
Bioinformatics Accelerates the Major Tetrad: A Real Boost for the Pharmaceutical Industry

 , , ,
, , , 
Abstract
:1. Introduction
2. Methodology
3. Bioinformatics as an Asset of the Drug Discovery and Development (DDD) Paradigm
3.1. Translational Bioinformatics and Systems Biology in DDD
3.2. Discussion of Methods, Tools, and Databases Based on Translational Bioinformatics
4. Optimization of Anti-Microbial Research by Bioinformatics Approaches
5. An Overview of Bioinformatics in Clinical Genomic Sequencing and MicroRNA Research
5.1. Bioinformatics Tools in MicroRNA Research
5.2. Bioinformatics Process in Clinical Genomic Sequencing
- A reference genome based upon graphical elucidation, which will promote a complete genome pathway with greater accuracy in alignment, especially for ethnicities that are under-represented.
- A longer read sequencing, which will promote greater genome resolution with greater accuracy in alignment.
- A software based upon a graphics processing unit (GPU), which will promote elevated parallelization and rapid computation.
6. The COVID-19 Pandemic Giving an Impulse to the Bioinformatics Approaches
6.1. Bioinformatics Tools and Techniques for COVID-19 Research
6.1.1. Next-Generation Sequencing (NGS)
6.1.2. Genome-Wide Association Study (GWAS)
6.1.3. Computer-Aided Drug Design (CADD)
7. Future Prospects and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gilbert, D. Bioinformatics software resources. Brief. Bioinform. 2004, 5, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teufel, A.; Krupp, M.; Weinmann, A.; Galle, P.R. Current bioinformatics tools in genomic biomedical research. Int. J. Mol. Med. 2006, 17, 967–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscombe, N.; Greenbaum, D.; Gerstein, M. Review: What is bioinformatics. An introduction and overview. Yearb. Med. Inform. 2001, 1, 2. [Google Scholar]
- Mandal, S.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Katara, P. Role of bioinformatics and pharmacogenomics in drug discovery and development process. Netw. Model. Anal. Health Inform. Bioinform. 2013, 2, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Thibaut, U. Bioinformatics and rational drug design: Tools for discovery and better understanding of biological targets and mode of action of drugs. Scand. J. Gastroenterol. 2002, 37, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Costa, C.; Orozco, M.; de la Cruz, X. Use of bioinformatics tools for the annotation of disease-associated mutations in animal models. Proteins Struct. Funct. Bioinform. 2005, 61, 878–887. [Google Scholar] [CrossRef]
- Moore, J.H.; Rhodes, C.H. Integration of molecular and cellular pathogenesis: A bioinformatics approach. In Molecular Pathology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 219–224. [Google Scholar]
- Lyall, A. Bioinformatics in the pharmaceutical industry. Trends Biotechnol. 1996, 14, 308–312. [Google Scholar] [CrossRef]
- WHO (World Health Organization). The Importance of Pharmacovigilance. Available online: http://apps.who.int/medicinedocs/en/d/Js4893e/ (accessed on 20 April 2021).
- Dal Pan, G.J. Ongoing challenges in pharmacovigilance. Drug Saf. 2014, 37, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Definition of Biomedical Informatics. American Medical Informatics Association. Available online: https://www.amia.org/biomedical-informatics-core-competencies (accessed on 15 March 2021).
- Bhangale, R.; Vaity, S.; Kulkarni, N. A day in the life of a pharmacovigilance case processor. Perspect. Clin. Res. 2017, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Bungau, C.; Blaga, F.; Gherghea, C. Kaizen Implementation for Cost Reduction in Manufacturing Process Product “Driver Control Board”; University of Oradea: Oradea, Romania, 2014; pp. 55–58. [Google Scholar]
- Daina, L.G.; Sabau, M.; Daina, C.M.; Neamțu, C.; Tit, D.M.; Buhaș, C.L.; Bungau, C.; Aleya, L.; Bungau, S. Improving performance of a pharmacy in a Romanian hospital through implementation of an internal management control system. Sci. Total Environ. 2019, 675, 51–61. [Google Scholar] [CrossRef]
- Grewal, A.; Kataria, H.; Drawan, I. Literature search for research planning and identification of research problem. Indian J. Anaesth. 2016, 60, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Murray-Rust, P. Bioinformatics and drug discovery. Curr. Opin. Biotechnol. 1994, 5, 648–653. [Google Scholar] [CrossRef]
- Gaasterland, T.; Selkov, E. Reconstruction of metabolic networks using incomplete information. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995, 3, 127–135. [Google Scholar] [PubMed]
- Bishop, M.J.; Plank, G.; Burton, R.A.B.; Schneider, J.E.; Gavaghan, D.J.; Grau, V.; Kohl, P. Development of an anatomically detailed MRI-derived rabbit ventricular model and assessment of its impact on simulations of electrophysiological function. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H699–H718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q. Translational Bioinformatics and Systems Biology Methods for Personalized Medicine; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Boland, M.R.; Jacunski, A.; Lorberbaum, T.; Romano, J.D.; Moskovitch, R.; Tatonetti, N.P. Systems biology approaches for identifying adverse drug reactions and elucidating their underlying biological mechanisms. Wiley Interdiscip. Rev. 2016, 8, 104–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prathipati, P.; Mizuguchi, K. Systems biology approaches to a rational drug discovery paradigm. Curr. Top. Med. Chem. 2016, 16, 1009–1025. [Google Scholar] [CrossRef]
- Chautard, E.; Thierry-Mieg, N.; Ricard-Blum, S. Interaction networks: From protein functions to drug discovery. A review. Pathol. Biol. 2009, 57, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Vlasblom, J.; Jin, K.; Kassir, S.; Babu, M. Exploring mitochondrial system properties of neurodegenerative diseases through interactome mapping. J. Proteom. 2014, 100, 8–24. [Google Scholar] [CrossRef]
- A Dunn, D.; Apanovitch, D.; Follettie, M.; He, T.; Ryan, T. Taking a systems approach to the identification of novel therapeutic targets and biomarkers. Curr. Pharm. Biotechnol. 2010, 11, 721–734. [Google Scholar] [CrossRef]
