.; Bilonick, R.A.; et al. Cardiac CD47 drives left ventricular heart failure through Ca2+-CaMKII-regulated induction of HDAC3. J. Am. Heart Assoc. 2014, 3, e000670. [Google Scholar] [CrossRef] [Green Version]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharifi-Sanjani, M.; Berman, M.; Goncharov, D.; Alhamaydeh, M.; Avolio, T.G.; Baust, J.; Chang, B.; Kobir, A.; Ross, M.; St. Croix, C.; et al. Yes-Associated Protein (Yap) Is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation. Int. J. Mol. Sci. 2021, 22, 6164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116164
Sharifi-Sanjani M, Berman M, Goncharov D, Alhamaydeh M, Avolio TG, Baust J, Chang B, Kobir A, Ross M, St. Croix C, et al. Yes-Associated Protein (Yap) Is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation. International Journal of Molecular Sciences. 2021; 22(11):6164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116164
Chicago/Turabian StyleSharifi-Sanjani, Maryam, Mariah Berman, Dmitry Goncharov, Mohammad Alhamaydeh, Theodore Guy Avolio, Jeffrey Baust, Baojun Chang, Ahasanul Kobir, Mark Ross, Claudette St. Croix, and et al. 2021. "Yes-Associated Protein (Yap) Is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation" International Journal of Molecular Sciences 22, no. 11: 6164. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116164