
Neuronal Cytoskeleton in Intellectual Disability: From Systems Biology and Modeling to Therapeutic Opportunities

 ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. From Genetics to Core Regulatory Modules
2.1. Chromatin Modification and Transcriptional Regulation
2.2. Signal Transduction
2.3. Ubiquitination System
2.4. Metabolism
2.5. Synaptic Function
2.6. Cytoskeleton Dynamics and Rho GTPases Signaling

3. Cytoskeleton Functions in Neuronal Development
3.1. The Core Regulation of Actin Dynamics
3.1.1. Rho GTPases and Effectors
3.1.2. GAPs and GEFs
3.1.3. Actin Binding Proteins

3.2. Synaptogenesis and Synaptic Plasticity
3.3. The Role of Microtubules in ID
4. Cytoskeleton in Non-Neuronal Cells and ID

5. In Silico Modeling of Cytoskeleton Regulation
- ‑
- simultaneously consider a large number of interacting proteins and link their relationships with emerging phenotypes
- ‑
- elaborate hypotheses and design new experiments
- ‑
- search for biomarkers and druggable targets for translational purposes
6. Therapeutic Opportunities for Cytoskeleton-Related Forms of ID and Related Conditions
- (1)
- Starting from the hypothesis that learning deficits in NF1 (neurofibromatosis 1) are caused by an excess of RAS activity and by the consequent increase in GABA-mediated inhibition, Nf1+/− mice (a model of NF1) were treated with both farnesyl-transferase and HMG-CoA reductase inhibitors to decrease RAS activity; the treatment was successful in rescuing spatial memory and attention deficits [385,386].
- (2)
- The observation that the hippocampal signaling through postsynaptic GABA receptors was significantly increased in Ts65Dn mice (a model of DS) prompted the testing of selective GABAB and GABAA receptor antagonists; both treatments rescued memory in novel place and object recognition tests and contextual fear conditioning tasks [387,388,389]. It was later shown that GABAA receptor signaling is excitatory rather than inhibitory in Ts65Dn mice and DS patients, because of an increased hippocampal expression of the cation chloride cotransporter SLC12A2 (solute carrier family 12 member 2). Its inhibitor, bumetanide, a common diuretic, was able to restore synaptic plasticity and hippocampus-dependent memory in adult Ts65Dn mice [390]. Recently, the discovery that the over-activation of microglia plays a role in the DS phenotype widened our knowledge about this pathology, as it has resulted in successful testing of anti-inflammatory drugs to rescue cognitive impairments [391].
- (3)
- As the mutation in CREBBP (CREB-binding protein) is considered the most significant mutation in Rubinstein–Taybi syndrome, pharmacological strategies to enhance CREBBP-dependent gene expression were investigated. Crebbp+/− mice (a model of Rubinstein–Taybi syndrome) treated with either PDE4 inhibitor (to enhance cAMP signaling) or HDAC inhibitor (to halt the counterpart of the histone acetylation function of CREBBP) were rescued for long-term memory deficit [392,393]. Similarly, Kmt2d+/βGeo mice (a model of Kabuki syndrome) were rescued by the treatment with HDAC inhibitors [394].
- (4)
- Hyperactivity of MTOR signaling is observed in several neurodevelopmental disorders, the so-called “mTORopathies”; therefore, it is not surprising that MTOR inhibitors were extensively tested. Heterozygous mutations in either TSC1 or TSC2 that form an MTOR-inhibiting complex can cause tuberous sclerosis by hyperactivating MTOR signaling. Tsc2+/− and Tsc1 homozygous mutant mice (a model of tuberous sclerosis), were treated with rapamycin, rescuing spatial learning and context discrimination deficits together with neurological findings [395,396]. Rapamycin prevented seizures and rescued defective cortical lamination and heterotopia in Strada-KO model, an upstream inhibitor of MTORC1, in a rare NDD called Pretzel syndrome [397]. Interestingly, MTOR inhibitors are currently in clinical trials as antiepileptic agents [398]. This class of drugs was also tested to revert neuronal hypertrophy caused by PTEN deficiency in Lhermitte–Duclose and Cowden syndromes [27] and is seen as a promising approach for the treatment of ASD [399].
- (5)
- Fmr1−/− mice (a model of FXS) were used to study the GABAAergic deficit that underlies FXS; treatment with a mGluR5 antagonist rescued associative learning [400], as well as treatments with positive allosteric modulators of GABAA receptors in animal models [401] and GABAB receptor agonists, which, in patients, seemed to rescue behavioral functions [20]. Additionally, the antibiotic minocycline, a metalloproteinase inhibitor, appeared to be effective in patients [402].
6.1. Pharmacological Stabilization of Microtubules
6.2. Pharmacological Modulation of Actin Dynamics
6.3. Modulation of Small GTPases Activity
7. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ID | Intellectual Disability |
| SB | Systems Biology |
| NDD | Neurodevelopmental Disorder |
| XLID | X-linked Intellectual Disability |
| PPI | Protein::Protein Interaction |
| GO | Gene Ontology |
| GDD | Global Developmental Delay |
| ASD | Autism Spectrum Disorder |
| LOF | Loss Of Function |
| FXS | Fragile X Syndrome |
| LTP | Long Term Potentiation |
| LTD | Long Term Depression, |
| DS | Down syndrome |
| G-actin | Glomerular actin |
| F-actin | Fibrous actin |
| GAP | GTPase Activating Protein |
| GEF | Guanine Exchange Factor |
| KD | Knock-Down |
| ABP | Actin Binding Protein |
| NPF | Nucleation Promoting Factor |
| WRC | WAVE Regulatory Complex |
| PSD | Postsynaptic Density |
| +TIP | Microtubule Plus End Tracking Protein |
| CLIP | Cytoplasmic Linker Protein |
References
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.; Kleefstra, T.; Kramer, J.; et al. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, Y.R.; Kuchay, R.A.H. Advances in identification of genes involved in autosomal recessive intellectual disability: A brief review. J. Med. Genet. 2019, 56, 567–573. [Google Scholar] [CrossRef]
- Van Bokhoven, H. Genetic and Epigenetic Networks in Intellectual Disabilities. Annu. Rev. Genet. 2011, 45, 81–104. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, K.; Milton, M.; Smith, G.; Ouellette-Kuntz, H. Systematic Review of the Prevalence and Incidence of Intellectual Disabilities: Current Trends and Issues. Curr. Dev. Disord. Rep. 2016, 3, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Mottron, L.; Belleville, S.; Rouleau, G.A.; Collignon, O. Linking neocortical, cognitive, and genetic variability in autism with alterations of brain plasticity: The Trigger-Threshold-Target model. Neurosci. Biobehav. Rev. 2014, 47, 735–752. [Google Scholar] [CrossRef] [Green Version]
- Ba, W.; van der Raadt, J.; Kasri, N.N. Rho GTPase signaling at the synapse: Implications for intellectual disability. Exp. Cell Res. 2013, 319, 2368–2374. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Buonanno, A.; Passafarro, M.; Sala, C.; Sweet, R.A. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J. Neurochem. 2013, 126, 165–182. [Google Scholar] [CrossRef]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nat. Cell Biol. 2011, 472, 100–104. [Google Scholar] [CrossRef]
- Tejada-Simon, M.V. Modulation of actin dynamics by Rac1 to target cognitive function. J. Neurochem. 2015, 133, 767–779. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Hartline, E.; et al. The Gene Ontology resource: Enriching a Gold mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, N.; Zhang, Y.; Du, Y.; Zhang, T.; Li, Z.; Wu, J.; Wang, X. Prioritized High-Confidence Risk Genes for Intellectual Disability Reveal Molecular Convergence During Brain Development. Front. Genet. 2018, 9, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- InanlooRahatloo, K.; Peymani, F.; Kahrizi, K.; Najmabadi, H. Whole-Transcriptome Analysis Reveals Dysregulation of Actin-Cytoskeleton Pathway in Intellectual Disability Patients. Neuroscience 2019, 404, 423–444. [Google Scholar] [CrossRef] [PubMed]
- Piergiorge, R.M.; De Vasconcelos, A.T.R.; Pimentel, M.M.G.; Santos-Rebouças, C.B. Strict network analysis of evolutionary conserved and brain-expressed genes reveals new putative candidates implicated in Intellectual Disability and in Global Development Delay. World J. Biol. Psychiatry 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone Methylation Regulates Memory Formation. J. Neurosci. 2010, 30, 3589–3599. [Google Scholar] [CrossRef]
- Baker-Andresen, D.; Ratnu, V.S.; Bredy, T.W. Dynamic DNA methylation: A prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci. 2013, 36, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Santen, G.W.E.; Aten, E.; Sun, Y.; Almomani, R.; Gilissen, C.; Nielsen, M.; Kant, S.G.; Snoeck, I.N.; Peeters, E.A.J.; Hilhorst-Hofstee, Y.; et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 2012, 44, 379–380. [Google Scholar] [CrossRef]
- Willemsen, M.; Silfhout, A.V.-V.; Nillesen, W.; Wissink-Lindhout, W.; Van Bokhoven, H.; Philip, N.; Berry-Kravis, E.; Kini, U.; Van Ravenswaaij-Arts, C.; Chiaie, B.D.; et al. Update on Kleefstra Syndrome. Mol. Syndr. 2012, 2, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Iwase, S.; Lan, F.; Bayliss, P.; De La Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-Linked Mental Retardation Gene SMCX/JARID1C Defines a Family of Histone H3 Lysine 4 Demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-H.; Yu, H.-I.; Tarn, W.-Y. DDX3 Modulates Neurite Development via Translationally Activating an RNA Regulon Involved in Rac1 Activation. J. Neurosci. 2016, 36, 9792–9804. [Google Scholar] [CrossRef]
- Gudenas, B.L.; Wang, L. Gene Coexpression Networks in Human Brain Developmental Transcriptomes Implicate the Association of Long Noncoding RNAs with Intellectual Disability. Bioinform. Biol. Insights 2015, 9, BBI-S29435. [Google Scholar] [CrossRef] [PubMed]
- Kuechler, A.; Willemsen, M.H.; Albrecht, B.; Bacino, C.A.; Bartholomew, D.W.; Van Bokhoven, H.; Boogaard, M.J.H.V.D.; Bramswig, N.; Buettner, C.; Cremer, K.; et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 2015, 134, 97–109. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Santini, E.; Huynh, T.; MacAskill, A.F.; Carter, A.G.; Pierre, P.; Ruggero, D.; Kaphzan, H.; Klann, E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013, 493, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.-H.; Zhu, X.; Zhang, J.; Baker, S.J. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 12923–12928. [Google Scholar] [CrossRef] [Green Version]
- Magdalon, J.; Sánchez-Sánchez, S.M.; Griesi-Oliveira, K.; Sertié, A.L. Dysfunctional mTORC1 Signaling: A Convergent Mechanism between Syndromic and Nonsyndromic Forms of Autism Spectrum Disorder? Int. J. Mol. Sci. 2017, 18, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, V.; Kleefstra, T.; Hardy, A.; Heise, I.; Maggi, S.; Willemsen, M.H.; Hilton, H.; Esapa, C.; Simon, M.; Buenavista, M.-T.; et al. Dominant β-catenin mutations cause intellectual disability with recognizable syndromic features. J. Clin. Investig. 2014, 124, 1468–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierpont, M.E.M.; Magoulas, P.L.; Adi, S.; Kavamura, M.I.; Neri, G.; Noonan, J.; Pierpont, E.I.; Reinker, K.; Roberts, A.E.; Shankar, S.; et al. Cardio-Facio-Cutaneous Syndrome: Clinical Features, Diagnosis, and Management Guidelines. Pediatrics 2014, 134, e1149–e1162. [Google Scholar] [CrossRef] [Green Version]
- Papale, A.; D’Isa, R.; Menna, E.; Cerovic, M.; Solari, N.; Hardingham, N.; Cambiaghi, M.; Cursi, M.; Barbacid, M.; Leocani, L.; et al. Severe Intellectual Disability and Enhanced Gamma-Aminobutyric Acidergic Synaptogenesis in a Novel Model of Rare RASopathies. Biol. Psychiatry 2017, 81, 179–192. [Google Scholar] [CrossRef]
- Yi, J.J.; Berrios, J.; Newbern, J.; Snider, W.D.; Philpot, B.D.; Hahn, K.M.; Zylka, M.J. An Autism-Linked Mutation Disables Phosphorylation Control of UBE3A. Cell 2015, 162, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Ding, H.; Lu, Z.; Li, Y.; Pan, Y.; Ning, T.; Ke, Y. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 2008, 13, 285–294. [Google Scholar] [CrossRef]
- Moortgat, S.; Berland, S.; Aukrust, I.; Maystadt, I.; Baker, L.; Benoit, V.; Caro-Llopis, A.; Cooper, N.S.; Debray, F.-G.; Faivre, L.; et al. HUWE1 variants cause dominant X-linked intellectual disability: A clinical study of 21 patients. Eur. J. Hum. Genet. 2018, 26, 64–74. [Google Scholar] [CrossRef] [Green Version]
- D’Arca, D.; Zhao, X.; Xu, W.; Ramirez-Martinez, N.C.; Iavarone, A.; Lasorella, A. Huwe1 ubiquitin ligase is essential to synchronize neuronal and glial differentiation in the developing cerebellum. Proc. Natl. Acad. Sci. USA 2010, 107, 5875–5880. [Google Scholar] [CrossRef] [Green Version]