- Vandamme, D.; Minke, B.A.; Fitzmaurice, W.; Kholodenko, B.N.; Kolch, W. Systems biology-embedded target validation: Improving efficacy in drug discovery. Wiley Interdiscip. Rev. 2014, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ryall, K.A.; Tan, A.C. Systems biology approaches for advancing the discovery of effective drug combinations. J. ChemInform. 2015, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Wathieu, H.; T Issa, N.; W Byers, S.; Dakshanamurthy, S. Harnessing polypharmacology with computer-aided drug design and systems biology. Curr. Pharm. Des. 2016, 22, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Liang, C.; Nilla, S.; Cecil, A.; Dandekar, T. The drug-minded protein interaction database (DrumPID) for efficient target analysis and drug development. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreyer, A.M.; Blundell, T.L. CREDO: A structural interactomics database for drug discovery. Database 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hu, G.; Wang, K.; Brylinski, M.; Xie, L.; Kurgan, L. PDID: Database of molecular-level putative protein–drug interactions in the structural human proteome. Bioinformatics 2016, 32, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanduri, R.; Bhutani, I.; Somavarapu, A.K.; Mahajan, S.; Parkesh, R.; Gupta, P. ONRLDB—Manually curated database of experimentally validated ligands for orphan nuclear receptors: Insights into new drug discovery. Database 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Taccioli, C.; Sorrentino, G.; Zannini, A.; Caroli, J.; Beneventano, D.; Anderlucci, L.; Lolli, M.; Bicciato, S.; Del Sal, G. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/TAZ in cancer cells. Oncotarget 2015, 6, 38854. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Achenbach, J.; Moser, D.; Proschak, E. VAMMPIRE: A matched molecular pairs database for structure-based drug design and optimization. J. Med. Chem. 2013, 56, 5203–5207. [Google Scholar] [CrossRef] [PubMed]
- Von Eichborn, J.; Murgueitio, M.S.; Dunkel, M.; Koerner, S.; Bourne, P.E.; Preissner, R. PROMISCUOUS: A database for network-based drug-repositioning. Nucleic Acids Res. 2010, 39, D1060–D1066. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Dudley, J. Translational bioinformatics approaches to drug development. Adv. Wound Care 2013, 2, 470–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Seoane, J.; Aguiar-Pulido, V.; R Munteanu, C.; Rivero, D.; R Rabunal, J.; Dorado, J.; Pazos, A. Biomedical data integration in computational drug design and bioinformatics. Curr. Comput. Aided Drug Des. 2013, 9, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Buchan, N.S.; Rajpal, D.K.; Webster, Y.; Alatorre, C.; Gudivada, R.C.; Zheng, C.; Sanseau, P.; Koehler, J. The role of translational bioinformatics in drug discovery. Drug Discov. Today 2011, 16, 426–434. [Google Scholar] [CrossRef]
- Yan, B.; Yin, F.; Wang, Q.; Zhang, W.; Li, L. Integration and bioinformatics analysis of DNA-methylated genes associated with drug resistance in ovarian cancer. Oncol. Lett. 2016, 12, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.; Li, Y.; Ma, Z.; Li, Y. Identification of drug candidate for osteoporosis by computational bioinformatics analysis of gene expression profile. Eur. J. Med. Res. 2013, 18, 5. [Google Scholar] [CrossRef] [Green Version]
- Roedder, S.; Kimura, N.; Okamura, H.; Hsieh, S.-C.; Gong, Y.; Sarwal, M.M. Significance and suppression of redundant IL17 responses in acute allograft rejection by bioinformatics based drug repositioning of fenofibrate. PLoS ONE 2013, 8, e56657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, P.M.; Dawany, N.; Dampier, W.; Byers, S.W.; Pestell, R.G.; Tozeren, A. Bioinformatics analysis reveals transcriptome and microRNA signatures and drug repositioning targets for IBD and other autoimmune diseases. Inflamm. Bowel Dis. 2012, 18, 2315–2333. [Google Scholar] [CrossRef] [PubMed]
- Abul Seoud, R.A. BioHCVKD: A bioinformatics knowledge discovery system for HCV drug discovery–identifying proteins, ligands and active residues, in biological literature. Int. J. Bioinform. Res. Appl. 2011, 7, 317–333. [Google Scholar]
- Nwankwo, N.; Seker, H. A signal processing-based Bioinformatics approach to assessing drug resistance: Human Immunodeficiency Virus as a case study. In Proceedings of the 2010 Annual International Conference of the IEEE Engineering in Medicine and Biology, Buenos Aires, Argentina, 31 August–4 September 2010; pp. 1836–1839. [Google Scholar]
- Shapshak, P.; Duncan, R.; Turchan, J.; Nath, A.; Minagar, A.; Kangueane, P.; Davis, W.; Chiappelli, F.; Elkomy, F.; Seth, R. Bioinformatics models in drug abuse and Neuro-AIDS: Using and developing databases. Bioinformation 2006, 1, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoain, J.; Franch, M.; Martínez, M.; Tabas-Madrid, D.; Sorzano, C.O.; Bakker, A.; Gonzalez-Couto, E.; Elvira, J.; Pascual-Montano, A. NFFinder: An online bioinformatics tool for searching similar transcriptomics experiments in the context of drug repositioning. Nucleic Acids Res. 2015, 43, W193–W199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Schneider, B.P.; Li, L. A bioinformatics approach for precision medicine off-label drug drug selection among triple negative breast cancer patients. J. Am. Med Inform. Assoc. 2016, 23, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki-Kinoshita, K.F.; Kanehisa, M. Bioinformatics approaches in glycomics and drug discovery. Curr. Opin. Mol. Ther. 2006, 8, 514–520. [Google Scholar] [PubMed]
- Koduru, S.K. The Impact of Bioinformatics Tools in the Development of Antimicrobial Drugs and Other Agents. In Recent Developments in Applied Microbiology and Biochemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 335–347. [Google Scholar]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.M.; Wilkening, J.; Wilke, A.; Antonopoulos, D.; Meyer, F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5368. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef]
- Dhusia, K.; Bajpai, A.; Ramteke, P. Overcoming antibiotic resistance: Is siderophore Trojan horse conjugation an answer to evolving resistance in microbial pathogens? J. Control. Release Off. J. Control. Release Soc. 2017, 269, 63–87. [Google Scholar] [CrossRef]
- Swaminathan, S.; Sundaramurthi, J.C.; Palaniappan, A.N.; Narayanan, S. Recent developments in genomics, bioinformatics and drug discovery to combat emerging drug-resistant tuberculosis. Tuberculosis 2016, 101, 31–40. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Zhu, T.; Gu, Q.; Li, D. Advanced tools in marine natural drug discovery. Curr. Opin. Biotechnol. 2016, 42, 13–23. [Google Scholar] [CrossRef]
- Freiberg, C. Novel computational methods in anti-microbial target identification. Drug Discov. Today 2001, 6, 72–80. [Google Scholar] [CrossRef]
- Lata, S.; Mishra, N.K.; Raghava, G.P. AntiBP2: Improved version of antibacterial peptide prediction. BMC Bioinform. 2010, 11, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.S.; Rasmussen, S.; Blom, N.; Deming, J.W.; Rysgaard, S.; Sicheritz-Ponten, T. Microbial community structure of Arctic multiyear sea ice and surface seawater by 454 sequencing of the 16S RNA gene. ISME J. 2012, 6, 11–20. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjell, C.D.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.; Cherkasov, A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar] [CrossRef]
- Mikut, R.; Hilpert, K. Interpretable features for the activity prediction of short antimicrobial peptides using fuzzy logic. Int. J. Pept. Res. Ther. 2009, 15, 129–137. [Google Scholar] [CrossRef]
- Hao, Y.; Weiss, G.M. Gene Selection from Microarray Data forAge-related Macular Degeneration by Data Mining. 2016. Available online: https://storm.cis.fordham.edu/~gweiss/papers/DMIN-2016.pdf (accessed on 20 April 2021).
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.T.; Petit, R.A., III; Crispell, E.K.; Thornton, T.A.; Conneely, K.N.; Jiang, Y.; Satola, S.W.; Read, T.D. Dissecting vancomycin-intermediate resistance in Staphylococcus aureus using genome-wide association. Genome Biol. Evol. 2014, 6, 1174–1185. [Google Scholar] [CrossRef]
- Ma, R.; Jiang, T.; Kang, X. Circulating microRNAs in cancer: Origin, function and application. J. Exp. Clin. Cancer Res. 2012, 31, 38. [Google Scholar] [CrossRef] [Green Version]
- Pourteymourfard-Tabrizi, Z.; Jami, M.-S. Selection of suitable bioinformatic tools in micro-RNA research. Gene Rep. 2020, 21, 100893. [Google Scholar] [CrossRef]
- Ghorai, A.; Ghosh, U. miRNA gene counts in chromosomes vary widely in a species and biogenesis of miRNA largely depends on transcription or post-transcriptional processing of coding genes. Front. Genet. 2014, 5, 100. [Google Scholar] [CrossRef] [Green Version]
- Mahmoudian-Sani, M.-R.; Jami, M.-S.; Mahdavinezhad, A.; Amini, R.; Farnoosh, G.; Saidijam, M. The effect of the microrna-183 family on hair cell-specific markers of human bone marrow-derived mesenchymal stem cells. Audiol. Neurotol. 2018, 23, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Mehri-Ghahfarrokhi, A.; Pourteymourfard-Tabrizi, Z.; Farrokhi, E.; Chaleshtori, M.H.; Jami, M.-S. Increased levels of miR-124 in human dental pulp stem cells alter the expression of neural markers. J. Otol. 2019, 14, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W. Circulating microRNA biomarker studies: Pitfalls and potential solutions. Clin. Chem. 2015, 61, 56–63. [Google Scholar] [CrossRef]
- Mahmoudian Sani, M.R.; Mehri-Ghahfarrokhi, A.; Hashemzadeh-Chaleshtori, M.; Saidijam, M.; Jami, M.-S. Comparison of three types of mesenchymal stem cells (bone marrow, adipose tissue, and umbilical cord-derived) as potential sources for inner ear regeneration. Int. Tinnitus J. 2017, 21, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Larrea, E.; Sole, C.; Manterola, L.; Goicoechea, I.; Armesto, M.; Arestin, M.; Caffarel, M.M.; Araujo, A.M.; Araiz, M.; Fernandez-Mercado, M. New concepts in cancer biomarkers: Circulating miRNAs in liquid biopsies. Int. J. Mol. Sci. 2016, 17, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhomia, M.; Balakathiresan, N.S.; Wang, K.K.; Papa, L.; Maheshwari, R.K. A panel of serum MiRNA biomarkers for the diagnosis of severe to mild traumatic brain injury in humans. Sci. Rep. 2016, 6, 28148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zang, J.; Jing, X.; Sun, Z.; Yan, W.; Yang, D.; Guo, F.; Shen, B. Identification of candidate miRNA biomarkers from miRNA regulatory network with application to prostate cancer. J. Transl. Med. 2014, 12, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panwar, B.; Omenn, G.S.; Guan, Y. miRmine: A database of human miRNA expression profiles. Bioinformatics 2017, 33, 1554–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestdagh, P.; Lefever, S.; Pattyn, F.; Ridzon, D.; Fredlund, E.; Fieuw, A.; Ongenaert, M.; Vermeulen, J.; De Paepe, A.; Wong, L. The microRNA body map: Dissecting microRNA function through integrative genomics. Nucleic Acids Res. 2011, 39, e136. [Google Scholar] [CrossRef] [PubMed]
- Juzenas, S.; Venkatesh, G.; Hübenthal, M.; Hoeppner, M.P.; Du, Z.G.; Paulsen, M.; Rosenstiel, P.; Senger, P.; Hofmann-Apitius, M.; Keller, A. A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res. 2017, 45, 9290–9301. [Google Scholar] [CrossRef] [Green Version]
- Antonov, A.; Knight, R.; Melino, G.; Barlev, N.; Tsvetkov, P. MIRUMIR: An online tool to test microRNAs as biomarkers to predict survival in cancer using multiple clinical data sets. Cell Death Differ. 2013, 20, 367. [Google Scholar] [CrossRef]
- Li, Y.; Huo, C.; Pan, T.; Li, L.; Jin, X.; Lin, X.; Chen, J.; Zhang, J.; Guo, Z.; Xu, J. Systematic review regulatory principles of non-coding RNAs in cardiovascular diseases. Brief. Bioinform. 2019, 20, 66–76. [Google Scholar] [CrossRef]
- Schmitz, U.; Lai, X.; Winter, F.; Wolkenhauer, O.; Vera, J.; Gupta, S.K. Cooperative gene regulation by microRNA pairs and their identification using a computational workflow. Nucleic Acids Res. 2014, 42, 7539–7552. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xie, W.-B.; Xiao, P.-P.; Zhao, X.-M.; Yan, H. mTD: A database of microRNAs affecting therapeutic effects of drugs. J. Genet. Genom. 2017, 44, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.; Miao, Z.; Bi, X.; Wu, D.; Jin, N.; Wang, L.; Wu, H.; Qian, K.; Li, C. ViRBase: A resource for virus–host ncRNA-associated interactions. Nucleic Acids Res. 2015, 43, D578–D582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, V.J.; Bandrowski, A.E.; Pepin, A.-S.; Gonzalez, B.J.; Desfeux, A. OMICtools: An informative directory for multi-omic data analysis. Database 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Lukasik, A.; Wójcikowski, M.; Zielenkiewicz, P. Tools4miRs–one place to gather all the tools for miRNA analysis. Bioinformatics 2016, 32, 2722–2724. [Google Scholar] [CrossRef]
- Reczko, M.; Maragkakis, M.; Alexiou, P.; Grosse, I.; Hatzigeorgiou, A.G. Functional microRNA targets in protein coding sequences. Bioinformatics 2012, 28, 771–776. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Weinstein, E.G.; Abdelhakim, A.; Yekta, S.; Rhoades, M.W.; Burge, C.B.; Bartel, D.P. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003, 17, 991–1008. [Google Scholar] [CrossRef] [Green Version]
- Thadani, R.; Tammi, M.T. MicroTar: Predicting microRNA targets from RNA duplexes. In BMC Bioinformatics; BioMed Centra: London, UK, 2006; pp. 1–9. [Google Scholar]
- Jha, A.; Shankar, R. miReader: Discovering novel miRNAs in species without sequenced genome. PLoS ONE 2013, 8, e66857. [Google Scholar]
- Vejnar, C.E.; Blum, M.; Zdobnov, E.M. miRmap web: Comprehensive microRNA target prediction online. Nucleic Acids Res. 2013, 41, W165–W168. [Google Scholar] [CrossRef] [PubMed]
- Rueda, A.; Barturen, G.; Lebrón, R.; Gómez-Martín, C.; Alganza, Á.; Oliver, J.L.; Hackenberg, M. sRNAtoolbox: An integrated collection of small RNA research tools. Nucleic Acids Res. 2015, 43, W467–W473. [Google Scholar] [CrossRef] [Green Version]
- Chiromatzo, A.; Oliveira, T.; Pereira, G.; Costa, A.; Montesco, C.; Gras, D.; Yosetake, F.; Vilar, J.; Cervato, M.; Prado, P. miRNApath: A database of miRNAs, target genes and metabolic pathways. Genet. Mol. Res. 2007, 6, 859–865. [Google Scholar]
- Jacobsen, A.; Krogh, A.; Kauppinen, S.; Lindow, M. miRMaid: A unified programming interface for microRNA data resources. BMC Bioinform. 2010, 11, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA–target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef] [PubMed]
- Rukov, J.L.; Wilentzik, R.; Jaffe, I.; Vinther, J.; Shomron, N. Pharmaco-miR: Linking microRNAs and drug effects. Brief. Bioinform. 2014, 15, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N. miRWalk2. 0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.W.-C.; Lin, L.-Z.; Hsu, S.-D.; Hsu, J.B.-K.; Huang, H.-D. ViTa: Prediction of host microRNAs targets on viruses. Nucleic Acids Res. 2007, 35, D381–D385. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, W.; Lian, B.; Liu, X.; Zhang, Y.; Dai, E.; Yu, X.; Meng, F.; Jiang, W.; Li, X. TM REC: A Database of Transcription Factor and MiRNA Regulatory Cascades in Human Diseases. PLoS ONE 2015, 10, e0125222. [Google Scholar]
- Bhattacharya, A.; Cui, Y. miR2GO: Comparative functional analysis for microRNAs. Bioinformatics 2015, 31, 2403–2405. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Kowarsch, A.; Schmidl, D.; Buggenthin, F.; Brauner, B.; Dunger, I.; Fobo, G.; Frishman, G.; Montrone, C.; Theis, F.J. PhenomiR: A knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010, 11, R6. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA–cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Friard, O.; Re, A.; Taverna, D.; De Bortoli, M.; Corá, D. CircuitsDB: A database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinform. 2010, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, S.; Bhattacharyya, M. PuTmiR: A database for extracting neighboring transcription factors of human microRNAs. BMC Bioinform. 2010, 11, 190. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Jang, I.; Jun, Y.; Yoon, S.; Ko, M.; Kwon, Y.; Choi, I.; Chang, H.; Ryu, D.; Lee, B. MiRGator v3. 0: A microRNA portal for deep sequencing, expression profiling and mRNA targeting. Nucleic Acids Res. 2012, 41, D252–D257. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, I.S.; Vergoulis, T.; Paraskevopoulou, M.D.; Lykokanellos, F.; Georgakilas, G.; Georgiou, P.; Chatzopoulos, S.; Karagkouni, D.; Christodoulou, F.; Dalamagas, T. DIANA-mirExTra v2.0: Uncovering microRNAs and transcription factors with crucial roles in NGS expression data. Nucleic Acids Res. 2016, 44, W128–W134. [Google Scholar] [CrossRef] [PubMed]
- Lebo, M.S.; Hao, L.; Lin, C.-F.; Singh, A. Bioinformatics in clinical genomic sequencing. Adv. Mol. Pathol. 2018, 1, 9–26. [Google Scholar] [CrossRef]
- Poplin, R.; Newburger, D.; Dijamco, J.; Nguyen, N.; Loy, D.; Gross, S.S.; McLean, C.Y.; DePristo, M.A. Creating a universal SNP and small indel variant caller with deep neural networks. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- NovoAlign | Novocraft. Available online: http://www.novocraft.com/products/novoalign/ (accessed on 15 March 2021).