- Londin, E.R.; Adijanto, J.; Philp, N.; Novelli, A.; Vitale, E.; Perria, C.; Serra, G.; Alesi, V.; Surrey, S.; Fortina, P. Donor splice-site mutation in CUL4Bis likely cause of X-linked intellectual disability. Am. J. Med. Genet. Part A 2014, 164, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Tsai, M.-S.; Lin, C.-Y.; Yu, I.-S.; Chen, Y.-T.; Lin, S.-R.; Juan, L.-W.; Chen, Y.-T.; Hsu, H.-M.; Lee, L.-J.; et al. Rescue of the genetically engineered Cul4b mutant mouse as a potential model for human X-linked mental retardation. Hum. Mol. Genet. 2012, 21, 4270–4285. [Google Scholar] [CrossRef] [Green Version]
- García-Cazorla, A.; Wolf, N.I.; Serrano, M.; Pérez-Dueñas, B.; Pineda, M.; Campistol, J.; Fernández-Alvarez, E.; Colomer, J.; DiMauro, S.; Hoffmann, G.F. Inborn errors of metabolism and motor disturbances in children. J. Inherit. Metab. Dis. 2009, 32, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Salomons, G.S.; van Dooren, S.J.; Verhoeven, N.M.; Cecil, K.M.; Ball, W.S.; Degrauw, T.J.; Jakobs, C. X-Linked Creatine-Transporter Gene (SLC6A8) Defect: A New Creatine-Deficiency Syndrome. Am. J. Hum. Genet. 2001, 68, 1497–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, E.H.; Almeida, L.S.; Kleefstra, T.; Degrauw, R.S.; Yntema, H.G.; Bahi, N.; Moraine, C.; Ropers, H.-H.; Fryns, J.-P.; Degrauw, T.J.; et al. High Prevalence of SLC6A8 Deficiency in X-Linked Mental Retardation. Am. J. Hum. Genet. 2004, 75, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knerr, I.; Gibson, K.M.; Jakobs, C.; Pearl, P.L. Neuropsychiatric morbidity in adolescent and adult succinic semialdehyde dehydrogenase deficiency patients. CNS Spectr. 2008, 13, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Nicolaides, P.; Liebsch, D.; Dale, N.; Leonard, J.; Surtees, R. Neurological outcome of patients with ornithine carbamoyltransferase deficiency. Arch. Dis. Child. 2002, 86, 54–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Adamo, P.; Menegon, A.; Nigro, C.L.; Grasso, M.; Gulisano, M.; Tamanini, F.; Bienvenu, T.; Gedeon, A.K.; Oostra, B.; Wu, S.-K.; et al. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat. Genet. 1998, 19, 134–139. [Google Scholar] [CrossRef]
- Bianchi, V.; Farisello, P.; Baldelli, P.; Meskenaite, V.; Milanese, M.; Vecellio, M.; Mühlemann, S.; Lipp, H.P.; Bonanno, G.; Benfenati, F.; et al. Cognitive impairment in Gdi1-deficient mice is associated with altered synaptic vesicle pools and short-term synaptic plasticity, and can be corrected by appropriate learning training. Hum. Mol. Genet. 2008, 18, 105–117. [Google Scholar] [CrossRef]
- Blumkin, L.; Michelson, M.; Leshinsky-Silver, E.; Kivity, S.; Lev, D.; Lerman-Sagie, T. Congenital Ataxia, Mental Retardation, and Dyskinesia Associated with a Novel CACNA1A Mutation. J. Child Neurol. 2010, 25, 892–897. [Google Scholar] [CrossRef]
- Luo, X.; Rosenfeld, J.A.; Yamamoto, S.; Harel, T.; Zuo, Z.; Hall, M.; Wierenga, K.J.; Pastore, M.T.; Bartholomew, D.; Delgado, M.R.; et al. Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 2017, 13, e1006905. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Daoud, H.; Piton, A.; Gauthier, J.; Dobrzeniecka, S.; Krebs, M.-O.; Joober, R.; Lacaille, J.-C.; Nadeau, A.; Milunsky, J.M.; et al. De Novo SYNGAP1 Mutations in Nonsyndromic Intellectual Disability and Autism. Biol. Psychiatry 2011, 69, 898–901. [Google Scholar] [CrossRef]
- Holder, J.L., Jr.; Hamdan, F.F.; Michaud, J.L. SYNGAP1-Related Intellectual Disability; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hotulainen, P.; Hoogenraad, C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Lee, C.W.; Fan, Y.; Komlos, D.; Tang, X.; Sun, C.; Yu, K.; Hartzell, H.C.; Chen, G.; Bamburg, J.R.; et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci. 2010, 13, 1208–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriroopreddy, R.; Sajeed, R.; Raghuraman, P.; Sudandiradoss, C. Differentially expressed gene (DEG) based protein-protein interaction (PPI) network identifies a spectrum of gene interactome, transcriptome and correlated miRNA in nondisjunction Down syndrome. Int. J. Biol. Macromol. 2019, 122, 1080–1089. [Google Scholar] [CrossRef]
- Bhambhvani, H.P.; Mueller, T.M.; Simmons, M.S.; Meador-Woodruff, J.H. Actin polymerization is reduced in the anterior cingulate cortex of elderly patients with schizophrenia. Transl. Psychiatry 2017, 7, 1278. [Google Scholar] [CrossRef] [Green Version]
- Michaelsen-Preusse, K.; Feuge, J.; Korte, M. Imbalance of synaptic actin dynamics as a key to fragile X syndrome? J. Physiol. 2018, 596, 2773–2782. [Google Scholar] [CrossRef] [Green Version]
- Bhattacherjee, A.; Mu, Y.; Winter, M.K.; Knapp, J.R.; Eggimann, L.S.; Gunewardena, S.S.; Kobayashi, K.; Kato, S.; Krizsan-Agbas, D.; Smith, P.G. Neuronal cytoskeletal gene dysregulation and mechanical hypersensitivity in a rat model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E6952–E6961. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Yang, X.; Shi, L. Rho GTPase Regulators and Effectors in Autism Spectrum Disorders: Animal Models and Insights for Therapeutics. Cells 2020, 9, 835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesi-Oliveira, K.; Suzuki, A.M.; Alves, A.Y.; Mafra, A.C.C.N.; Yamamoto, G.L.; Ezquina, S.; Magalhães, Y.T.; Forti, F.L.; Sertie, A.L.; Zachi, E.C.; et al. Actin cytoskeleton dynamics in stem cells from autistic individuals. Sci. Rep. 2018, 8, 11138. [Google Scholar] [CrossRef]
- Barón-Mendoza, I.; García, O.; Calvo-Ochoa, E.; Rebollar-García, J.O.; Garzón-Cortés, D.; Haro, R.; González-Arenas, A. Alterations in neuronal cytoskeletal and astrocytic proteins content in the brain of the autistic-like mouse strain C58/J. Neurosci. Lett. 2018, 682, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mantilla, A.J.; Moreno-De-Luca, A.; Ledbetter, D.H.; Martin, C.L. A Cross-Disorder Method to Identify Novel Candidate Genes for Developmental Brain Disorders. JAMA Psychiatry 2016, 73, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. McKusick’s Online Mendelian Inheritance in Man (OMIM®). Nucleic Acids Res. 2009, 37, D793–D796. [Google Scholar] [CrossRef] [PubMed]
- Kahn, O.I.; Baas, P.W. Microtubules and Growth Cones: Motors Drive the Turn. Trends Neurosci. 2016, 39, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Lowery, L.A.; Van Vactor, D. The trip of the tip: Understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 2009, 10, 332–343. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, E.; Sabry, J. Making the connection: Cytoskeletal rearrangements during growth cone guidance. Cell 1995, 83, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.A.; Tian, G.; Cowan, N.J. The α- and β-tubulin folding pathways. Trends Cell Biol. 1997, 7, 479–484. [Google Scholar] [CrossRef]
- Kunze, D.; Rüstow, B. Pathobiochemical Aspects of Cytoskeleton Components. Clin. Chem. Lab. Med. 1993, 31, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.W.; Jope, R.S. The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. J. Neurosci. Res. 1992, 33, 505–512. [Google Scholar] [CrossRef]
- Winder, S.J.; Ayscough, K.R. Actin-binding proteins. J. Cell Sci. 2005, 118, 651–654. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, T.J.; Kirschner, M.W. Dynamic instability of microtubule growth. Nat. Cell Biol. 1984, 312, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Haydon, P.G.; Drapeau, P. From contact to connection: Early events during synaptogenesis. Trends Neurosci. 1995, 18, 196–201. [Google Scholar] [CrossRef]
- Korobova, F.; Svitkina, T. Molecular Architecture of Synaptic Actin Cytoskeleton in Hippocampal Neurons Reveals a Mechanism of Dendritic Spine Morphogenesis. Mol. Biol. Cell 2010, 21, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. Towards an Understanding of Synapse Formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, Y.; De Roo, M.; Muller, D. Dendritic spine formation and stabilization. Curr. Opin. Neurobiol. 2009, 19, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Spence, E.F.; Soderling, S.H. Actin Out: Regulation of the Synaptic Cytoskeleton. J. Biol. Chem. 2015, 290, 28613–28622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusifov, R.; Tippmann, A.; Staiger, J.F.; Schlüter, O.M.; Löwel, S. Spine dynamics of PSD-95-deficient neurons in the visual cortex link silent synapses to structural cortical plasticity. Proc. Natl. Acad. Sci. USA 2021, 118, e2022701118. [Google Scholar] [CrossRef]
- Sala, C.; Piëch, V.; Wilson, N.R.; Passafaro, M.; Liu, G.; Sheng, M. Regulation of Dendritic Spine Morphology and Synaptic Function by Shank and Homer. Neuron 2001, 31, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Repetto, D.; Camera, P.; Melani, R.; Morello, N.; Russo, I.; Calcagno, E.; Tomasoni, R.; Bianchi, F.T.; Berto, G.; Giustetto, M.; et al. P140cap Regulates Memory and Synaptic Plasticity through Src-Mediated and Citron-N-Mediated Actin Reorganization. J. Neurosci. 2014, 34, 1542–1553. [Google Scholar] [CrossRef] [Green Version]
- Huganir, R.L.; Nicoll, R.A. AMPARs and Synaptic Plasticity: The Last 25 Years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef] [Green Version]
- Allison, D.W.; Gelfand, V.I.; Spector, I.; Craig, A.M. Role of Actin in Anchoring Postsynaptic Receptors in Cultured Hippocampal Neurons: Differential Attachment of NMDA versus AMPA Receptors. J. Neurosci. 1998, 18, 2423–2436. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-H.; Lisman, J.E. A Role of Actin Filament in Synaptic Transmission and Long-Term Potentiation. J. Neurosci. 1999, 19, 4314–4324. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Xiao, M.-Y.; Nicoll, R.A. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 1261–1266. [Google Scholar] [CrossRef]
- Duffney, L.J.; Wei, J.; Cheng, J.; Liu, W.; Smith, K.R.; Kittler, J.T.; Yan, Z. Shank3 Deficiency Induces NMDA Receptor Hypofunction via an Actin-Dependent Mechanism. J. Neurosci. 2013, 33, 15767–15778. [Google Scholar] [CrossRef] [Green Version]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef]
- Kozma, R.; Sarner, S.; Ahmed, S.; Lim, L. Rho family GTPases and neuronal growth cone remodelling: Relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 1997, 17, 1201–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.L. Cytoskeletal movements and substrate interactions during initiation of neurite outgrowth by sympathetic neurons in vitro. J. Neurosci. 1994, 14, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, A.; Minden, A.; Yuste, R. Regulation of dendritic spine morphology by the rho family of small GTPases: Antagonistic roles of Rac and Rho. Cereb. Cortex 2000, 10, 927–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiens, K.M.; Lin, H.; Liao, D. Rac1 Induces the Clustering of AMPA Receptors during Spinogenesis. J. Neurosci. 2005, 25, 10627–10636. [Google Scholar] [CrossRef]
- Martinez, L.A.; Tejada-Simon, M.V. Pharmacological inactivation of the small GTPase Rac1 impairs long-term plasticity in the mouse hippocampus. Neuropharmacology 2011, 61, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.; Gomez, T.M. Rac1 and RhoA Promote Neurite Outgrowth through Formation and Stabilization of Growth Cone Point Contacts. J. Neurosci. 2006, 26, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Nakamura, T.; Matsuda, M. Spatio-temporal Regulation of Rac1 and Cdc42 Activity during Nerve Growth Factor-induced Neurite Outgrowth in PC12 Cells. J. Biol. Chem. 2004, 279, 713–719. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.-J.; De Vries, P.; De Vries, B.B.A.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef]
- Reijnders, M.R.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Kay, Y.; Sadybekov, A.; Rao, S.; Katritch, V.; Herring, B.E. An Intellectual Disability-Related Missense Mutation in Rac1 Prevents LTP Induction. Front. Mol. Neurosci. 2018, 11, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liao, G.; Waclaw, R.R.; Burns, K.A.; Linquist, D.; Campbell, K.; Zheng, Y.; Kuan, C.-Y. Rac1 Controls the Formation of Midline Commissures and the Competency of Tangential Migration in Ventral Telencephalic Neurons. J. Neurosci. 2007, 27, 3884–3893. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Ren, X.-R.; Huang, Y.-Z.; Xie, Y.; Liu, G.; Saito, H.; Tang, H.; Wen, L.; Brady-Kalnay, S.M.; Mei, L.; et al. Signal Transduction in Neuronal Migration. Cell 2001, 107, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Garvalov, B.K.; Flynn, K.C.; Neukirchen, D.; Meyn, L.; Teusch, N.; Wu, X.; Brakebusch, C.; Bamburg, J.R.; Bradke, F. Cdc42 Regulates Cofilin during the Establishment of Neuronal Polarity. J. Neurosci. 2007, 27, 13117–13129. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Zhu, H.-M.; Chen, Y.; Yan, F.; Liu, Z.-Y.; Li, Z.-L.; Dong, W.-R.; Zhang, L.; Wang, H.-H. Cdc42 Facilitates Axonogenesis by Enhancing Microtubule Stabilization in Primary Hippocampal Neurons. Cell. Mol. Neurobiol. 2021, 1–12. [Google Scholar] [CrossRef]