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, C.; Dong, S. Pipe MEM: A Framework to Speed up BWA-MEM in Spark with Low Overhead. Genes 2019, 10, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics—FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 April 2021).
- Picard Tools—By Broad Institute. Available online: https://broadinstitute.github.io/picard/ (accessed on 16 April 2021).
- Handsaker, R.E.; Van Doren, V.; Berman, J.R.; Genovese, G.; Kashin, S.; Boettger, L.M.; McCarroll, S.A. Large multiallelic copy number variations in humans. Nat. Genet. 2015, 47, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Jun, G.; Flickinger, M.; Hetrick, K.N.; Romm, J.M.; Doheny, K.F.; Abecasis, G.R.; Boehnke, M.; Kang, H.M. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 2012, 91, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Abbott, T.E.; Larson, D.; Chen, K. BreakDancer: Identification of genomic structural variation from paired-end read mapping. Curr. Protoc. Bioinform. 2014, 45, 15–16. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.G.; Braithwaite, R.; Gaastra, K.; Hilbush, B.S.; Inglis, S.; Irvine, S.A.; Jackson, A.; Littin, R.; Rathod, M.; Ware, D. Comparing variant call files for performance benchmarking of next-generation sequencing variant calling pipelines. bioRxiv 2015, 023754. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Amr, S.S.; Bowser, M.J.; Gowrisankar, S.; Hynes, E.; Mahanta, L.M.; Rehm, H.L.; Funke, B.; Lebo, M.S. VisCap: Inference and visualization of germ-line copy-number variants from targeted clinical sequencing data. Genet. Med. 2016, 18, 712–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromer, M.; Purcell, S.M. Using XHMM software to detect copy number variation in whole-exome sequencing data. Curr. Protoc. Hum. Genet. 2014, 81, 7–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; White, S.; Peng, B.; Johnson, A.D.; Brody, J.A.; Li, A.H.; Huang, Z.; Carroll, A.; Wei, P.; Gibbs, R. WGSA: An annotation pipeline for human genome sequencing studies. J. Med. Genet. 2016, 53, 111–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Ramos, A.H.; Lichtenstein, L.; Gupta, M.; Lawrence, M.S.; Pugh, T.J.; Saksena, G.; Meyerson, M.; Getz, G. Oncotator: Cancer variant annotation tool. Hum. Mutat. 2015, 36, E2423–E2429. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Forni, G.; Mantovani, A.; Moretta, L.; Rappuoli, R.; Rezza, G.; Bagnasco, A.; Barsacchi, G.; Bussolati, G.; Cacciari, M.; Cappuccinelli, P.; et al. COVID-19 vaccines: Where we stand and challenges ahead. Cell Death Differ. 2021, 28, 626–639. [Google Scholar] [CrossRef]
- Ray, M.; Sable, M.N.; Sarkar, S.; Hallur, V.K. Essential interpretations of bioinformatics in COVID-19 pandemic. Meta Gene 2020, 100844. [Google Scholar]
- Cannataro, M.; Harrison, A. Bioinformatics helping to mitigate the impact of COVID-19–Editorial. Brief. Bioinform. 2021, 22, 613–615. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Weber, G.M.; Hong, C.; Palmer, N.P.; Avillach, P.; Murphy, S.N.; Gutiérrez-Sacristán, A.; Xia, Z.; Serret-Larmande, A.; Neuraz, A.; Omenn, G.S.; et al. International comparisons of harmonized laboratory value trajectories to predict severe covid-19: Leveraging the 4ce collaborative across 342 hospitals and 6 countries: A retrospective cohort study. medRxiv 2021. [Google Scholar] [CrossRef]
- Gautam, A.; Tiwari, A.; Malik, Y.S. Bioinformatics applications in advancing animal virus research. In Recent Advances in Animal Virology; Springer: Berlin, Germany, 2019; pp. 447–471. [Google Scholar]
- Bah, S.Y.; Morang’a, C.M.; Kengne-Ouafo, J.A.; Amenga-Etego, L.; Awandare, G.A. Highlights on the application of genomics and bioinformatics in the fight against infectious diseases: Challenges and opportunities in Africa. Front. Genet. 2018, 9, 575. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Messina, F.; Giombini, E.; Agrati, C.; Vairo, F.; Bartoli, T.A.; Al Moghazi, S.; Piacentini, M.; Locatelli, F.; Kobinger, G.; Maeurer, M. COVID-19: Viral–host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection. J. Transl. Med. 2020, 18, 233. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Bungau, S.; Kumar, A.; Uddin, M.S.; Kumar, C.; Pal, G.; Shrivastava, K.; Zengin, G.; Arora, S. The dual impact of ACE2 in COVID-19 and ironical actions in geriatrics and pediatrics with possible therapeutic solutions. Life Sci. 2020, 257, 118075. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.T.; Uddin, M.S.; Hossain, M.F.; Abdulhakim, J.A.; Alam, M.A.; Ashraf, G.M.; Bungau, S.G.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Aleya, L. nCOVID-19 Pandemic: From Molecular Pathogenesis to Potential Investigational Therapeutics. Front. Cell Dev. Biol. 2020, 8, 616. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M.; Collaboration, I.N.S.D. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Raney, B.; Paten, B.; Pham, S. Ragout—a reference-assisted assembly tool for bacterial genomes. Bioinformatics 2014, 30, i302–i309. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The gene expression omnibus database. In Statistical Genomics; Springer: Berlin, Germany, 2016; pp. 93–110. [Google Scholar]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Karolchik, D.; Hinrichs, A.S.; Kent, W.J. The UCSC genome browser. Curr. Protoc. Bioinform. 2012, 40, 1–4. [Google Scholar] [CrossRef]
- Ray, M.; Mishra, J.; Priyadarshini, A.; Sahoo, S. In silico identification of potential drug target and analysis of effective single nucleotide polymorphisms for autism spectrum disorder. Gene Rep. 2019, 16, 100420. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Ray, M.; Sarkar, S.; Rath, S.N. Druggability for COVID-19: In silico discovery of potential drug compounds against nucleocapsid (N) protein of SARS-CoV-2. Genom. Inform. 2020, 18, e43. [Google Scholar] [CrossRef]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R. Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Wilgenbusch, J.; Swofford, D. Inferring evolutionary trees with PAUP*. Curr. Protoc. Bioinform. 2003. [Google Scholar] [CrossRef]
- Consortium, U. The universal protein resource (UniProt). Nucleic Acids Res. 2007, 36, D190–D195. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Cheng, T.; Yu, B.; Shoemaker, B.A.; Wang, J.; Bolton, E.E.; Wang, Y.; Bryant, S.H. Literature information in PubChem: Associations between PubChem records and scientific articles. J. Cheminform. 2016, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.; Eramian, D.; Shen, M.y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Hsin, J.; Arkhipov, A.; Yin, Y.; Stone, J.; Schulten, K. Using VMD: An Introductory Tutorial. Curr. Protoc. Bioinform. 2008, 24, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peddu, V.; Shean, R.C.; Xie, H.; Shrestha, L.; Perchetti, G.A.; Minot, S.S.; Roychoudhury, P.; Huang, M.-L.; Nalla, A.; Reddy, S.B. Metagenomic analysis reveals clinical SARS-CoV-2 infection and bacterial or viral superinfection and colonization. Clin. Chem. 2020, 66, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, W.; Zhang, Q.; Xu, K.; Ye, G.; Wu, W.; Sun, Z.; Liu, F.; Wu, K.; Zhong, B. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 2020, 9, 313–319. [Google Scholar] [CrossRef]
- Manning, J.E.; Bohl, J.A.; Lay, S.; Chea, S.; Sovann, L.; Sengdoeurn, Y.; Heng, S.; Vuthy, C.; Kalantar, K.; Ahyong, V. Rapid metagenomic characterization of a case of imported COVID-19 in Cambodia. bioRxiv 2020. [Google Scholar] [CrossRef]
- Van Tan, L.; Man, D.; Hang, V.; Khanh, P.; Xuan, T.; Phong, N.; Tu, T.; Hien, T.; Hung, L.; Truong, N. SARS-CoV-2 and co-infections detection in nasopharyngeal throat swabs of COVID-19 patients by metagenomics. J. Infect. 2020, 81, e175–e177. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.-Y.; Jia, N.; Zhang, Y.-W.; Shum, M.H.-H.; Jiang, J.-F.; Zhu, H.-C.; Tong, Y.-G.; Shi, Y.-X.; Ni, X.-B.; Liao, Y.-S. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [Green Version]
- Wahba, L.; Jain, N.; Fire, A.Z.; Shoura, M.J.; Artiles, K.L.; McCoy, M.J.; Jeong, D.-E. An extensive meta-metagenomic search identifies SARS-CoV-2-homologous sequences in pangolin lung viromes. MSphere 2020, 5, e00160-20. [Google Scholar] [CrossRef]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.-Y.; Tso, E.Y.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 2021, 70, 276–284. [Google Scholar]
- Kremer, F.S.; McBride, A.J.A.; Pinto, L.D.S. Approaches for in silico finishing of microbial genome sequences. Genet. Mol. Biol. 2017, 40, 553–576. [Google Scholar] [CrossRef] [Green Version]