- Takenouchi, T.; Okamoto, N.; Ida, S.; Uehara, T.; Kosaki, K. Further evidence of a mutation in CDC42 as a cause of a recognizable syndromic form of thrombocytopenia. Am. J. Med. Genet. Part A 2015, 170, 852–855. [Google Scholar] [CrossRef]
- Takenouchi, T.; Kosaki, R.; Niizuma, T.; Hata, K.; Kosaki, K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay. Am. J. Med. Genet. Part A 2015, 167, 2822–2825. [Google Scholar] [CrossRef]
- Martinelli, S.; Krumbach, O.H.; Pantaleoni, F.; Coppola, S.; Amin, E.; Pannone, L.; Nouri, K.; Farina, L.; Dvorsky, R.; Lepri, F.; et al. Functional Dysregulation of CDC42 Causes Diverse Developmental Phenotypes. Am. J. Hum. Genet. 2018, 102, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Motokawa, M.; Watanabe, S.; Nakatomi, A.; Kondoh, T.; Matsumoto, T.; Morifuji, K.; Sawada, H.; Nishimura, T.; Nunoi, H.; Yoshiura, K.I.; et al. A hot-spot mutation in CDC42 (p.Tyr64Cys) and novel phenotypes in the third patient with Takenouchi-Kosaki syndrome. J. Hum. Genet. 2018, 63, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.H.; Wang, H.; Soderling, S.H.; Yasuda, R. Loss of Cdc42 leads to defects in synaptic plasticity and remote memory recall. Elife 2014, 3, e02839. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Katoh, H.; Yasui, H.; Mori, K.; Negishi, M. RhoA Inhibits the Nerve Growth Factor-induced Rac1 Activation through Rho-associated Kinase-dependent Pathway. J. Biol. Chem. 2001, 276, 18977–18983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalink, K.; Van Corven, E.J.; Hengeveld, T.; Morii, N.; Narumiya, S.; Moolenaar, W.H. Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J. Cell Biol. 1994, 126, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Hirose, M.; Ishizaki, T.; Watanabe, N.; Uehata, M.; Kranenburg, O.; Moolenaar, W.H.; Matsumura, F.; Maekawa, M.; Bito, H.; Narumiya, S. Molecular dissection of the Rho-associated protein kinase (p160ROCK)- regulated neurite remodeling in neuroblastoma N1E-115 cells. J. Cell Biol. 1998, 141, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Endo, M.; Hata, K.; Taniguchi, J.; Kitajo, K.; Tomura, S.; Yamaguchi, A.; Mueller, B.K.; Yamashita, T. Myosin IIA is required for neurite outgrowth inhibition produced by repulsive guidance molecule. J. Neurochem. 2008, 105, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Pilpel, Y.; Segal, M. Activation of PKC induces rapid morphological plasticity in dendrites of hippocampal neurons via Rac and Rho-dependent mechanisms. Eur. J. Neurosci. 2004, 19, 3151–3164. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin Mediates Degradation of RhoA through Evolutionarily Conserved BTB Adaptors to Control Actin Cytoskeleton Structure and Cell Movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef]
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.F.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316. [Google Scholar] [CrossRef]
- Lin, G.N.; Corominas, R.; Lemmens, I.; Yang, X.; Tavernier, J.; Hill, D.E.; Vidal, M.; Sebat, J.; Iakoucheva, L.M. Spatiotemporal 16p11.2 Protein Network Implicates Cortical Late Mid-Fetal Brain Development and KCTD13-Cul3-RhoA Pathway in Psychiatric Diseases. Neuron 2015, 85, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escamilla, C.O.; Filonova, I.; Walker, A.K.; Xuan, Z.X.; Holehonnur, R.; Espinosa, F.; Liu, S.; Thyme, S.; López-García, I.A.; Mendoza, D.B.; et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 2017, 551, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Martin Lorenzo, S.; Nalesso, V.; Chevalier, C.; Birling, M.C.; Herault, Y. Targeting the RHOA pathway improves learning and memory in adult Kctd13 and 16p11.2 deletion mouse models. Mol. Autism 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, Y.; Fan, T.; Zeng, C.; Sun, Z.S. The p21-activated kinases in neural cytoskeletal remodeling and related neurological disorders. Protein Cell 2020. [Google Scholar] [CrossRef]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, G.; Hostinova, E.; Rudolph, M.G.; Kraemer, A.; Sickmann, A.; Meyer, H.E.; Scheffzek, K.; Wittinghofer, A. Conformational Switch and Role of Phosphorylation in PAK Activation. Mol. Cell. Biol. 2001, 21, 5179–5189. [Google Scholar] [CrossRef] [Green Version]
- Reeder, M.K.; Serebriiskii, I.G.; Golemis, E.A.; Chernoff, J. Analysis of Small GTPase Signaling Pathways Using p21-activated Kinase Mutants that Selectively Couple to Cdc42. J. Biol. Chem. 2001, 276, 40606–40613. [Google Scholar] [CrossRef] [Green Version]
- Asrar, S.; Meng, Y.; Zhou, Z.; Todorovski, Z.; Huang, W.W.; Jia, Z. Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 2009, 56, 73–80. [Google Scholar] [CrossRef]
- Causeret, F.; Terao, M.; Jacobs, T.; Nishimura, Y.V.; Yanagawa, Y.; Obata, K.; Hoshino, M.; Nikolić, M. The p21-activated kinase is required for neuronal migration in the cerebral cortex. Cereb. Cortex 2009, 19, 861–875. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Au, M.; Basel-Salmon, L.; Bayrak-Toydemir, P.; Chapin, A.; Cohen, L.; Elting, M.W.; Graham, J.M.; Gonzaga-Jauregui, C.; Konen, O.; et al. De novo variants in PAK1 lead to intellectual disability with macrocephaly and seizures. Brain 2019, 142, 3351–3359. [Google Scholar] [CrossRef]
- Ohori, S.; Mitsuhashi, S.; Ben-Haim, R.; Heyman, E.; Sengoku, T.; Ogata, K.; Matsumoto, N. A novel PAK1 variant causative of neurodevelopmental disorder with postnatal macrocephaly. J. Hum. Genet. 2020, 65, 481–485. [Google Scholar] [CrossRef]
- Huo, H.Q.; Qu, Z.Y.; Yuan, F.; Ma, L.; Yao, L.; Xu, M.; Hu, Y.; Ji, J.; Bhattacharyya, A.; Zhang, S.C.; et al. Modeling Down Syndrome with Patient iPSCs Reveals Cellular and Migration Deficits of GABAergic Neurons. Stem Cell Rep. 2018, 10, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Kreis, P.; Thévenot, E.; Rousseau, V.; Boda, B.; Muller, D.; Barnier, J.V. The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J. Biol. Chem. 2007, 282, 21497–21506. [Google Scholar] [CrossRef] [Green Version]
- Duarte, K.; Heide, S.; Poëa-Guyon, S.; Rousseau, V.; Depienne, C.; Rastetter, A.; Nava, C.; Attié-Bitach, T.; Razavi, F.; Martinovic, J.; et al. PAK3 mutations responsible for severe intellectual disability and callosal agenesis inhibit cell migration. Neurobiol. Dis. 2020, 136, 104709. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Wu, B.; Lu, Y.; Zhou, W.; Wang, S.; Wang, H. Novel PAK3 gene missense variant associated with two Chinese siblings with intellectual disability: A case report. BMC Med. Genet. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillon, C.; Gonzalez, L.; Domenichini, F.; Guyon, S.; Da Silva, K.; Durand, C.; Lestaevel, P.; Vaillend, C.; Laroche, S.; Barnier, J.V.; et al. The intellectual disability PAK3 R67C mutation impacts cognitive functions and adult hippocampal neurogenesis. Hum. Mol. Genet. 2020, 29, 1950–1968. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Meng, Y.; Hanna, A.; Janus, C.; Jia, Z. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J. Neurosci. 2005, 25, 6641–6650. [Google Scholar] [CrossRef] [Green Version]
- Okano, I.; Hiraoka, J.; Otera, H.; Nunoue, K.; Ohashi, K.; Iwashita, S.; Hirai, M.; Mizuno, K. Identification and characterization of a novel family of serine/threonine kinases containing two N-terminal LIM motifs. J. Biol. Chem. 1995, 270, 31321–31330. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.C.; Sanders, L.C.; Bokoch, G.M.; Gill, G.N. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1999, 1, 253–259. [Google Scholar] [CrossRef]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofflin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Matsumoto, K.; Takai, Y.; Nakamura, T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by Rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 1999, 147, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Hoogenraad, C.C.; Akhmanova, A.; Galjart, N.; De Zeeuw, C.I. LIMK1 and CLIP-115: Linking cytoskeletal defects to Williams Syndrome. BioEssays 2004, 26, 141–150. [Google Scholar] [CrossRef]
- Todorovski, Z.; Asrar, S.; Liu, J.; Saw, N.M.N.; Joshi, K.; Cortez, M.A.; Snead, O.C.; Xie, W.; Jia, Z. LIMK1 Regulates Long-Term Memory and Synaptic Plasticity via the Transcriptional Factor CREB. Mol. Cell. Biol. 2015, 35, 1316–1328. [Google Scholar] [CrossRef] [Green Version]
- Proschel, C.; Blouin, M.J.; Gutowski, N.J.; Ludwig, R.; Noble, M. Limk1 is predominantly expressed in neural tissues and phosphorylates serine, threonine and tyrosine residues in vitro. Oncogene 1995, 11, 1271–1281. [Google Scholar]
- Wang, J.Y.; Wigston, D.J.; Rees, H.D.; Levey, A.I.; Falls, D.L. LIM kinase 1 accumulates in presynaptic terminals during synapse maturation. J. Comp. Neurol. 2000, 416, 319–334. [Google Scholar] [CrossRef]
- Tastet, J.; Vourc’h, P.; Laumonnier, F.; Vallée, B.; Michelle, C.; Duittoz, A.; Bénédetti, H.; Andres, C.R. LIMK2d, a truncated isoform of Lim kinase 2 regulates neurite growth in absence of the LIM kinase domain. Biochem. Biophys. Res. Commun. 2012, 420, 247–252. [Google Scholar] [CrossRef]
- Andrews, W.D.; Zito, A.; Memi, F.; Jones, G.; Tamamaki, N.; Parnavelas, J.G. Limk2 mediates semaphorin signalling in cortical interneurons migrating through the subpallium. Biol. Open 2013, 2, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Amano, M.; Yamamoto, T.; Chihara, K.; Nakafuku, M.; Ito, M.; Nakano, T.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for the small GTP binding protein Rho. EMBO J. 1996, 15, 2208–2216. [Google Scholar] [CrossRef]
- Ishizaki, T.; Maekawa, M.; Fujisawa, K.; Okawa, K.; Iwamatsu, A.; Fujita, A.; Watanabe, N.; Saito, Y.; Kakizuka, A.; Morii, N.; et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996, 15, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K.; Narumiya, S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, K.; Nagata, K.; Maekawa, M.; Ishizaki, T.; Narumiya, S.; Mizuno, K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 2000, 275, 3577–3582. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho- kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho- kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef]
- Katoh, K.; Kano, Y.; Amano, M.; Kaibuchi, K.; Fujiwara, K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280. [Google Scholar] [CrossRef] [Green Version]
- Bowling, K.M.; Thompson, M.L.; Amaral, M.D.; Finnila, C.R.; Hiatt, S.M.; Engel, K.L.; Cochran, J.N.; Brothers, K.B.; East, K.M.; Gray, D.E.; et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Kollins, K.M.; Hy, J.; Bridgman, P.C.; Hyang, Y.Q.; Gallo, G. Myosin-II negatvely regulates minor process extension and the temporal development of neuronal polarity. Dev. Neurobiol. 2009, 69, 279–298. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, N.A.; Burnette, D.T.; Forscher, P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat. Cell Biol. 2006, 8, 215–226. [Google Scholar] [CrossRef]
- Sanders, L.C.; Matsumura, F.; Bokoch, G.M.; De Lanerolle, P. Inhibition of myosin light chain kinase by p21-activated kinase. Science 1999, 283, 2083–2085. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.H.; Delalle, I.; Caviness, V.S.; Chae, T.; Harlow, E. P35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef]
- Tang, D.; Yeung, J.; Lee, K.Y.; Matsushita, M.; Matsui, H.; Tomizawa, K.; Hatase, O.; Wang, J.H. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J. Biol. Chem. 1995, 270, 26897–26903. [Google Scholar] [CrossRef] [Green Version]
- Hisanaga, S.I.; Endo, R. Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J. Neurochem. 2010, 115, 1309–1321. [Google Scholar] [CrossRef]
- Hawasli, A.H.; Benavides, D.R.; Nguyen, C.; Kansy, J.W.; Hayashi, K.; Chambon, P.; Greengard, P.; Powell, C.M.; Cooper, D.C.; Bibb, J.A. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat. Neurosci. 2007, 10, 880–886. [Google Scholar] [CrossRef]
- He, L.; Zhang, Z.; Yu, Y.; Ahmed, S.; Cheung, N.S.; Qi, R.Z. The neuronal p35 activator of Cdk5 is a novel F-actin binding and bundling protein. Cell. Mol. Life Sci. 2011, 68, 1633–1643. [Google Scholar] [CrossRef]
- Fu, W.Y.; Chen, Y.; Sahin, M.; Zhao, X.S.; Shi, L.; Bikoff, J.B.; Lai, K.O.; Yung, W.H.; Fu, A.K.Y.; Greenberg, M.E.; et al. Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat. Neurosci. 2007, 10, 67–76. [Google Scholar] [CrossRef]
- Kawauchi, T.; Chihama, K.; Nabeshima, Y.I.; Hoshino, M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat. Cell Biol. 2006, 8, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Gurian-West, M.; Schmidt, A.; Hall, A.; Roberts, J.M. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004, 18, 862–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, X.; Wang, Y.; Ma, X.M.; Rompolas, P.; Keutmann, H.T.; Mains, R.E.; Eipper, B.A. Regulation of Kalirin by Cdk5. J. Cell Sci. 2008, 121, 2601–2611. [Google Scholar] [CrossRef] [Green Version]