- Sah, R.; Rodriguez-Morales, A.J.; Jha, R.; Chu, D.K.; Gu, H.; Peiris, M.; Bastola, A.; Lal, B.K.; Ojha, H.C.; Rabaan, A.A. Complete genome sequence of a 2019 novel coronavirus (SARS-CoV-2) strain isolated in Nepal. Microbiol. Resour. Announc. 2020, 9, e00169-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, P.D.; Potdar, V.A.; Choudhary, M.L.; Nyayanit, D.A.; Agrawal, M.; Jadhav, S.M.; Majumdar, T.D.; Shete-Aich, A.; Basu, A.; Abraham, P.; et al. Full-genome sequences of the first two SARS-CoV-2 viruses from India. Indian J. Med. Res. 2020, 151, 200–209. [Google Scholar]
- Caly, L.; Druce, J.; Roberts, J.; Bond, K.; Tran, T.; Kostecki, R.; Yoga, Y.; Naughton, W.; Taiaroa, G.; Seemann, T. Isolation and rapid sharing of the 2019 novel coronavirus (SARS-CoV-2) from the first patient diagnosed with COVID-19 in Australia. Med. J. Aust. 2020, 212, 459–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, K.-B.; Lee, I.-H.; Li, H.; Won, D.-G.; Hernandez-Ferrer, C.; Negron, J.A.; Kong, S.W. Comparative analysis of whole-genome sequencing pipelines to minimize false negative findings. Sci. Rep. 2019, 9, 3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [PubMed]
- Patron, J.; Serra-Cayuela, A.; Han, B.; Li, C.; Wishart, D.S. Assessing the performance of genome-wide association studies for predicting disease risk. PLoS ONE 2019, 14, e0220215. [Google Scholar]
- Ray, M.; Sarkar, S.; Rath, S.N.; Sable, M.N. Elucidation of Genome Polymorphisms in Emerging SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ramírez, J.D.; Muñoz, M.; Hernández, C.; Flórez, C.; Gomez, S.; Rico, A.; Pardo, L.; Barros, E.C.; Paniz-Mondolfi, A.E. Genetic diversity among SARS-CoV2 strains in South America may impact performance of molecular detection. Pathogens 2020, 9, 580. [Google Scholar] [CrossRef]
- Fang, B.; Liu, L.; Yu, X.; Li, X.; Ye, G.; Xu, J.; Zhang, L.; Zhan, F.; Liu, G.; Pan, T. Genome-wide data inferring the evolution and population demography of the novel pneumonia coronavirus (SARS-CoV-2). bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Paul, S. Phylogenetic analysis of the novel coronavirus reveals important variants in Indian strains. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhang, S.; Chen, J.; Wan, C.; Zhao, W.; Zhang, B. Analysis of variation and evolution of SARS-CoV-2 genome. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 2020, 40. [Google Scholar] [CrossRef]
- Lopes, L.R.; de Mattos Cardillo, G.; Paiva, P.B. Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host. Braz. J. Microbiol. 2020, 51, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Lanza, D.C.; Lima, J.P.; Jerônimo, S.M. Design and in silico validation of polymerase chain reaction primers to detect severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Bioinformatics 2020. [Google Scholar] [CrossRef]
- Toms, D.; Li, J.; Cai, H.Y. Evaluation of WHO listed COVID-19 qPCR primers and probe in silico with 375 SERS-CoV-2 full genome sequences. medRxiv 2020. [Google Scholar] [CrossRef]
- Bisht, N.; Singh, B. Role of computer aided drug design in drug development and drug discovery. Int. J. Pharm. Sci. Res. 2018, 9, 1405–1415. [Google Scholar]
- Choudhury, M.; Saikia, R. Essential basic protocol in computer aided drug designing: Efficiency and challenges. Int. J. Biotechnol. Bioeng. 2018, 4, 77–80. [Google Scholar]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar]
- Beninger, P.; Ibara, M.A. Pharmacovigilance and biomedical informatics: A model for future development. Clin. Ther. 2016, 38, 2514–2525. [Google Scholar] [CrossRef] [Green Version]






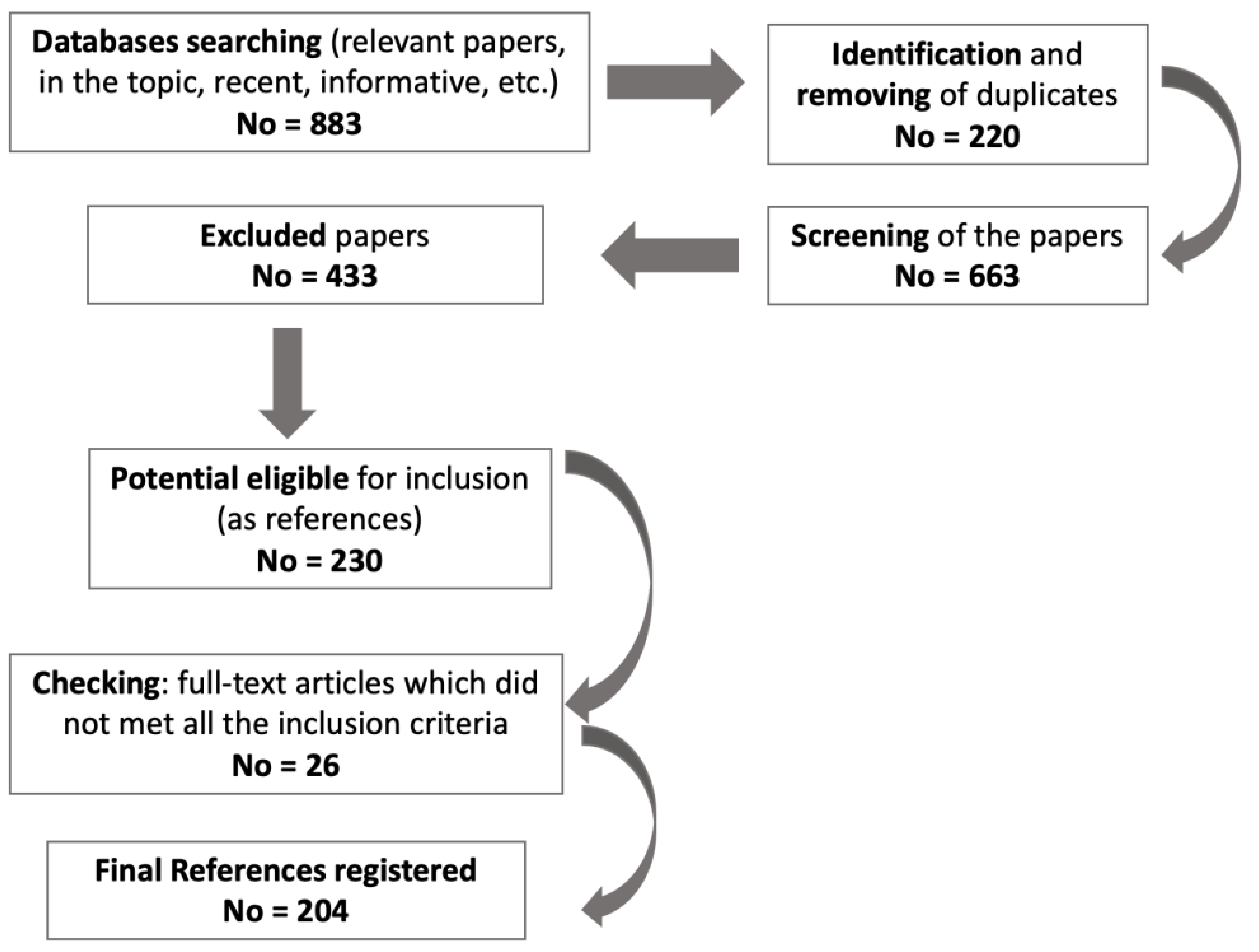{kind=link}
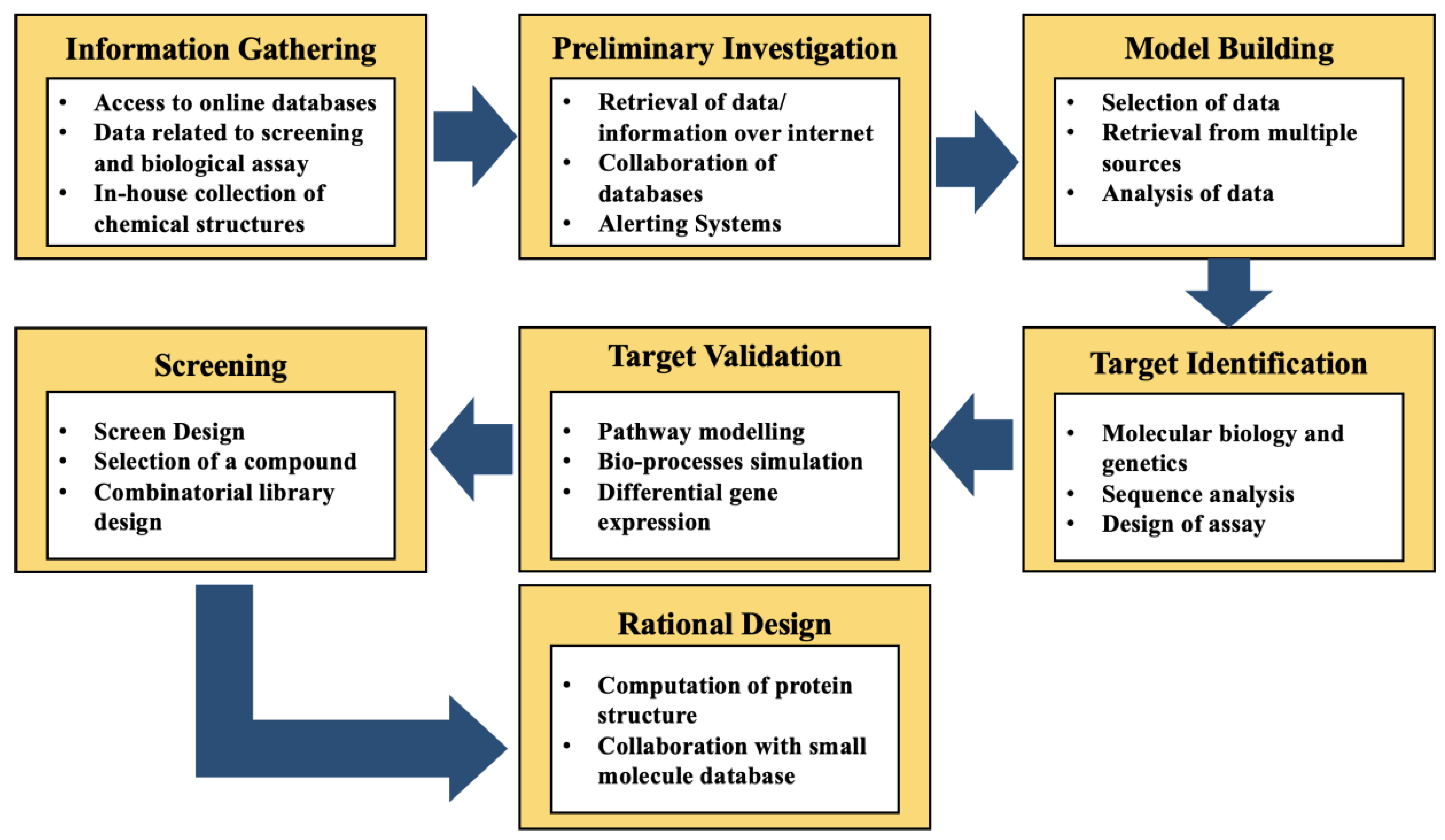{kind=link}
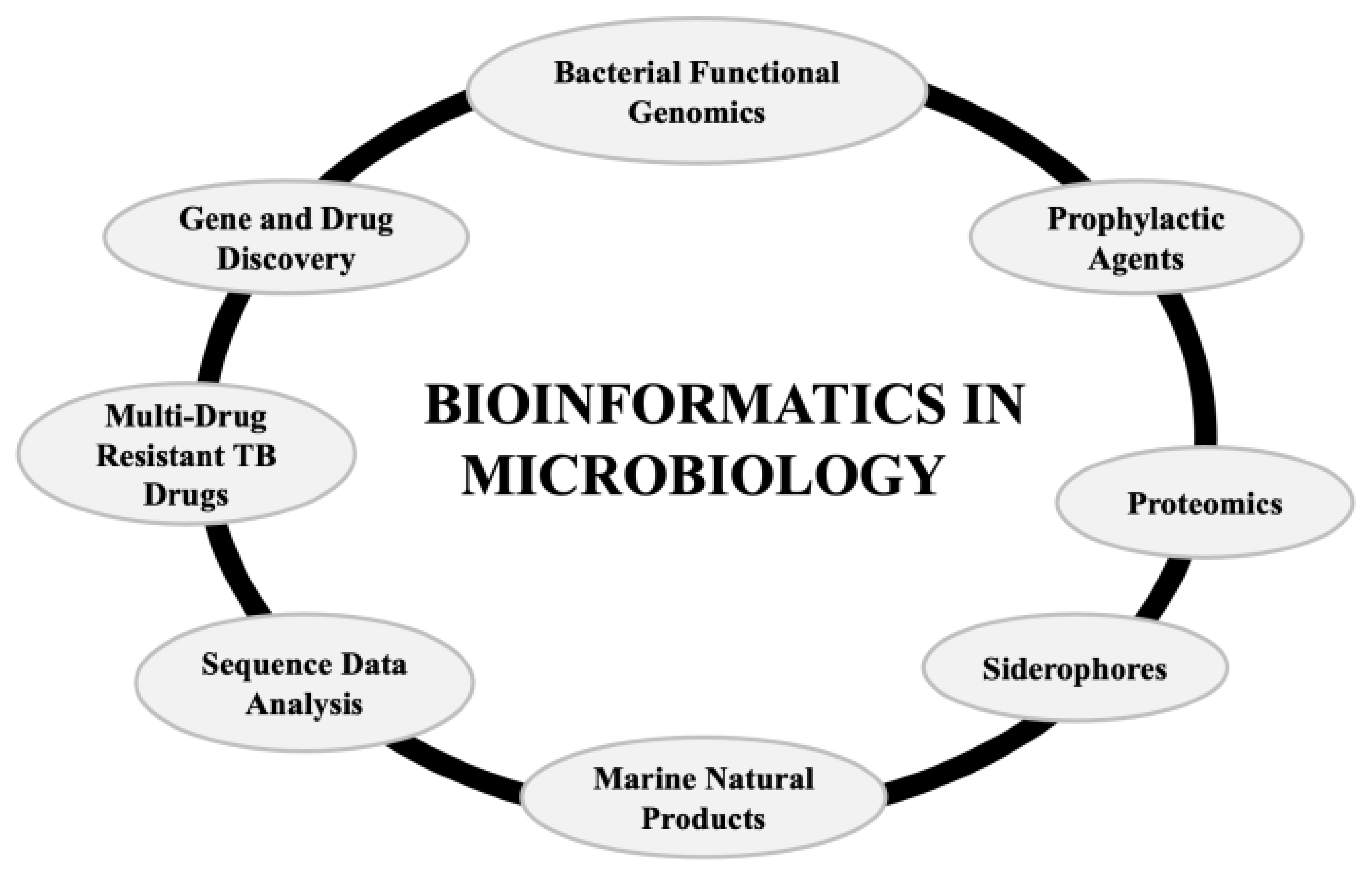{kind=link}
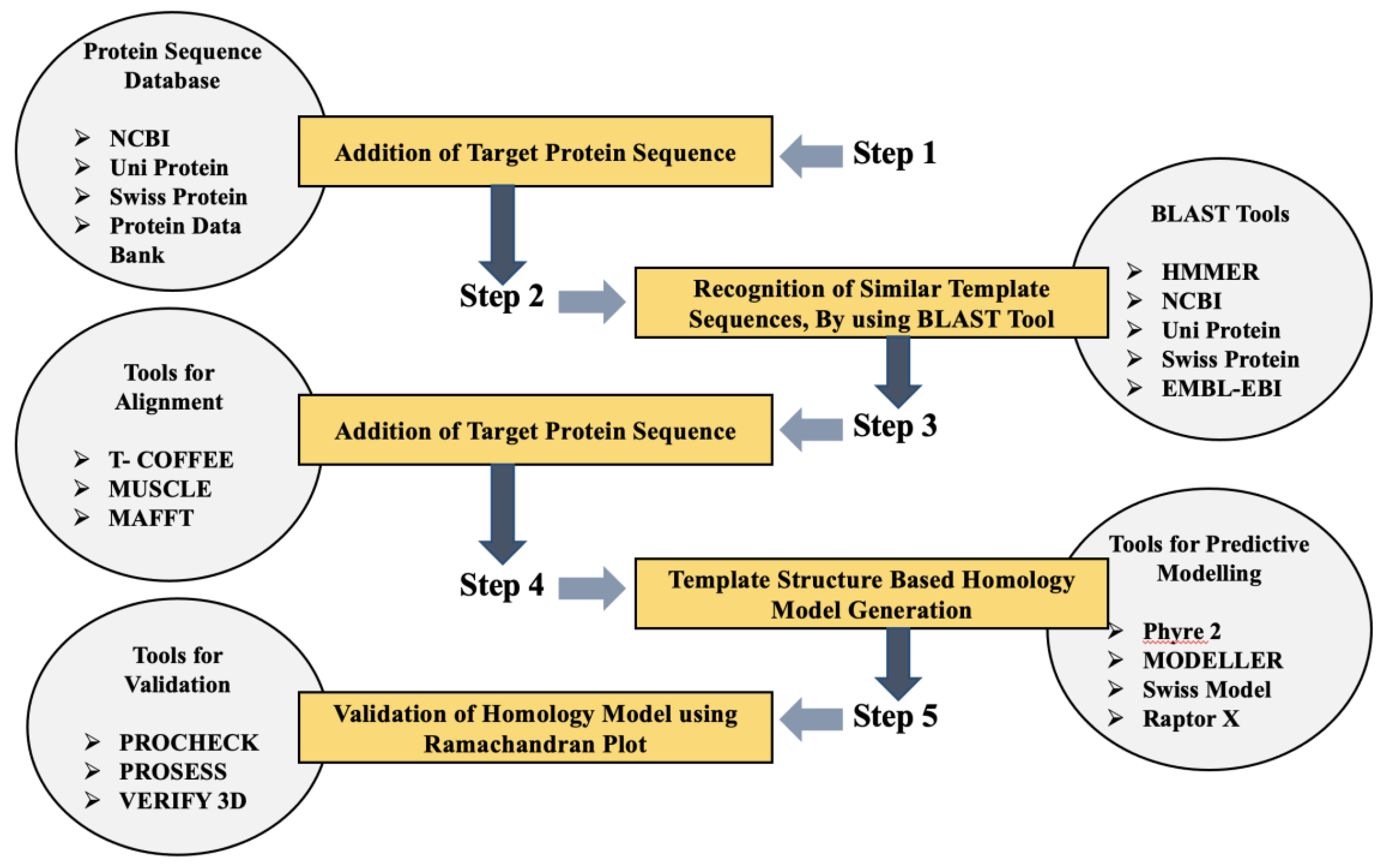{kind=link}
{kind=link}
| Diseased States | Translational Bioinformatics Techniques | Refs. |
|---|---|---|
| Osteoporosis drug targets | Functional pathway enrichment, genetic expression profiles from GEO, dysfunctional pathways | [41] |
| Neuro AIDS and drug abuse | A public domain database, molecular relationship-evaluating database | [46] |
| Repositioning of drugs | Evaluation of transcriptomic information for relationship between drugs and disease | [47] |
| Drug resistance in ovarian cancer therapy | Evaluation of protein interactions, evaluation of methylated genes, related to drug resistance, enrichment of biological process | [40] |
| Drugs for AIDS and drug resistance | Calculator of resistance to drugs, evaluation of residues of protein with digital signaling processing | [45] |
| Repositioning of drugs in transplantation of organs | Microarray dataset profiling, meta-analysis of genomic drugs as well as information, recognizing redundant molecular processes | [42] |
| HCV drug discovery | Collation of filtering, based upon dictionary, and gene mention tagger, knowledge discovery process for literature mining | [44] |
| Off-label selection of drugs for TNBC | Evaluation of TNBC patient information, collaboration of databases of cancer drugs and respective targets, evaluation of personal molecular profiles | [48] |
| Glycomics and drug targets | Tree-based algorithmic models for glycan structure data, collation of data, public databases for glycome informatics, such as KEGG | [49] |
| Bioinformatics Tools | Type | Genotyping |
|---|---|---|
| PhyTB | Online | SNP |
| CASTB | Online | 4a |
| TGS-TB | Online | 4b |
| KvarQ | Stand-alone | SNP/Spol |
| TB-Profiler | Online | SNP |
| PhyResSE | Online | SNP |
| Bioinformatics Tools | Role | Refs. |
|---|---|---|
| AntiBP server | Anti-bacterial peptides prediction in a protein sequence | [61] |
| BACTIBASE incorporated with MODELER | Prediction of 3D structure of user peptide by homology to known bacteriocins; such relational database is applicable for in silico designing of newer AMPs. | [50] |
| Titanium | Capable of making 400–600 million bases/run with 400 base pairs’ read lengths | [50] |
| 454 sequencing technology | Employed in studies related to 16S profile of microbiomes | [62] |
| Illumina sequencing platform | Rapid outputs due to more accurate outcomes and read lengths | [50] |
| MiSeq and HiSeq | Production of sequencing data of 100 GB in just 6 days | [63] |
| ANNs | Mathematical modeling algorithms, which provide an effective and reliable method for in silico detection of newer AMPs | [64] |
| Fuzzy logic modeling | Accurate evaluation of AMPs, enables antimicrobial research and development in quantitative structure–activity relationship (QSAR) | [65] |
| Machine learning | Evaluation of high-dimensional gene expression datasets for selection of gene | [66] |
| CHARMM | Program employed to facilitate activation of AMP interactions with lipid bilayers | [67] |
| GROMACS | Freeware tool employed in MD studies to develop trajectories | [68] |
| Tool | Category | Significance | Refs. |
|---|---|---|---|
| DIANAmicroT-CDS | Predicted miRNA target evaluation | A web-based application that facilitates data interpretation, evaluates functions exhibited by ncRNAs in body processes and diseases, and scrutinizes expression regulation datasets and miRNA regulatory elements. | [90] |
| MiRBase | Search for miRNAs | Introduces miRNA-based novel genes, provides comprehensive data on immature and mature miRNAs, provides immediate access to all the miRNA-related published data and resources | [91] |
| MiRDB | Predicted miRNA target evaluation | Predicts and evaluates the role of target genes, offers comprehensive data, provides screening options to facilitate role-based anticipation of different miRNAs, facilitates alignment of sequences | [92] |
| MiRscan | Search for miRNAs | A web-based application that recognizes and contrasts miRNA genes in the genetic sequence of greater than one organism | [93] |
| microTar | Predicted miRNA target evaluation | A windows application that evaluates the effect of miRNA binding on the whole mRNA molecule | [94] |
| MiReader | Search for miRNAs | A Linux and Windows application that recognizes the sequences of mature miRNAs without the requirement for reference genome sequences | [95] |
| miRmap | Predicted miRNA target evaluation | A windows/web-based application that is a mixture of characteristics from PITA, TargetScan, PACMIT, and miRanda, offers user-friendly approaches for operating precomputed predictions and modeling of miRNA targets | [96] |
| MiRanalyzer | Search for miRNAs | A windows/web-based application that is an sRNA toolbox, which recognizes or evaluates miRNA features based upon the outcome of the next-generation sequencing approaches | [97] |
| MiRNAPath | miRNAs and metabolic pathway | Depicts the inter-association between gene, miRNA, and metabolic pathway inputs, thus used to study the miRNA-based metabolic pathways | [98] |