- Kesavapany, S.; Amin, N.; Zheng, Y.L.; Nijhara, R.; Jaffe, H.; Sihag, R.; Gutkind, J.S.; Takahashi, S.; Kulkarni, A.; Grant, P.; et al. p35/Cyclin-Dependent Kinase 5 Phosphorylation of Ras Guanine Nucleotide Releasing Factor 2 (RasGRF2) Mediates Rac-Dependent Extracellular Signal-Regulated Kinase 1/2 Activity, Altering RasGRF2 and Microtubule-Associated Protein 1b Distribution in Neurons. J. Neurosci. 2004, 24, 4421–4431. [Google Scholar] [CrossRef] [PubMed]
- Causeret, F.; Jacobs, T.; Terao, M.; Heath, O.; Hoshino, M.; Nikolić, M. Neurabin-I is phosphorylated by Cdk5: Implications for neuronal morphogenesis and cortical migration. Mol. Biol. Cell 2007, 18, 4327–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, Z.H.; Chin, W.H.; Chen, Y.; Ng, Y.P.; Ip, N.Y. Cdk5 is involved in BDNF-stimulated dendritic growth in hippocampal neurons. PLoS Biol. 2007, 5, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Moncini, S.; Castronovo, P.; Murgia, A.; Russo, S.; Bedeschi, M.F.; Lunghi, M.; Selicorni, A.; Bonati, M.T.; Riva, P.; Venturin, M. Functional characterization of CDK5 and CDK5R1 mutations identified in patients with non-syndromic intellectual disability. J. Hum. Genet. 2016, 61, 283–293. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [Green Version]
- Fauchereau, F.; Herbrand, U.; Chafey, P.; Eberth, A.; Koulakoff, A.; Vinet, M.C.; Ahmadian, M.R.; Chelly, J.; Billuart, P. The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol. Cell. Neurosci. 2003, 23, 574–586. [Google Scholar] [CrossRef]
- Khelfaoui, M.; Denis, C.; Van Galen, E.; De Bock, F.; Schmitt, A.; Houbron, C.; Morice, E.; Giros, B.; Ramakers, G.; Fagni, L.; et al. Loss of X-linked mental retardation gene oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J. Neurosci. 2007, 27, 9439–9450. [Google Scholar] [CrossRef]
- Billuart, P.; Bienvenu, T.; Roncet, N.; Des Portes, V.; Vinet, M.C.; Zemni, R.; Crollius, H.R.; Carrié, A.; Fauchereau, F.; Cherry, M.; et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 1998, 392, 923–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuovo, S.; Brankovic, V.; Caputi, C.; Casella, A.; Nigro, V.; Leuzzi, V.; Valente, E.M. Novel unconventional variants expand the allelic spectrum of OPHN1 gene. Am. J. Med. Genet. Part A 2021, 185, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Zerres, K.; Senderek, J.; Rudnik-Schöneborn, S.; Eggermann, T.; Häusler, M.; Mull, M.; Ramaekers, V.T. Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 2003, 126, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Bogliş, A.; Cosma, A.S.; Tripon, F.; Bãnescu, C. Exon 21 deletion in the OPHN1 gene in a family with syndromic X-linked intellectual disability: Case report. Medicine 2020, 99, e21632. [Google Scholar] [CrossRef]
- Busti, I.; Allegra, M.; Spalletti, C.; Panzi, C.; Restani, L.; Billuart, P.; Caleo, M. ROCK/PKA inhibition rescues hippocampal hyperexcitability and GABAergic neuron alterations in a oligophrenin-1 knock-out mouse model of X-linked intellectual disability. J. Neurosci. 2020, 40, 2776–2788. [Google Scholar] [CrossRef]
- Compagnucci, C.; Barresi, S.; Petrini, S.; Billuart, P.; Piccini, G.; Chiurazzi, P.; Alfieri, P.; Bertini, E.; Zanni, G. Rho Kinase Inhibition Is Essential During In Vitro Neurogenesis and Promotes Phenotypic Rescue of Human Induced Pluripotent Stem Cell-Derived Neurons With Oligophrenin-1 Loss of Function. Stem Cells Transl. Med. 2016, 5, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Zamboni, V.; Armentano, M.; Saró, G.; Ciraolo, E.; Ghigo, A.; Germena, G.; Umbach, A.; Valnegri, P.; Passafaro, M.; Carabelli, V.; et al. Disruption of ArhGAP15 results in hyperactive Rac1, affects the architecture and function of hippocampal inhibitory neurons and causes cognitive deficits. Sci. Rep. 2016, 6, 34877. [Google Scholar] [CrossRef] [PubMed]
- Mulatinho, M.V.; De Carvalho Serao, C.L.; Scalco, F.; Hardekopf, D.; Pekova, S.; Mrasek, K.; Liehr, T.; Weise, A.; Rao, N.; Llerena, J.C. Severe intellectual disability, omphalocele, hypospadia and high blood pressure associated to a deletion at 2q22.1q22.3: Case report. Mol. Cytogenet. 2012, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Smigiel, R.; Szafranska, A.; Czyzewska, M.; Rauch, A.; Zweier, C.; Patkowski, D. Severe clinical course of Hirschsprung disease in a Mowat-Wilson syndrome patient. J. Appl. Genet. 2010, 51, 111–113. [Google Scholar] [CrossRef] [Green Version]
- Rosário, M.; Franke, R.; Bednarski, C.; Birchmeier, W. The neurite outgrowth multiadaptor RhoGAP, NOMA-GAP, regulates neurite extension through SHP2 and Cdc42. J. Cell Biol. 2007, 178, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Rosário, M.; Schuster, S.; Jüttner, R.; Parthasarathy, S.; Tarabykin, V.; Birchmeier, W. Neocortical dendritic complexity is controlled during development by NOMA-GAP-dependent inhibition of Cdc42 and activation of cofilin. Genes Dev. 2012, 26, 1743–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debant, A.; Serra-PagèS, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Pengelly, R.J.; Greville-Heygate, S.; Schmidt, S.; Seaby, E.G.; Jabalameli, M.R.; Mehta, S.G.; Parker, M.J.; Goudie, D.; Fagotto-Kaufmann, C.; Mercer, C.; et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J. Med. Genet. 2016, 53, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Debant, A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 2014, 5, e983880. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.P.; Seipel, K.; Medley, Q.G.; Bronson, R.; Segal, R.; Streuli, M. Skeletal muscle deformity and neuroanl disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 2000, 97, 12074–12078. [Google Scholar] [CrossRef] [Green Version]
- Katrancha, S.M.; Shaw, J.E.; Zhao, A.Y.; Myers, S.A.; Cocco, A.R.; Jeng, A.T.; Zhu, M.; Pittenger, C.; Greer, C.A.; Carr, S.A.; et al. Trio Haploinsufficiency Causes Neurodevelopmental Disease-Associated Deficits. Cell Rep. 2019, 26, 2805–2817. [Google Scholar] [CrossRef] [Green Version]
- Ba, W.; Yan, Y.; Reijnders, M.R.F.; Schuurs-Hoeijmakers, J.H.M.; Feenstra, I.; Bongers, E.M.H.F.; Bosch, D.G.M.; De Leeuw, N.; Pfundt, R.; Gilissen, C.; et al. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 2016, 25, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Ten Klooster, J.P.; Jaffer, Z.M.; Chernoff, J.; Hordijk, P.L. Targeting and activation of Rac1 are mediated by the exchange factor β-Pix. J. Cell Biol. 2006, 172, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Ramakers, G.J.A.; Wolfer, D.; Rosenberger, G.; Kuchenbecker, K.; Kreienkamp, H.J.; Prange-kiel, J.; Rune, G.; Richter, K.; Langnaese, K.; Masneuf, S.; et al. Dysregulation of Rho GTPases in the αPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum. Mol. Genet. 2012, 21, 268–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manser, E.; Loo, T.H.; Koh, C.G.; Zhao, Z.S.; Chen, X.Q.; Tan, L.; Tan, I.; Leung, T.; Lim, L. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell 1998, 1, 183–192. [Google Scholar] [CrossRef]
- Santiago-Medina, M.; Gregus, K.A.; Gomez, T.M. PAK-PIX interactions regulate adhesion dynamics and membrane protrusion to control neurite outgrowth. J. Cell Sci. 2013, 126, 1122–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Na, M.; Choi, J.; Kim, S.; Lee, J.R.; Yoon, J.; Park, D.; Sheng, M.; Kim, E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the βPIX guanine nucleotide exchange factor for Rac1 and Cdc42. J. Biol. Chem. 2003, 278, 19220–19229. [Google Scholar] [CrossRef] [Green Version]
- Kutsche, K.; Yntema, H.; Brandt, A.; Jantke, I.; Nothwang, H.G.; Orth, U.; Boavida, M.G.; David, D.; Chelly, J.; Fryns, J.P.; et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat. Genet. 2000, 26, 247–250. [Google Scholar] [CrossRef]
- Yntema, H.G.; Hamel, B.C.J.; Smits, A.P.T.; Van Roosmalen, T.; Van Den Helm, B.; Kremer, H.; Ropers, H.H.; Smeets, D.F.C.M.; Van Bokhoven, H. Localisation of a gene for non-specific X linked mental retardation (MRX46) to Xq25-q26. J. Med. Genet. 1998, 35, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Piton, A.; Redin, C.; Mandel, J.L. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 2013, 93, 368–383. [Google Scholar] [CrossRef] [Green Version]
- Orsini, A.; Bonuccelli, A.; Striano, P.; Azzara, A.; Costagliola, G.; Consolini, R.; Peroni, D.G.; Valetto, A.; Bertini, V. Generalized epilepsy and mild intellectual disability associated with 13q34 deletion: A potential role for SOX1 and ARHGEF7. Seizure 2018, 59, 38–40. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Lee, S.J.; Lee, E.; Kim, D.; Park, D. βPix heterozygous mice have defects in neuronal morphology and social interaction. Biochem. Biophys. Res. Commun. 2019, 516, 1204–1210. [Google Scholar] [CrossRef]
- López Tobón, A.; Suresh, M.; Jin, J.; Vitriolo, A.; Pietralla, T.; Tedford, K.; Bossenz, M.; Mahnken, K.; Kiefer, F.; Testa, G.; et al. The guanine nucleotide exchange factor Arhgef7/βPix promotes axon formation upstream of TC10. Sci. Rep. 2018, 8, 8811. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Bathoorn, A.; Ahmadian, M.R.; Collard, J.G. Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J. Biol. Chem. 1999, 274, 33587–33593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagarajan, S.K.; Ghosh, H.; Harvey, K.; Fritschy, J.M. Collybistin splice variants differentially interact with gephyrin and Cdc42 to regulate gephyrin clustering at GABAergic synapses. J. Cell Sci. 2011, 124, 2786–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzarelli, R.; Griguoli, M.; Zacchi, P.; Petrini, E.M.; Barberis, A.; Cattaneo, A.; Cherubini, E. Tuning GABAergic Inhibition: Gephyrin Molecular Organization and Functions. Neuroscience 2020, 439, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, T.; Korte, M.; Eulenburg, V.; Kubota, H.; Retiounskaia, M.; Harvey, R.J.; Harvey, K.; O’Sullivan, G.A.; Laube, B.; Hülsmann, S.; et al. Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 2007, 26, 3888–3899. [Google Scholar] [CrossRef]
- Alber, M.; Kalscheuer, V.M.; Marco, E.; Sherr, E.; Lesca, G.; Till, M.; Gradek, G.; Wiesener, A.; Korenke, C.; Mercier, S.; et al. ARHGEF9 disease. Neurol. Genet. 2017, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Y.; Zhou, P.; Wang, J.; Tang, B.; Su, T.; Liu, X.R.; Li, B.M.; Meng, H.; Shi, Y.W.; Yi, Y.H.; et al. ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: Toward understanding the mechanism underlying phenotypic variation. Neurogenetics 2018, 19, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Zhang, Y.; Liu, J.; Wang, J.; Xu, Y.; Li, N.; Wang, J.; Yu, T. Clinical and Molecular Characterization of Three Novel ARHGEF9 Mutations in Patients with Developmental Delay and Epilepsy. J. Mol. Neurosci. 2020, 70, 908–915. [Google Scholar] [CrossRef]
- Michiels, F.; Habets, G.G.M.M.; Stam, J.C.; Habets, G.G.M.; Van Der Kammen, R.A.; Collard, J.G. A role for Rac in Tiaml-induced membrane ruffling and invasion. Nature 1995, 375, 338–340. [Google Scholar] [CrossRef]
- Ehler, E.; Van Leeuwen, F.; Collard, J.G.; Salinas, P.C. Expression of Tiam-1 in the developing brain suggests a role for the Tiam-1-Rac signaling pathway in cell migration and neurite outgrowth. Mol. Cell. Neurosci. 1997, 9, 1–12. [Google Scholar] [CrossRef]
- Van Leeuwen, F.N.; Kain, H.E.T.; Van Der Kammen, R.A.; Michiels, F.; Kranenburg, O.W.; Collard, J.G. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J. Cell Biol. 1997, 139, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Yamauchi, J.; Tanoue, A.; Wu, C.; Mobley, W.C. TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 10444–10449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunda, P.; Paglini, G.; Quiroga, S.; Kosik, K.; Cáceres, A. Evidence for the involvement of Tiam1 in axon formation. J. Neurosci. 2001, 21, 2361–2372. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, T.; Chihama, K.; Nabeshima, Y.I.; Hoshino, M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003, 22, 4190–4201. [Google Scholar] [CrossRef] [Green Version]
- Tolias, K.F.; Bikoff, J.B.; Kane, C.G.; Tolias, C.S.; Hu, L.; Greenberg, M.E. The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptor-dependent dendritic spine development. Proc. Natl. Acad. Sci. USA 2007, 104, 7265–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saneyoshi, T.; Matsuno, H.; Suzuki, A.; Murakoshi, H.; Hedrick, N.G.; Agnello, E.; O’Connell, R.; Stratton, M.M.; Yasuda, R.; Hayashi, Y. Reciprocal Activation within a Kinase-Effector Complex Underlying Persistence of Structural LTP. Neuron 2019, 102, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Rosendale, M.; Sugiyama, Y.; Hayashi, M.; Horiguchi, Y.; Yoshihara, T.; Ikegaya, Y.; Saneyoshi, T.; Hayashi, Y. The role of CaMKII-Tiam1 complex on learning and memory. Neurobiol. Learn. Mem. 2019, 166, 107070. [Google Scholar] [CrossRef]
- Rao, S.; Kay, Y.; Herring, B.E. Tiam1 is Critical for Glutamatergic Synapse Structure and Function in the Hippocampus. J. Neurosci. 2019, 39, 9306–9315. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Scala, F.; Blanco, F.A.; Niu, S.; Firozi, K.; Keehan, L.; Mulherkar, S.; Froudarakis, E.; Li, L.; Duman, J.G.; et al. The Rac-GEF Tiam1 Promotes Dendrite and Synapse Stabilization of Dentate Granule Cells and Restricts Hippocampal-Dependent Memory Functions. J. Neurosci. 2021, 41, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schnelder, C.; Stanyon, C.A.; Bernards, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM- kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef]
- Lappalainen, P.; Drubin, D.G. Cofilin promotes rapid actin filament turnover in vivo. Nature 1997, 388, 78–82. [Google Scholar] [CrossRef]
- Maciver, S.K.; Hussey, P.J. The ADF/cofilin family: Actin-remodeling proteins. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Agnew, B.J.; Minamide, L.S.; Bamburg, J.R. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J. Biol. Chem. 1995, 270, 17582–17587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svitkina, T.M.; Borisy, G.G. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 1999, 145, 1009–1026. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Magalhaes, M.A.O.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Meberg, P.J.; Bamburg, J.R. Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J. Neurosci. 2000, 20, 2459–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Z.; Han, L.; Bamburg, J.R.; Shim, S.; Ming, G.L.; Zheng, J.Q. BMP gradients steer nerve growth cones by a balancing act of LIM kinase and Slingshot phosphatase on ADF/cofilin. J. Cell Biol. 2007, 178, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meberg, P.J.; Ono, S.; Minamide, L.S.; Takahashi, M.; Bamburg, J.R. Actin depolymerizing factor and cofilin phosphorylation dynamics: Response to signals that regulate neurite extension. Cell Motil. Cytoskelet. 1998, 39, 172–190. [Google Scholar] [CrossRef]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Ohashi, K.; Mizuno, K. LIM kinase and slingshot are critical for neurite extension. J. Biol. Chem. 2007, 282, 13692–13702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohla, A.; Bokoch, G.M. 14-3-3 Regulates actin dynamics by stabilizing phosphorylated cofilin. Curr. Biol. 2002, 12, 1704–1710. [Google Scholar] [CrossRef] [Green Version]
- Soosairajah, J.; Maiti, S.; Wiggan, O.; Sarmiere, P.; Moussi, N.; Sarcevic, B.; Sampath, R.; Bamburg, J.R.; Bernard, O. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 2005, 24, 473–486. [Google Scholar] [CrossRef]
- Toyo-Oka, K.; Wachi, T.; Hunt, R.F.; Baraban, S.C.; Taya, S.; Ramshaw, H.; Kaibuchi, K.; Schwarz, Q.P.; Lopez, A.F.; Wynshaw-Boris, A. 14-3-3ε and ζ regulate neurogenesis and differentiation of neuronal progenitor cells in the developing brain. J. Neurosci. 2014, 34, 12168–12181. [Google Scholar] [CrossRef] [Green Version]
- Taniuchi, K.; Nakagawa, H.; Hosokawa, M.; Nakamura, T.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Katagiri, T.; Nakamura, Y. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating Rho-family GTPases. Cancer Res. 2005, 65, 3092–3099. [Google Scholar] [CrossRef] [Green Version]
- Noren, N.K.; Liu, B.P.; Burridge, K.; Kreft, B. p120 Catenin regulates the actin cytoskeleton via RHO family GTPases. J. Cell Biol. 2000, 150, 567–579. [Google Scholar] [CrossRef]
- Anastasiadis, P.Z.; Moon, S.Y.; Thoreson, M.A.; Mariner, D.J.; Crawford, H.C.; Zheng, Y.; Reynolds, A.B. Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2000, 2, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jaehne, E.J.; Greenberg, Z.; McCarthy, P.; Saleh, E.; Parish, C.L.; Camera, D.; Heng, J.; Haas, M.; Baune, B.T.; et al. 14-3-3ζ deficient mice in the BALB/c background display behavioural and anatomical defects associated with neurodevelopmental disorders. Sci. Rep. 2015, 5, 12434. [Google Scholar] [CrossRef] [PubMed]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef]
- Pollard, T.D. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 451–477. [Google Scholar] [CrossRef] [PubMed]
- Schlau, M.; Terheyden-Keighley, D.; Theis, V.; Mannherz, H.G.; Theiss, C. VEGF triggers the activation of cofilin and the Arp2/3 complex within the growth cone. Int. J. Mol. Sci. 2018, 19, 384. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Bridgman, P.C. Disruption of the cytoskeleton during Semaphorin 3A induced growth cone collapse correlates with differences in actin organization and associated binding proteins. Dev. Neurobiol. 2009, 69, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Chou, F.-S.; Wang, P.-S. The Arp2/3 complex is essential at multiple stages of neural development. Neurogenesis 2016, 3, e1261653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.H.; Racz, B.; Wang, H.; Burianek, L.; Weinberg, R.; Yasuda, R.; Wetsel, W.C.; Soderling, S.H. Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J. Neurosci. 2013, 33, 6081–6092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, E.F.; Kanak, D.J.; Carlson, B.R.; Soderling, S.H. The Arp2/3 complex is essential for distinct stages of spine synapse maturation, including synapse unsilencing. J. Neurosci. 2016, 36, 9696–9709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadlamudi, R.K.; Li, F.; Barnes, C.J.; Bagheri-Yarmand, R.; Kumar, R. p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Rep. 2004, 5, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Borek, D.; Padrick, S.B.; Gomez, T.S.; Metlagel, Z.; Ismail, A.M.; Umetani, J.; Billadeau, D.D.; Otwinowski, Z.; Rosen, M.K. Structure and control of the actin regulatory WAVE complex. Nature 2010, 468, 533–538. [Google Scholar] [CrossRef]
- Kim, A.S.; Kakalis, L.T.; Abdul-Manan, N.; Liu, G.A.; Rosen, M.K. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature 2000, 404, 151–158. [Google Scholar] [CrossRef]
- Tomasevic, N.; Jia, Z.; Russell, A.; Fujii, T.; Hartman, J.J.; Clancy, S.; Wang, M.; Beraud, C.; Wood, K.W.; Sakowicz, R. Differential regulation of WASP and N-WASP by Cdc42, Rac1, Nck, and PI(4,5)P2. Biochemistry 2007, 46, 3494–3502. [Google Scholar] [CrossRef]
- Eden, S.; Rohatgi, R.; Podtelejnikov, A.V.; Mann, M.; Kirschner, M.W. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 2002, 418, 790–793. [Google Scholar] [CrossRef]
- Miki, H.; Suetsugu, S.; Takenawa, T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998, 17, 6932–6941. [Google Scholar] [CrossRef] [Green Version]
- Abekhoukh, S.; Bardoni, B. CYFIP family proteins between autism and intellectual disability: Links with fragile X syndrome. Front. Cell. Neurosci. 2014, 8, 81. [Google Scholar] [CrossRef]
- Davenport, E.C.; Szulc, B.R.; Drew, J.; Taylor, J.; Morgan, T.; Higgs, N.F.; López-Doménech, G.; Kittler, J.T. Autism and Schizophrenia-Associated CYFIP1 Regulates the Balance of Synaptic Excitation and Inhibition. Cell Rep. 2019, 26, 2037–2051. [Google Scholar] [CrossRef] [Green Version]
- Oguro-Ando, A.; Rosensweig, C.; Herman, E.; Nishimura, Y.; Werling, D.; Bill, B.R.; Berg, J.M.; Gao, F.; Coppola, G.; Abrahams, B.S.; et al. Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR. Mol. Psychiatry 2015, 20, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Bozdagi, O.; Sakurai, T.; Dorr, N.; Pilorge, M.; Takahashi, N.; Buxbaum, J.D. Haploinsufficiency of Cyfip1 produces fragile X-like phenotypes in mice. PLoS ONE 2012, 7, e42422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begemann, A.; Sticht, H.; Begtrup, A.; Vitobello, A.; Faivre, L.; Banka, S.; Alhaddad, B.; Asadollahi, R.; Becker, J.; Bierhals, T.; et al. New insights into the clinical and molecular spectrum of the novel CYFIP2-related neurodevelopmental disorder and impairment of the WRC-mediated actin dynamics. Genet. Med. 2020, 23, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Schaks, M.; Reinke, M.; Witke, W.; Rottner, K. Molecular Dissection of Neurodevelopmental Disorder-Causing Mutations in CYFIP2. Cells 2020, 9, 1355. [Google Scholar] [CrossRef] [PubMed]
- Ropers, F.; Derivery, E.; Hu, H.; Garshasbi, M.; Karbasiyan, M.; Herold, M.; Nürnberg, G.; Ullmann, R.; Gautreau, A.; Sperling, K.; et al. Identification of a novel candidate gene for non-syndromic autosomal recessive intellectual disability: The WASH complex member swip. Hum. Mol. Genet. 2011, 20, 2585–2590. [Google Scholar] [CrossRef] [Green Version]
- Assoum, M.; Bruel, A.L.; Crenshaw, M.L.; Delanne, J.; Wentzensen, I.M.; McWalter, K.; Dent, K.M.; Vitobello, A.; Kuentz, P.; Thevenon, J.; et al. Novel KIAA1033/WASHC4 mutations in three patients with syndromic intellectual disability and a review of the literature. Am. J. Med. Genet. Part A 2020, 182, 792–797. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, Q.; Dai, R.; Yu, B.; Hoekzema, K.; Tan, J.; Tan, S.; Jia, X.; Chung, W.K.; Hernan, R.; et al. NCKAP1 Disruptive Variants Lead to a Neurodevelopmental Disorder with Core Features of Autism. Am. J. Hum. Genet. 2020, 107, 963–976. [Google Scholar] [CrossRef]
- Harripaul, R.; Vasli, N.; Mikhailov, A.; Rafiq, M.A.; Mittal, K.; Windpassinger, C.; Sheikh, T.I.; Noor, A.; Mahmood, H.; Downey, S.; et al. Mapping autosomal recessive intellectual disability: Combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol. Psychiatry 2018, 23, 973–984. [Google Scholar] [CrossRef]
- Elliott, A.M.; Simard, L.R.; Coghlan, G.; Chudley, A.E.; Chodirker, B.N.; Greenberg, C.R.; Burch, T.; Ly, V.; Hatch, G.M.; Zelinski, T. A novel mutation in KIAA0196: Identification of a gene involved in Ritscher-Schinzel/3C syndrome in a First Nations cohort. J. Med. Genet. 2013, 50, 819–822. [Google Scholar] [CrossRef]
- Feldt, J.; Schicht, M.; Garreis, F.; Welss, J.; Schneider, U.W.; Paulsen, F. Structure, regulation and related diseases of the actin-binding protein gelsolin. Expert Rev. Mol. Med. 2019, 20. [Google Scholar] [CrossRef]
- Lu, M.; Witke, W.; Kwiatkowski, D.J.; Kosik, K.S. Delayed retraction of filopodia in gelsolin null mice. J. Cell Biol. 1997, 138, 1279–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlushchenko, I.; Hotulainen, P. Chemical LTD, but not LTP, induces transient accumulation of gelsolin in dendritic spines. Biol. Chem. 2019, 400, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Khaitlina, S.; Hinssen, H. Ca-dependent binding of actin to gelsolin. FEBS Lett. 2002, 521, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Sahasrabudhe, A.; Ghate, K.; Mutalik, S.; Jacob, A.; Ghose, A. Formin 2 regulates the stabilization of filopodial tip adhesions in growth cones and affects neuronal outgrowth and pathfinding in vivo. Development 2016, 143, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, R.; Dixon-Salazar, T.; Jerber, J.; Cai, N.; Abbasi, A.A.; Zaki, M.S.; Mittal, K.; Gabriel, S.B.; Rafiq, M.A.; Khan, V.; et al. Biallelic truncating mutations in FMN2, encoding the actin-regulatory protein formin 2, cause nonsyndromic autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2014, 95, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D. Actin and actin-binding proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D.; Cooper, J.A. Quantitative Analysis of the Effect of Acanthamoeba Profilin on Actin Filament Nucleation and Elongation. Biochemistry 1984, 23, 6631–6641. [Google Scholar] [CrossRef]
- Carlsson, L.; Nyström, L.E.; Sundkvist, I.; Markey, F.; Lindberg, U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J. Mol. Biol. 1977, 115, 465–483. [Google Scholar] [CrossRef]
- Tobacman, L.S.; Korn, E.D. The regulation of actin polymerization and the inhibition of monomeric actin ATPase activity by Acanthamoeba profilin. J. Biol. Chem. 1982, 257, 4166–4170. [Google Scholar] [CrossRef]
- Borovac, J.; Bosch, M.; Okamoto, K. Regulation of actin dynamics during structural plasticity of dendritic spines: Signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 2018, 91, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, A.; Jonckheere, V.; Peleman, C.; Polet, D.; De Vos, W.; Vandekerckhove, J.; Ampe, C. Profilin-I-ligand interactions influence various aspects of neuronal differentiation. J. Cell Sci. 2006, 119, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Toriyama, M.; Kozawa, S.; Sakumura, Y.; Inagaki, N. Conversion of a signal into forces for axon outgrowth through pak1-mediated shootin1 phosphorylation. Curr. Biol. 2013, 23, 529–534. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Forscher, P. Growth cone advance is inversely proportional to retrograde F-actin flow. Neuron 1995, 14, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Toriyama, M.; Uemura, K.; Kamiguchi, H.; Sugiura, T.; Watanabe, N.; Inagaki, N. Shootin1 interacts with actin retrograde flow and L1-CAM to promote axon outgrowth. J. Cell Biol. 2008, 181, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Huang, J.; Wang, Y.; Eipper, B.A.; Mains, R.E. Kalirin, a Multifunctional Rho Guanine Nucleotide Exchange Factor, Is Necessary for Maintenance of Hippocampal Pyramidal Neuron Dendrites and Dendritic Spines. J. Neurosci. 2003, 23, 10593–10603. [Google Scholar] [CrossRef]
- Ma, X.M.; Kiraly, D.D.; Gaier, E.D.; Wang, Y.; Kim, E.J.; Levine, E.S.; Eipper, B.A.; Mains, R.E. Kalirin-7 is required for synaptic structure and function. J. Neurosci. 2008, 28, 12368–12382. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Nakanishi, H.; Obaishi, H.; Wada, M.; Takahashi, K.; Satoh, K.; Hirao, K.; Nishioka, H.; Hata, Y.; Mizoguchi, A.; et al. Neurabin-II/spinophilin: An actin filament-binding protein with one PDZ domain localized at cadherin-based cell-cell adhesion sites. J. Biol. Chem. 1998, 273, 3470–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, S.D.; Hsieh-Wilson, L.C.; Allen, P.B.; Nairn, A.C.; Greengard, P. The actin-binding domain of spinophilin is necessary and sufficient for targeting to dendritic spines. Neuro Mol. Med. 2002, 2, 61–69. [Google Scholar] [CrossRef]
- Feng, J.; Yan, Z.; Ferreira, A.; Tomizawa, K.; Liauw, J.A.; Zhuo, M.; Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. USA 2000, 97, 9287–9292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, X.P.; Alldritt, J.; Svenningsson, P.; Allen, P.B.; Wu, G.Y.; Nairn, A.C.; Greengard, P. The Rho-specific GEF Lfc interacts with neurabin and spinophilin to regulate dendritic spine morphology. Neuron 2005, 47, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, E.; Hu, H.; Yuzwa, S.A.; Hernandez-Miranda, L.R.; Kraemer, N.; Ninnemann, O.; Musante, L.; Boltshauser, E.; Schindler, D.; Hübner, A.; et al. Homozygous ARHGEF2 mutation causes intellectual disability and midbrain-hindbrain malformation. PLoS Genet. 2017, 13, e1006746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qualmann, B.; Boeckers, T.M.; Jeromin, M.; Gundelfinger, E.D.; Kessels, M.M. Linkage of the Actin Cytoskeleton to the Postsynaptic Density via Direct Interactions of Abp1 with the ProSAP/Shank Family. J. Neurosci. 2004, 24, 2481–2495. [Google Scholar] [CrossRef] [Green Version]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Holder, J.L.; Schaaf, C.P.; Lu, H.; Chen, H.; Kang, H.; Tang, J.; Wu, Z.; Hao, S.; Cheung, S.W.; et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 2013, 503, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Sarowar, T.; Grabrucker, A.M. Actin-Dependent Alterations of Dendritic Spine Morphology in Shankopathies. Neural Plast. 2016, 2016, 8051861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaglia, M.C.; Giorda, R.; Mani, E.; Aceti, G.; Anderlid, B.M.; Baroncini, A.; Pramparo, T.; Zuffardi, O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J. Med. Genet. 2006, 43, 822–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaglia, M.C.; Giorda, R.; Borgatti, R.; Felisari, G.; Gagliardi, C.; Selicorni, A.; Zuffardi, O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 2001, 69, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Teruel, M.N.; Subramanian, K.; Meyer, T. CaMKIIβ functions as an F-actin targeting module that localizes CaMKIIα/β heterooligomers to dendritic spines. Neuron 1998, 21, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Küry, S.; van Woerden, G.M.; Besnard, T.; Proietti Onori, M.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Lasser, M.; Tiber, J.; Lowery, L.A. The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Witte, H.; Neukirchen, D.; Bradke, F. Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 2008, 180, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Firestein, B.L.; Zheng, J.Q. Microtubules in dendritic spine development. J. Neurosci. 2008, 28, 12120–12124. [Google Scholar] [CrossRef]
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; Di Stefano, P.; Demmers, J.; et al. Dynamic Microtubules Regulate Dendritic Spine Morphology and Synaptic Plasticity. Neuron 2009, 61, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, E.W. Of microtubules and memory: Implications for microtubule dynamics in dendrites and spines. Mol. Biol. Cell 2017, 28, 1–8. [Google Scholar] [CrossRef]
- Karabay, A.; Yu, W.; Solowska, J.M.; Baird, D.H.; Baas, P.W. Axonal growth is sensitive to the levels of katanin, a protein that severs microtubules. J. Neurosci. 2004, 24, 5778–5788. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in differentiated mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Neukirchen, D.; Bradke, F. Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J. Neurosci. 2011, 31, 1528–1538. [Google Scholar] [CrossRef]
- Swiech, L.; Blazejczyk, M.; Urbanska, M.; Pietruszka, P.; Dortland, B.R.; Malik, A.R.; Wulf, P.S.; Hoogenraad, C.C.; Jaworski, J. CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J. Neurosci. 2011, 31, 4555–4568. [Google Scholar] [CrossRef]
- Yonekawa, V.; Harada, A.; Okada, Y.; Funakoshi, T.; Kanai, Y.; Takei, Y.; Terada, S.; Noda, T.; Hirokawa, N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 1998, 141, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdan, F.F.; Gauthier, J.; Araki, Y.; Lin, D.T.; Yoshizawa, Y.; Higashi, K.; Park, A.R.; Spiegelman, D.; Dobrzeniecka, S.; Piton, A.; et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011, 88, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Esmaeeli Nieh, S.; Madou, M.R.Z.; Sirajuddin, M.; Fregeau, B.; Mcknight, D.; Lexa, K.; Strober, J.; Spaeth, C.; Hallinan, B.E.; Smaoui, N.; et al. De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy. Ann. Clin. Transl. Neurol. 2015, 2, 623–635. [Google Scholar] [CrossRef]
- Willemsen, M.H.; Ba, W.; Wissink-Lindhout, W.M.; de Brouwer, A.P.M.; Haas, S.A.; Bienek, M.; Hu, H.; Vissers, L.E.L.M.; van Bokhoven, H.; Kalscheuer, V.; et al. Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 2014, 51, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Saint Pierre, B.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef]
- Putoux, A.; Thomas, S.; Coene, K.L.M.; Davis, E.E.; Alanay, Y.; Ogur, G.; Uz, E.; Buzas, D.; Gomes, C.; Patrier, S.; et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 2011, 43, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Kevenaar, J.T.; Bianchi, S.; Van Spronsen, M.; Olieric, N.; Lipka, J.; Frias, C.P.; Mikhaylova, M.; Harterink, M.; Keijzer, N.; Wulf, P.S.; et al. Kinesin-Binding Protein Controls Microtubule Dynamics and Cargo Trafficking by Regulating Kinesin Motor Activity. Curr. Biol. 2016, 26, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Hempel, M.; Cremer, K.; Ockeloen, C.W.; Lichtenbelt, K.D.; Herkert, J.C.; Denecke, J.; Haack, T.B.; Zink, A.M.; Becker, J.; Wohlleber, E.; et al. De Novo Mutations in CHAMP1 Cause Intellectual Disability with Severe Speech Impairment. Am. J. Hum. Genet. 2015, 97, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidor, B.; Küry, S.; Rosenfeld, J.A.; Besnard, T.; Schmitt, S.; Joss, S.; Davies, S.J.; Roger Lebel, R.; Henderson, A.; Schaaf, C.P.; et al. De Novo Truncating Mutations in the Kinetochore-Microtubules Attachment Gene CHAMP1 Cause Syndromic Intellectual Disability. Hum. Mutat. 2016, 37, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.J.; Cho, M.T.; Retterer, K.; Jones, J.R.; Nowak, C.; Douglas, J.; Jiang, Y.-H.; McConkie-Rosell, A.; Schaefer, G.B.; Kaylor, J.; et al. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Mol. Case Stud. 2016, 2, a000661. [Google Scholar] [CrossRef] [Green Version]
- Larti, F.; Kahrizi, K.; Musante, L.; Hu, H.; Papari, E.; Fattahi, Z.; Bazazzadegan, N.; Liu, Z.; Banan, M.; Garshasbi, M.; et al. Erratum: A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur. J. Hum. Genet. 2015, 23, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholdi, D.; Stray-Pedersen, A.; Azzarello-Burri, S.; Kibaek, M.; Kirchhoff, M.; Oneda, B.; Rødningen, O.; Schmitt-Mechelke, T.; Rauch, A.; Kjaergaard, S. A newly recognized 13q12.3 microdeletion syndrome characterized by intellectual disability, microcephaly, and eczema/atopic dermatitis encompassing the HMGB1 and KATNAL1 genes. Am. J. Med. Genet. Part A 2014, 164, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Cainarca, S.; Messali, S.; Ballabio, A.; Meroni, G. Functional characterization of the Opitz syndrome gene product (midin): Evidence for homodimerization and association with microtubules throughout the cell cycle. Hum. Mol. Genet. 1999, 8, 1387–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geetha, T.S.; Michealraj, K.A.; Kabra, M.; Kaur, G.; Juyal, R.C.; Thelma, B.K. Targeted Deep Resequencing Identifies MID2 Mutation for X-Linked Intellectual Disability with Varied Disease Severity in a Large Kindred from India. Hum. Mutat. 2014, 35, 41–44. [Google Scholar] [CrossRef]
- Barbiero, I.; Peroni, D.; Tramarin, M.; Chandola, C.; Rusconi, L.; Landsberger, N.; Kilstrup-Nielsen, C. The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5-IQGAP1 complex. Hum. Mol. Genet. 2017, 26, 3520–3530. [Google Scholar] [CrossRef]
- Baltussen, L.L.; Negraes, P.D.; Silvestre, M.; Claxton, S.; Moeskops, M.; Christodoulou, E.; Flynn, H.R.; Snijders, A.P.; Muotri, A.R.; Ultanir, S.K. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 2018, 37, e99763. [Google Scholar] [CrossRef]
- Nawaz, M.S.; Giarda, E.; Bedogni, F.; La Montanara, P.; Ricciardi, S.; Ciceri, D.; Alberio, T.; Landsberger, N.; Rusconi, L.; Kilstrup-Nielsen, C. CDKL5 and shootin1 interact and concur in regulating neuronal polarization. PLoS ONE 2016, 11, e0148634. [Google Scholar] [CrossRef] [PubMed]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Abraham, W.C. Astrocytes and synaptic plasticity in health and disease. Exp. Brain Res. 2017, 235, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamsky, A.; Kol, A.; Kreisel, T.; Doron, A.; Ozeri-Engelhard, N.; Melcer, T.; Refaeli, R.; Horn, H.; Regev, L.; Groysman, M.; et al. Astrocytic Activation Generates De Novo Neuronal Potentiation and Memory Enhancement. Cell 2018, 174, 59–71.e14. [Google Scholar] [CrossRef] [Green Version]
- Molotkov, D.; Zobova, S.; Arcas, J.M.; Khiroug, L. Calcium-induced outgrowth of astrocytic peripheral processes requires actin binding by Profilin-1. Cell Calcium 2013, 53, 338–348. [Google Scholar] [CrossRef]
- Ghézali, G.; Dallérac, G.; Rouach, N. Perisynaptic astroglial processes: Dynamic processors of neuronal information. Brain Struct. Funct. 2016, 221, 2427–2442. [Google Scholar] [CrossRef]
- Renault-Mihara, F.; Mukaino, M.; Shinozaki, M.; Kumamaru, H.; Kawase, S.; Baudoux, M.; Ishibashi, T.; Kawabata, S.; Nishiyama, Y.; Sugai, K.; et al. Regulation of RhoA by STAT3 coordinates glial scar formation. J. Cell Biol. 2017, 216, 2533–2550. [Google Scholar] [CrossRef] [Green Version]
- Boukhelifa, M.; Hwang, S.J.; Valtschanoff, J.G.; Meeker, R.B.; Rustioni, A.; Otey, C.A. A critical role for palladin in astrocyte morphology and response to injury. Mol. Cell. Neurosci. 2003, 23, 661–668. [Google Scholar] [CrossRef]
- Potokar, M.; Kreft, M.; Li, L.; Andersson, J.D.; Pangršič, T.; Chowdhury, H.H.; Pekny, M.; Zorec, R. Cytoskeleton and vesicle mobility in astrocytes. Traffic 2007, 8, 12–20. [Google Scholar] [CrossRef]
- Pillet, L.E.; Cresto, N.; Saillour, Y.; Ghézali, G.; Bemelmans, A.P.; Livet, J.; Bienvenu, T.; Rouach, N.; Billuart, P. The intellectual disability protein Oligophrenin-1 controls astrocyte morphology and migration. Glia 2020, 68, 1729–1742. [Google Scholar] [CrossRef]
- Ali, A.A.H.; Schwarz-Herzke, B.; Rollenhagen, A.; Anstötz, M.; Holub, M.; Lübke, J.; Rose, C.R.; Schnittler, H.; Gall, C. Bmal1-deficiency affects glial synaptic coverage of the hippocampal mossy fiber synapse and the actin cytoskeleton in astrocytes. Glia 2020, 68, 947–962. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Morrison, B.M.; Lee, Y.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol. 2013, 23, 644–651. [Google Scholar] [CrossRef] [Green Version]
- Xin, W.; Chan, J.R. Myelin plasticity: Sculpting circuits in learning and memory. Nat. Rev. Neurosci. 2020, 21, 682–694. [Google Scholar] [CrossRef]
- O’Rourke, M.; Gasperini, R.; Young, K.M. Adult myelination: Wrapping up neuronal plasticity. Neural Regen. Res. 2014, 9, 1261–1264. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Fu, M.M.; Sloan, S.A.; Ibrahim, A.; Olson, A.; Zaremba, A.; Dugas, J.C.; Wienbar, S.; Caprariello, A.V.; Kantor, C.; et al. CNS Myelin Wrapping Is Driven by Actin Disassembly. Dev. Cell 2015, 34, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Muggironi, M.; Marin-Husstege, M.; Casaccia-Bonnefil, P. Oligodendrocyte Process Outgrowth In Vitro Is Modulated by Epigenetic Regulation of Cytoskeletal Severing Proteins. Glia 2003, 44, 264–274. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Ábrahám, H.; Vincze, A.; Veszprémi, B.; Kravják, A.; Gömöri, É.; Kovács, G.G.; Seress, L. Impaired myelination of the human hippocampal formation in Down syndrome. Int. J. Dev. Neurosci. 2012, 30, 147–158. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Kang, H.J.; Tyler, W.A.; Silbereis, J.C.; Cheng, F.; Zhu, Y.; Pletikos, M.; Jankovic-Rapan, L.; Cramer, N.P.; Galdzicki, Z.; et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron 2016, 89, 1208–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, K.M.; Gleeson, J.G.; Bagrodia, S.; Partington, M.W.; MacMillan, J.C.; Cerione, R.A.; Mulley, J.C.; Walsh, C.A. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 1998, 20, 25–30. [Google Scholar] [CrossRef]
- Gedeon, A.K.; Nelson, J.; Gécz, J.; Mulley, J.C. X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. Am. J. Med. Genet. 2003, 120 A, 509–517. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Dugas, J.C.; Tai, Y.C.; Speed, T.P.; Ngai, J.; Barres, B.A. Functional genomic analysis of oligodendrocyte differentiation. J. Neurosci. 2006, 26, 10967–10983. [Google Scholar] [CrossRef]
- Renkilaraj, M.R.L.M.; Baudouin, L.; Wells, C.M.; Doulazmi, M.; Wehrlé, R.; Cannaya, V.; Bachelin, C.; Barnier, J.-V.; Jia, Z.; Oumesmar, B.N.; et al. The intellectual disability protein PAK3 regulates oligodendrocyte precursor cell differentiation. Neurobiol. Dis. 2017, 98, 137–148. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Molbioltools. Available online: https://www.molbiotools.com/index.html (accessed on 4 June 2021).
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Contino, S.; Bertolazzi, G.; Calì, F.; Cantone, M.; Vera, J.; Romano, V. Axon Growth and Guidance in ASD: From Static Pathway Analysis to Dynamic Boolean Modeling. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780128012383. [Google Scholar]
- Müssel, C.; Hopfensitz, M.; Kestler, H.A. BoolNet-an R package for generation, reconstruction and analysis of Boolean networks. Bioinformatics 2010, 26, 1378–1380. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Costa, R.; Sousa, S.C.; Leite, S.C.; Nogueira-Rodrigues, J.; da Silva, T.F.; Machado, D.; Marques, J.; Costa, A.C.; Liz, M.A.; Bartolini, F.; et al. Profilin 1 delivery tunes cytoskeletal dynamics toward CNS axon regeneration. J. Clin. Investig. 2020, 130, 2024–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szikora, S.; Földi, I.; Tóth, K.; Migh, E.; Vig, A.; Bugyi, B.; Maléth, J.; Hegyi, P.; Kaltenecker, P.; Sanchez-Soriano, N.; et al. The formin DAAM is required for coordination of the actin and microtubule cytoskeleton in axonal growth cones. J. Cell Sci. 2017, 130, 2506–2519. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Chou, H.T.; Brautigam, C.A.; Xing, W.; Yang, S.; Henry, L.; Doolittle, L.K.; Walz, T.; Rosen, M.K. Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. eLife 2017, 6, e29795. [Google Scholar] [CrossRef]
- Carlier, M.F.; Laurent, V.; Santolini, J.; Melki, R.; Didry, D.; Xia, G.X.; Hong, Y.; Chua, N.H.; Pantaloni, D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: Implication in actin-based motility. J. Cell Biol. 1997, 136, 1307–1322. [Google Scholar] [CrossRef]
- Furukawa, K.; Fu, W.; Li, Y.; Witke, W.; Kwiatkowski, D.J.; Mattson, M.P. The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability to excitotoxicity in hippocampal neurons. J. Neurosci. 1997, 17, 8178–8186. [Google Scholar] [CrossRef] [Green Version]
- Koronakis, V.; Hume, P.J.; Humphreys, D.; Liu, T.; Hørning, O.; Jensen, O.N.; McGhie, E.J. WAVE regulatory complex activation by cooperating GTPases Arf and Rac1. Proc. Natl. Acad. Sci. USA 2011, 108, 14449–14454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaks, M.; Döring, H.; Kage, F.; Steffen, A.; Klünemann, T.; Blankenfeldt, W.; Stradal, T.; Rottner, K. RhoG and Cdc42 can contribute to Rac-dependent lamellipodia formation through WAVE regulatory complex-binding. Small GTPases 2021, 12, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Sung, J.Y.; Ceglia, I.; Lee, K.W.; Ahn, J.H.; Halford, J.M.; Kim, A.M.; Kwak, S.P.; Park, J.B.; Ho Ryu, S.; et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 2006, 442, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Kim, E.Y.; Connelly, J.J.; Nassar, N.; Kirsch, J.; Winking, J.; Schwarz, G.; Schindelin, H. The Crystal Structure of Cdc42 in Complex with Collybistin II, a Gephyrin-interacting Guanine Nucleotide Exchange Factor. J. Mol. Biol. 2006, 359, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Backer, S.; Lokmane, L.; Landragin, C.; Deck, M.; Garel, S.; Bloch-Gallego, E. Trio gef mediates rhoa activation downstream of slit2 and coordinates telencephalic wiring. Development 2018, 145, dev153692. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Aizenman, C.D.; Cline, H.T. Regulation of Rho GTPases by crosstalk and neuronal activity in vivo. Neuron 2002, 33, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Yamashita, T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Front. Neurosci. 2014, 8, 338. [Google Scholar] [CrossRef]
- Hayashi, K.; Ohshima, T.; Mikoshiba, K. Pak1 is involved in dendrite initiation as a downstream effector of Rac1 in cortical neurons. Mol. Cell. Neurosci. 2002, 20, 579–594. [Google Scholar] [CrossRef]
- Parrini, M.C.; Lei, M.; Harrison, S.C.; Mayer, B.J. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol. Cell 2002, 9, 73–83. [Google Scholar] [CrossRef]
- Radu, M.; Rawat, S.J.; Beeser, A.; Iliuk, A.; Tao, W.A.; Chernoff, J. ArhGAP15, a rac-specific gtpase-activating protein, plays a dual role in inhibiting small GTpase signaling. J. Biol. Chem. 2013, 288, 21117–21125. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, M.; Chou, M.M.; Lu, W.; Mayer, B.J.; Tsai, L.H. The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature 1998, 395, 194–198. [Google Scholar] [CrossRef]
- Cuberos, H.; Vallée, B.; Vourc’H, P.; Tastet, J.; Andres, C.R.; Bénédetti, H. Roles of LIM kinases in central nervous system function and dysfunction. FEBS Lett. 2015, 589, 3795–3806. [Google Scholar] [CrossRef] [Green Version]
- Kurita, S.; Watanabe, Y.; Gunji, E.; Ohashi, K.; Mizuno, K. Molecular dissection of the mechanisms of substrate recognition and F-actin-mediated activation of cofilin-phosphatase slingshot-1. J. Biol. Chem. 2008, 283, 32542–32552. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, S.A. The Origins of Order: Self-Organization and Selection in Evolution; Oxford University Press: Oxford, UK, 1992; pp. 61–100. [Google Scholar] [CrossRef] [Green Version]
- Dupraz, S.; Hilton, B.J.; Husch, A.; Santos, T.E.; Coles, C.H.; Stern, S.; Brakebusch, C.; Bradke, F. RhoA Controls Axon Extension Independent of Specification in the Developing Brain. Curr. Biol. 2019, 29, 3874–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, J.P.; Wang-Dunlop, J.; Gonzales, C.; Goad, M.E.P.; Mark, R.J.; Kwak, S.P. Characterization of the WAVE1 knock-out mouse: Implications for CNS development. J. Neurosci. 2003, 23, 3343–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic, M.; Dudek, H.; Kwon, Y.T.; Ramos, Y.F.M.; Tsai, L.H. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996, 10, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Svitkina, T. Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol. Biol. Cell 2008, 19, 1561–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, P.; Dey, C.S. P21-activated kinase 2 (PAK2) regulates glucose uptake and insulin sensitivity in neuronal cells. Mol. Cell. Endocrinol. 2016, 429, 50–61. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, Z.; Asrar, S.; Henkelman, M.; Xie, W.; Jia, Z. p21-Activated Kinases 1 and 3 Control Brain Size through Coordinating Neuronal Complexity and Synaptic Properties. Mol. Cell. Biol. 2011, 31, 388–403. [Google Scholar] [CrossRef] [Green Version]
- Greathouse, K.M.; Boros, B.D.; Deslauriers, J.F.; Henderson, B.W.; Curtis, K.A.; Gentry, E.G.; Herskowitz, J.H. Distinct and complementary functions of rho kinase isoforms ROCK1 and ROCK2 in prefrontal cortex structural plasticity. Brain Struct. Funct. 2018, 223, 4227–4241. [Google Scholar] [CrossRef]
- Zamboni, V.; Armentano, M.; Berto, G.; Ciraolo, E.; Ghigo, A.; Garzotto, D.; Umbach, A.; Dicunto, F.; Parmigiani, E.; Boido, M.; et al. Hyperactivity of Rac1-GTPase pathway impairs neuritogenesis of cortical neurons by altering actin dynamics. Sci. Rep. 2018, 8, 7254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.J.; He, W.Q.; Tang, J.; Tao, T.; Chen, C.; Gao, Y.Q.; Zhang, W.C.; He, X.Y.; Dai, Y.Y.; Zhu, N.C.; et al. Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J. Biol. Chem. 2010, 285, 24834–24844. [Google Scholar] [CrossRef] [Green Version]
- Powell, A.D.; Gill, K.K.; Saintot, P.P.; Jiruska, P.; Chelly, J.; Billuart, P.; Jefferys, J.G.R. Rapid reversal of impaired inhibitory and excitatory transmission but not spine dysgenesis in a mouse model of mental retardation. J. Physiol. 2012, 590, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Bellenchi, G.C.; Gurniak, C.B.; Perlas, E.; Middei, S.; Ammassari-Teule, M.; Witke, W. N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev. 2007, 21, 2347–2357. [Google Scholar] [CrossRef] [Green Version]
- Gombos, R.; Migh, E.; Antal, O.; Mukherjee, A.; Jenny, A.; Mihály, J. The formin DAAM functions as molecular effector of the planar cell polarity pathway during axonal development in Drosophila. J. Neurosci. 2015, 35, 10154–10167. [Google Scholar] [CrossRef] [Green Version]
- Sapir, T.; Levy, T.; Sakakibara, A.; Rabinkov, A.; Miyata, T.; Reiner, O. Shootin1 acts in concert with KIF20B to promote polarization of migrating neurons. J. Neurosci. 2013, 33, 11932–11948. [Google Scholar] [CrossRef] [Green Version]
- Albert, R.; Othmer, H.G. The topology of the regulatory interactions predicts the expression pattern of the segment polarity genes in Drosophila melanogaster. J. Theor. Biol. 2003, 223, 1–18. [Google Scholar] [CrossRef]
- Hetmanski, J.H.R.; Schwartz, J.M.; Caswell, P.T. Rationalizing Rac1 and RhoA GTPase signaling: A mathematical approach. Small GTPases 2018, 9, 224–229. [Google Scholar] [CrossRef]
- Huang, S. Gene expression profiling, genetic networks, and cellular states: An integrating concept for tumorigenesis and drug discovery. J. Mol. Med. 1999, 77, 469–480. [Google Scholar] [CrossRef]
- Spiridigliozzi, G.A.; Keeling, L.A.; Stefanescu, M.; Li, C.; Austin, S.; Kishnani, P.S. Cognitive and academic outcomes in long-term survivors of infantile-onset Pompe disease: A longitudinal follow-up. Mol. Genet. Metab. 2017, 121, 127–137. [Google Scholar] [CrossRef]
- Costa, R.M.; Federov, N.B.; Kogan, J.H.; Murphy, G.G.; Stern, J.; Ohno, M.; Kucherlapati, R.; Jacks, T.; Silva, A.J. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002, 415, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cui, Y.; Kushner, S.A.; Brown, R.A.M.; Jentsch, J.D.; Frankland, P.W.; Cannon, T.D.; Silva, A.J. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of Neurofibromatosis Type 1. Curr. Biol. 2005, 15, 1961–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleschevnikov, A.M.; Belichenko, P.V.; Faizi, M.; Jacobs, L.F.; Htun, K.; Shamloo, M.; Mobley, W.C. Deficits in cognition and synaptic plasticity in a mouse model of down syndrome ameliorated by GABA B receptor antagonists. J. Neurosci. 2012, 32, 9217–9227. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, F.; Morishita, W.; Zuniga, E.; Nguyen, J.; Blank, M.; Malenka, R.C.; Garner, C.C. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 2007, 10, 411–413. [Google Scholar] [CrossRef]
- Rueda, N.; Flórez, J.; Martínez-Cué, C. Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neurosci. Lett. 2008, 433, 22–27. [Google Scholar] [CrossRef]
- Deidda, G.; Parrini, M.; Naskar, S.; Bozarth, I.F.; Contestabile, A.; Cancedda, L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015, 21, 318–326. [Google Scholar] [CrossRef]
- Pinto, B.; Morelli, G.; Rastogi, M.; Savardi, A.; Fumagalli, A.; Petretto, A.; Bartolucci, M.; Varea, E.; Catelani, T.; Contestabile, A.; et al. Rescuing Over-activated Microglia Restores Cognitive Performance in Juvenile Animals of the Dp(16) Mouse Model of Down Syndrome. Neuron 2020, 108, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Bletsch, M.; Stanley, J.; Wheeler, D.; Scott, R.; Tully, T. The PDE4 inhibitor HT-0712 improves hippocampus-dependent memory in aged mice. Neuropsychopharmacology 2014, 39, 2938–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón, J.M.; Malleret, G.; Touzani, K.; Vronskaya, S.; Ishii, S.; Kandel, E.R.; Barco, A. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: A model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 2004, 42, 947–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornsson, H.T.; Benjamin, J.S.; Zhang, L.; Weissman, J.; Gerber, E.E.; Chen, Y.C.; Vaurio, R.G.; Potter, M.C.; Hansen, K.D.; Dietz, H.C. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci. Transl. Med. 2014, 6, 256ra135. [Google Scholar] [CrossRef] [Green Version]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Meikle, L.; Pollizzi, K.; Egnor, A.; Kramvis, I.; Lane, H.; Sahin, M.; Kwiatkowski, D.J. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: Effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 2008, 28, 5422–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, W.E.; Orlova, K.A.; Parker, W.H.; Birnbaum, J.F.; Krymskaya, V.P.; Goncharov, D.A.; Baybis, M.; Helfferich, J.; Okochi, K.; Strauss, K.A.; et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci. Transl. Med. 2013, 5, 182ra53. [Google Scholar] [CrossRef] [Green Version]
- U.N.L. of M.C. 2017. Available online: govhttps://clinicaltrials.gov/show/NCT0171394 (accessed on 27 February 2021).
- Veenstra-Vanderweele, J.; Blakely, R.D. Networking in autism: Leveraging genetic, biomarker and model system findings in the search for new treatments. Neuropsychopharmacology 2012, 37, 196–212. [Google Scholar] [CrossRef]
- Vinueza Veloz, M.F.; Buijsen, R.A.M.; Willemsen, R.; Cupido, A.; Bosman, L.W.J.; Koekkoek, S.K.E.; Potters, J.W.; Oostra, B.A.; De Zeeuw, C.I. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 2012, 11, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Braat, S.; Kooy, R.F. Fragile X syndrome neurobiology translates into rational therapy. Drug Discov. Today 2014, 19, 510–519. [Google Scholar] [CrossRef]
- Dziembowska, M.; Pretto, D.I.; Janusz, A.; Kaczmarek, L.; Leigh, M.J.; Gabriel, N.; Durbin-Johnson, B.; Hagerman, R.J.; Tassone, F. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am. J. Med. Genet. Part A 2013, 161, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Mastinu, A.; Ferrari-Toninelli, G.; Memo, M. Potential role of microtubule stabilizing agents in neurodevelopmental disorders. Int. J. Mol. Sci. 2017, 18, 1627. [Google Scholar] [CrossRef] [Green Version]
- Marchisella, F.; Coffey, E.T.; Hollos, P. Microtubule and microtubule associated protein anomalies in psychiatric disease. Cytoskeleton 2016, 73, 596–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, V.M.Y.; Daughenbaugh, R.; Trojanowski, J.Q. Microtubule stabilizing drugs for the treatment of Alzheimer’s disease. Neurobiol. Aging 1994, 15, 87–89. [Google Scholar] [CrossRef]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.M.L.; Xie, S.X.; Ballatore, C.; Smith, A.B.; et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef]
- Andrieux, A.; Salin, P.; Schweitzer, A.; Bégou, M.; Pachoud, B.; Brun, P.; Gory-Fauré, S.; Kujala, P.; Suaud-Chagny, M.F.; Höfle, G.; et al. Microtubule Stabilizer Ameliorates Synaptic Function and Behavior in a Mouse Model for Schizophrenia. Biol. Psychiatry 2006, 60, 1224–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowinsky, E.K. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu. Rev. Med. 1997, 48, 353–374. [Google Scholar] [CrossRef]
- Gozes, I. Activity-dependent neuroprotective protein (ADNP): From autism to Alzheimer’s disease. SpringerPlus 2015, 4, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Levine, J.; Cohen, D.; Herman, C.; Verloes, A.; Guinchat, V.; Diaz, L.; Cravero, C.; Mandel, A.; Gozes, I. Developmental Phenotype of the Rare Case of DJ Caused by a Unique ADNP Gene De Novo Mutation. J. Mol. Neurosci. 2019, 68, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Ivashko-Pachima, Y.; Sayas, C.L.; Malishkevich, A.; Gozes, I. ADNP/NAP dramatically increase microtubule end-binding protein-Tau interaction: A novel avenue for protection against tauopathy. Mol. Psychiatry 2017, 22, 1335–1344. [Google Scholar] [CrossRef]
- Chua, L.; Chong, S.A.; Pang, E.; Ng, Y.; Chan, Y.H.; Chua, L. The Effect of Risperidone on Cognitive Functioning in a Sample of Asian Patients with Schizophrenia in Singapore. Singap. Med. J 2001, 42, 243–246. [Google Scholar]
- Troost, P.W.; Lahuis, B.E.; Steenhuis, M.P.; Ketelaars, C.E.J.; Buitelaar, J.K.; Van Engeland, H.; Scahill, L.; Minderaa, R.B.; Hoekstra, P.J. Long-term effects of risperidone in children with autism spectrum disorders: A placebo discontinuation study. J. Am. Acad. Child Adolesc. Psychiatry 2005, 44, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.Y.; Smith, A.B. Non-Naturally Occurring Small Molecule Microtubule-Stabilizing Agents: A Potential Tactic for CNS-Directed Therapies. ACS Chem. Neurosci. 2017, 8, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Toba, S.; Tamura, Y.; Kumamoto, K.; Yamada, M.; Takao, K.; Hattori, S.; Miyakawa, T.; Kataoka, Y.; Azuma, M.; Hayasaka, K.; et al. Post-natal treatment by a blood-brain-barrier permeable calpain inhibitor, SNJ1945 rescued defective function in lissencephaly. Sci. Rep. 2013, 3, 1224. [Google Scholar] [CrossRef]
- Chen, Y.K.; Chen, C.Y.; Hu, H.T.; Hsueh, Y.P. CTTNBP2, but not CTTNBP2NL, regulates dendritic spinogenesis and synaptic distribution of the striatin-PP2A complex. Mol. Biol. Cell 2012, 23, 4383–4392. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Johnson, R.C.; Mains, R.E.; Eipper, B.A. Expression of Kalirin, a neuronal GDP/GTP exchange factor of the trio family, in the central nervous system of the adult rat. J. Comp. Neurol. 2001, 429, 388–402. [Google Scholar] [CrossRef]
- Chen, Y.; Derin, R.; Petralia, R.S.; Li, M. Actinfilin, a Brain-specific Actin-binding Protein in Postsynaptic Density. J. Biol. Chem. 2002, 277, 30495–30501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klenchin, V.A.; Allingham, J.S.; King, R.; Tanaka, J.; Marriott, G.; Rayment, I. Trisoxazole macrolide toxins mimic the binding of actin-capping proteins to actin. Nat. Struct. Biol. 2003, 10, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Crevenna, A.H.; Ugur, I.; Marion, A.; Antes, I.; Kazmaier, U.; Hoyer, M.; Lamb, D.C.; Gegenfurtner, F.; Kliesmete, Z.; et al. Actin stabilizing compounds show specific biological effects due to their binding mode. Sci. Rep. 2019, 9, 9731. [Google Scholar] [CrossRef]
- Moser, C.; Rüdiger, D.; Förster, F.; Von Blume, J.; Yu, P.; Kuster, B.; Kazmaier, U.; Vollmar, A.M.; Zahler, S. Persistent inhibition of pore-based cell migration by sub-toxic doses of miuraenamide, an actin filament stabilizer. Sci. Rep. 2017, 7, 16407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bongmba, O.Y.N.; Martinez, L.A.; Elhardt, M.E.; Butler, K.; Tejada-Simon, M.V. Modulation of dendritic spines and synaptic function by Rac1: A possible link to Fragile X syndrome pathology. Brain Res. 2011, 1399, 79–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; De La Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef] [Green Version]
- Zins, K.; Lucas, T.; Reichl, P.; Abraham, D.; Aharinejad, S. A Rac1/Cdc42 GTPase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS ONE 2013, 8, e74924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef] [Green Version]
- Contini, A.; Ferri, N.; Bucci, R.; Lupo, M.G.; Erba, E.; Gelmi, M.L.; Pellegrino, S. Peptide modulators of Rac1/Tiam1 protein-protein interaction: An alternative approach for cardiovascular diseases. Pept. Sci. 2018, 110, e23089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meziane, H.; Khelfaoui, M.; Morello, N.; Hiba, B.; Calcagno, E.; Reibel-Foisset, S.; Selloum, M.; Chelly, J.; Humeau, Y.; Riet, F.; et al. Fasudil treatment in adult reverses behavioural changes and brain ventricular enlargement in Oligophrenin-1 mouse model of intellectual disability. Hum. Mol. Genet. 2016, 25, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of rett syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef]
- Lemichez, E.; Flatau, G.; Bruzzone, M.; Boquet, P.; Gauthier, M. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 1997, 24, 1061–1070. [Google Scholar] [CrossRef]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gin 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef]
- De Viti, S.; Martino, A.; Musilli, M.; Fiorentini, C.; Diana, G. The Rho GTPase activating CNF1 improves associative working memory for object-in-place. Behav. Brain Res. 2010, 212, 78–83. [Google Scholar] [CrossRef]
- Diana, G.; Valentini, G.; Travaglione, S.; Falzano, L.; Pieri, M.; Zona, C.; Meschini, S.; Fabbri, A.; Fiorentini, C. Enhancement of learning and memory after activation of cerebral Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corti, S.; Faravelli, I.; Cardano, M.; Conti, L. Human pluripotent stem cells as tools for neurodegenerative and neurodevelopmental disease modeling and drug discovery. Expert Opin. Drug Discov. 2015, 10, 615–629. [Google Scholar] [CrossRef] [PubMed]


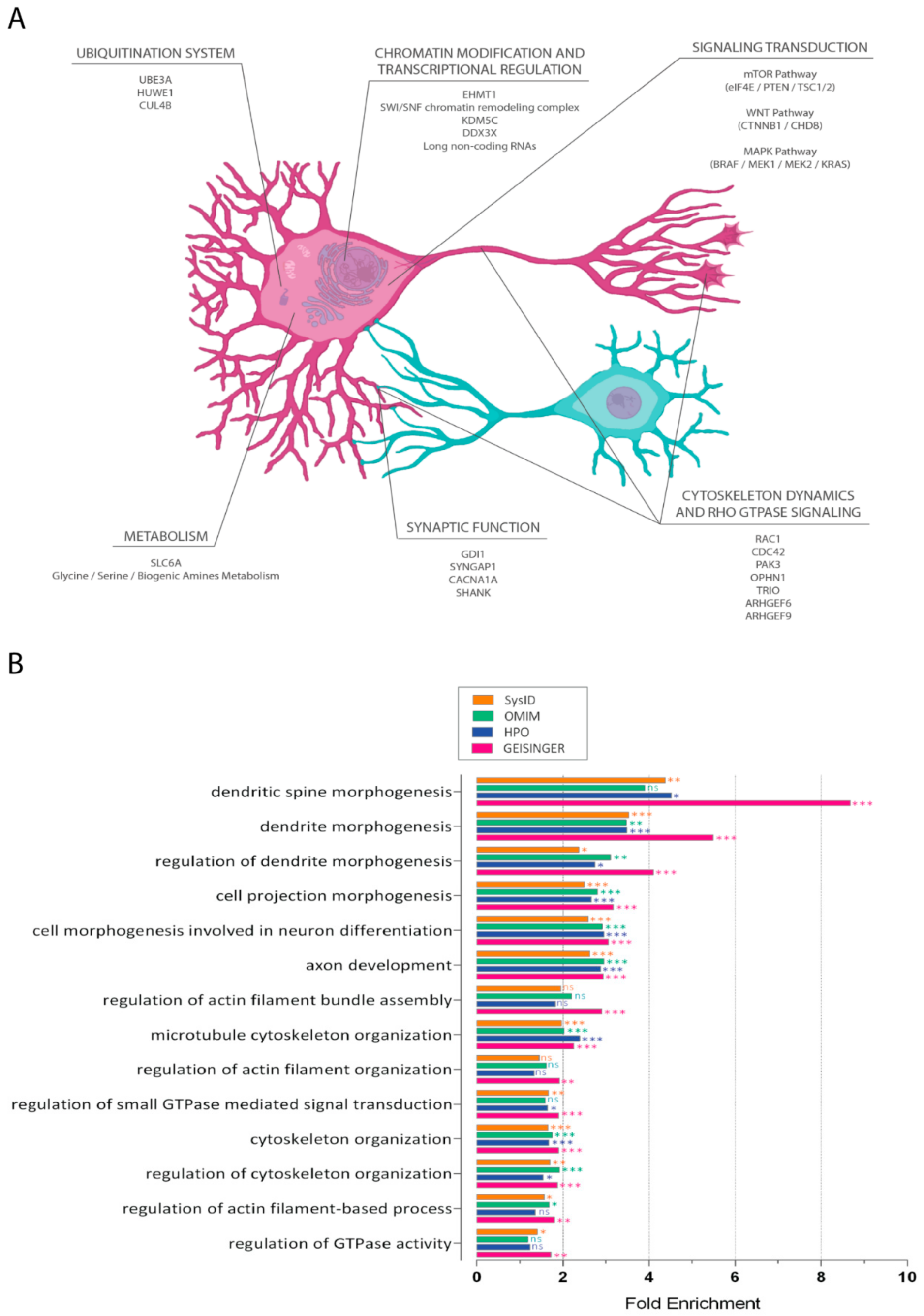{kind=link}
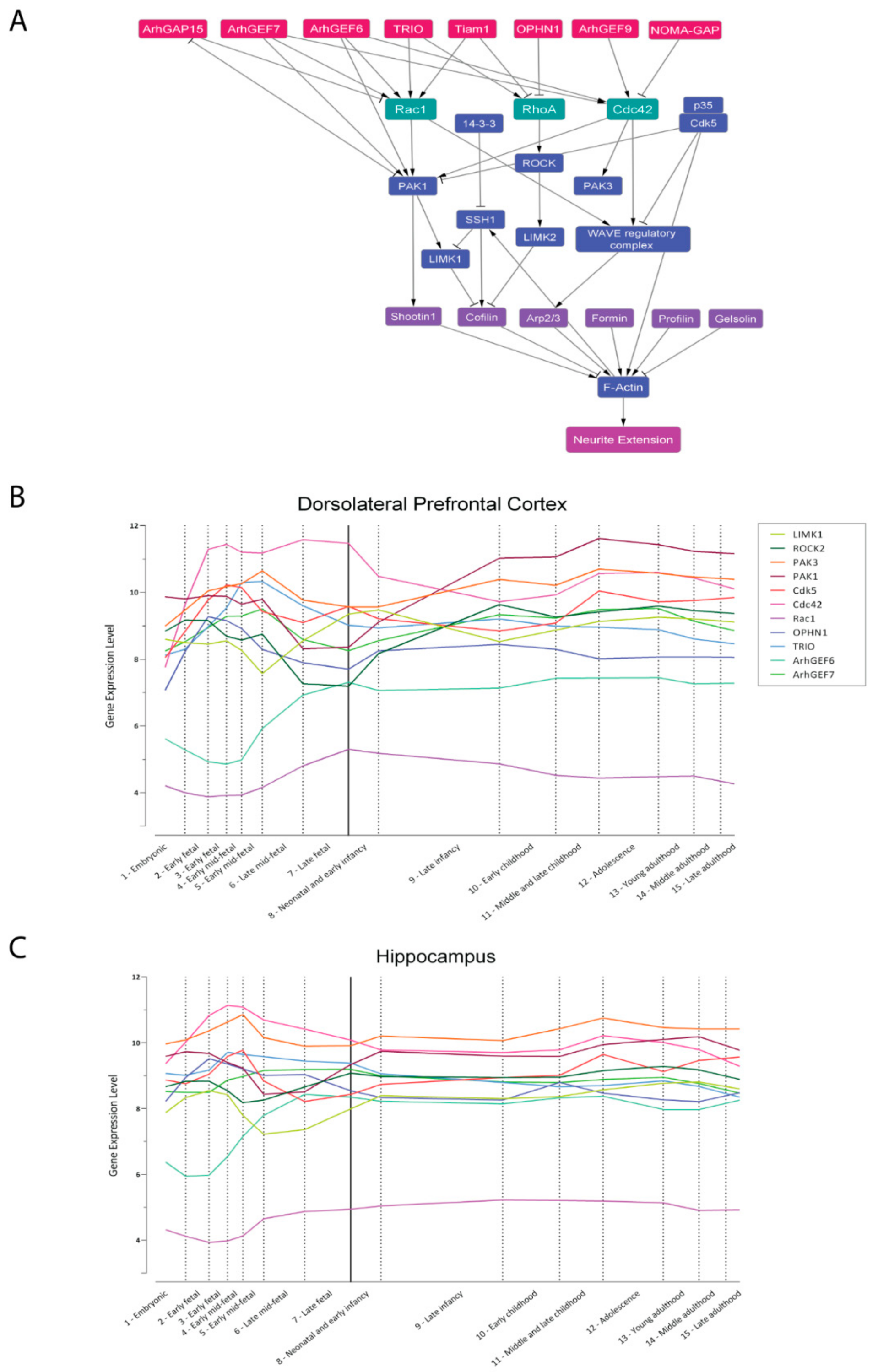{kind=link}
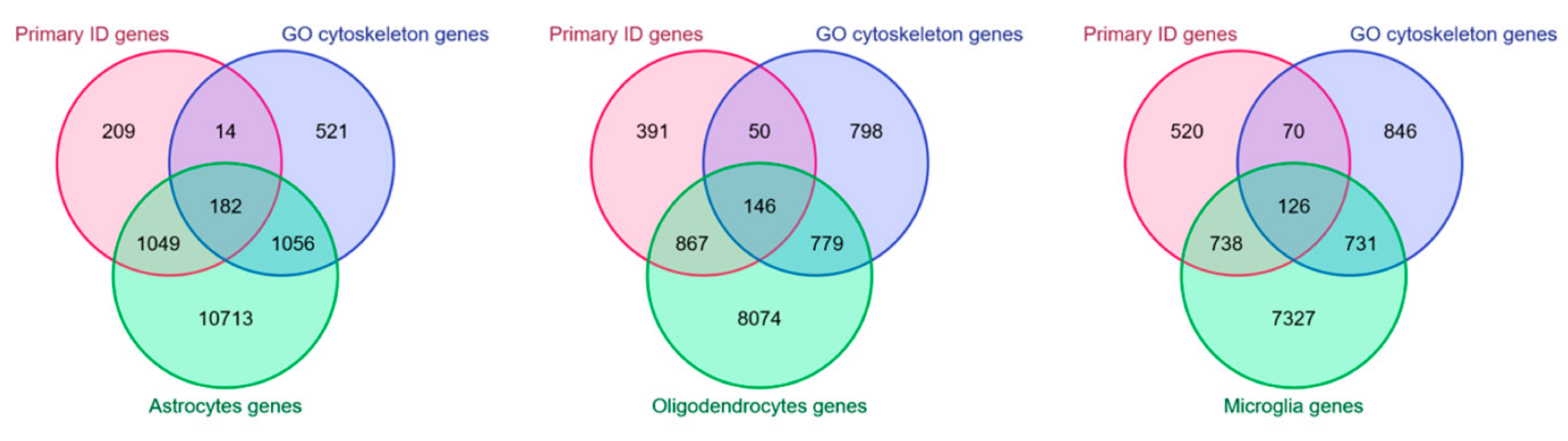{kind=link}
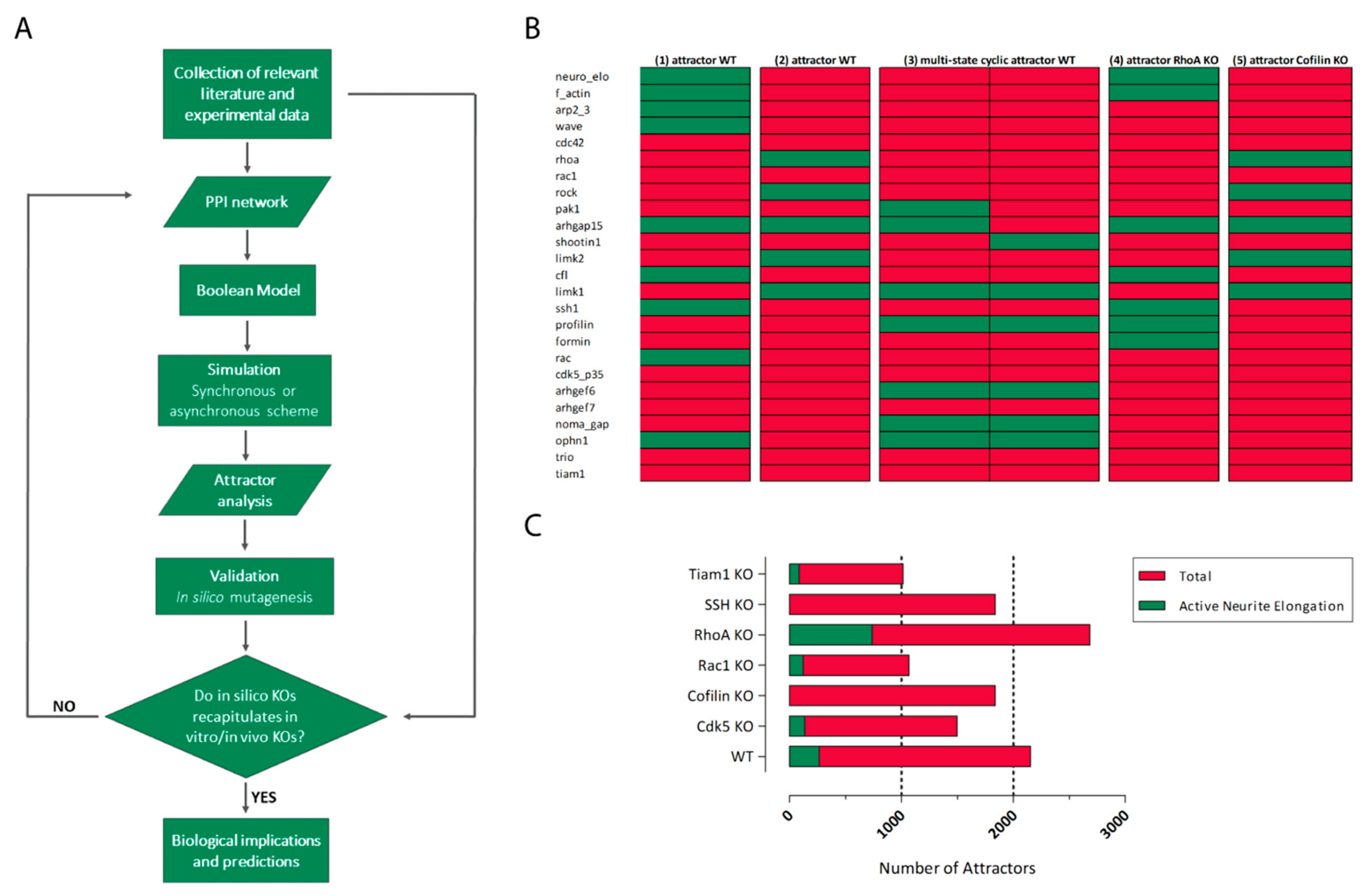{kind=link}
| Targets, Factors (1) | Reference |
|---|---|
| neuro_elo, f_actin | [68] |
| f_actin, (profilin | formin | arp2_3 | shootin1) & !cfl | [266,349,350,351,352,353] |
| arp2_3, wave | [351,354] |
| wave, (rac1 | cdc42) & !cdk5_p35 | [351,355,356] |
| cdc42, (arhgef6 | arhgef7) & !(noma_gap | ophn1) | [166,176,184,193,357] |
| rhoa, trio | !(ophn1 | tiam1) | [164,358,359] |
| rac1, (tiam1 | arhgef6 | arhgef7 | trio) & !(arhgap15 | ophn1) | [164,172,184,193,205,358] |
| rock, rhoa | [360] |
| pak1, (rac1 | cdc42 | arhgef6 | arhgef7) & !(arhgap15 | cdk5_p35) | [186,361,362,363,364] |
| arhgap15, !pak1 | [363] |
| shootin1, pak1 | [266] |
| cdk5_p35, f_actin | [154] |
| limk2, rock | [360] |
| cfl, ssh1 & !(limk2 | limk1) | [222,365] |
| limk1, pak1 | !ssh1 | [129,224] |
| ssh1, f_actin | [222,366] |
| Protein | Gene Mutation (1) | Phenotype | Reference |
|---|---|---|---|
| Rac1 | Forebrain-specific KO | Increased number of primary neurites and secondary branches | [95] |
| RhoA | KO | Increased axon length (significantly greater actin retrograde flow, fewer actin arcs, and substantially longer F-actin bundles) | [368] |
| Cdc42 | KO | Defective axon formation (disrupted cytoskeletal organization, enlargement of the growth cones, and inhibition of filopodia dynamics) | [97] |
| WAVE1 | KO | No effect on neurite growth | [369] |
| Cdk5 | Dominant-negative | Inhibition of neurite outgrowth | [370] |
| p35 | KD | Inhibition of neurite outgrowth | [370] |
| Arp2/3 | KD | Increased number of irregular, shorter, and broader neurites | [371] |
| PAK1 | Dominant-negative | Decreased number of dendrites | [361] |
| PAK2 | Dominant-negative | No effect on the neurite growth | [372] |
| PAK3 | KD | Increased elongation of neuronal processes | [373] |
| LIMK2 | KD | Reduced number of neurite-bearing cells and the mean neurite length | [222] |
| LIMK1 | KD | Reduced number of neurite-bearing cells and the mean neurite length | [222] |
| ROCK1 | Haploinsufficiency | Increased basal and apical dendritic length and dendritic intersections | [374] |
| ROCK2 | Haploinsufficiency | No effect on the neurite growth | [374] |
| SSH1/SSH2 | KD | Decreased neurite extension | [222] |
| ArhGEF6 | KO | Increased neurite length | [184] |
| ArhGEF7 | Cortex-specific KO | Impaired axon formation | [193] |
| ArhGAP15 | KO | Decreased neurite length and branching | [172,375] |
| TRIO | Neuron-specific KO | Decreased axon length | [376] |
| Tiam1 | KO | Decreased neurite length | [211] |
| NOMA-GAP | KO | Decreased dendritic branching | [176] |
| OPHN1 | KO | Decreased dendritic tree complexity, i.e., branching | [377] |
| Cofilin | KO | Inhibited neurite outgrowth | [378] |
| Profilin1 | KD | Impaired axon elongation | [349] |
| Profilin1 | Mutation of the actin-binding domain | Decreased of neurite length | [265] |
| Formin | KO | Impaired axon elongation | [379] |
| Shootin1 | KD | Inhibited polarization | [380] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liaci, C.; Camera, M.; Caslini, G.; Rando, S.; Contino, S.; Romano, V.; Merlo, G.R. Neuronal Cytoskeleton in Intellectual Disability: From Systems Biology and Modeling to Therapeutic Opportunities. Int. J. Mol. Sci. 2021, 22, 6167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116167
Liaci C, Camera M, Caslini G, Rando S, Contino S, Romano V, Merlo GR. Neuronal Cytoskeleton in Intellectual Disability: From Systems Biology and Modeling to Therapeutic Opportunities. International Journal of Molecular Sciences. 2021; 22(11):6167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116167
Chicago/Turabian StyleLiaci, Carla, Mattia Camera, Giovanni Caslini, Simona Rando, Salvatore Contino, Valentino Romano, and Giorgio R. Merlo. 2021. "Neuronal Cytoskeleton in Intellectual Disability: From Systems Biology and Modeling to Therapeutic Opportunities" International Journal of Molecular Sciences 22, no. 11: 6167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116167