| MiRmaid | Search for miRNAs | A web-based application that comprises all the characteristics of the miRNA database | [99] |
| MiRecords | Assessment of confirmed and estimated targets | Search and review of targets for miRNAs | [100] |
| Pharmaco-miR | MiRNAs, genes, and drugs | Significant in pharmacogenomics; integrates function status to the expression profile of miRNA via validated experimental proofs and computational techniques | [101] |
| MiRwalk | Assessment of confirmed and estimated targets | Validation of newer MiRNA targets, comprehensive database | [102] |
| ViTa | Viruses and miRNAs | Curates the target sites of miRNAs in chicken, mice, humans, or rats, with miRBase-derived known viral miRNA genes | [103] |
| TMREC | MiRNA regulatory network | Evaluation of role of regulatory processes, controlled by interactions between transcription factors and miRNAs in diseased states | [104] |
| MiR2GO | Mutations and miRNAs | Evaluation of impact of mutations and alterations in single nucleotide in central core miRNA sequence, coupling with target mRNA on their function; comparison and assessment of similarity percentage of miRNA pair functions at a cellular and molecular level | [105] |
| DAVID | MiRNA regulatory network | Effective interpretation of changes in a large number of genes; evaluation of procedure of functional product generation and genetic expression | [106] |
| PolymiRTS | Mutations and miRNAs | Evaluation of genetic polymorphisms in central core binding or target pairing site | [107] |
| PhenomiR | Diseases and miRNAs | Relationship between miRNA and disease | [108] |
| MiREnvironment | Environment and miRNAs | Interaction between environmental factors and miRNAs | [71] |
| MiRcancer | Diseases and miRNAs | Relationship between miRNA and cancer | [109] |
| CircuitsDB | Transcription factors and miRNAs | Interaction between miRNA and transcription factors to facilitate regulation of joint target gene | [110] |
| MiR2disease | Diseases and miRNAs | Portrays validated and estimated gene targets upon miRNA changes in diseases | [111] |
| PutmiR | Transcription factors and miRNAs | Interaction between miRNA and transcription factors to facilitate regulation of gene expression | [112] |
| MiRgator | MiRNA-miRNA interaction | Deep sequencing miRNA database that facilitates massive data analysis, comprehensive evaluation of target genes, expression profiles of miRNA–miRNA interactions, proper representation of miRNAs genes chromosomal region | [113] |
| DIANA-mirExTra | MiRNA-miRNA interaction | Functional evaluation of expression profiles and targets of miRNAs | [114] |
| Name | Application | Refs. |
|---|---|---|
| ||
| GATK-UnifiedGenotyper | Consistently proceeds towards HalotypeCaller | [117] |
| BWA-SW | Shorter reads alignment | [118] |
| Freebayes | Based upon Bayesian haplotype, efficient in evaluating genomes with specific properties | [119] |
| Novoalign | Alignment software for commercial use | [120] |
| BCF tools and VCF tools | Distinct analytical characteristics, yet some common functions | [119,121] |
| BWA-MEM | Longer reads alignment, with bp > 100 | [118,122] |
| FastQC | Evaluation of sequencing quality | [123] |
| Picard Tools | Provides QC evaluation for multiple secondary analysis stages | [124] |
| GenomeStrip | Evaluation of read length, read depth, and read mate pairing | [125] |
| verifybamID | Detection of sample contamination | [126] |
| BreakDancer | SV detection | [127] |
| Vcfeval | Comparison of two distinct variant call files, significant for validation | [128] |
| Pindel | Detection of large deletions and limited insertions | [121] |
| Bedtools | Manipulation of multiple bed files | [129] |
| Manta | Germline and somatic evaluation | [130] |
| VisCap | CNV calling for panel information | [131] |
| XHMM | CNV calling for exome information | [132] |
| ||
| VEP | Annotation and prediction of impact of variant on genes | [133] |
| WGSA | Integration of results from SnpEff, VEP, and ANNOVAR, for annotation based upon gene modeling, integration of numerous epigenomics projects, integration of conservation scores, database associated with the disease, multiple prediction scores, and allele frequencies for SNV-centric resources | [134] |
| DANN | Integration of multiple annotations into one metric, annotation of non-coding and coding variants | [135] |
| CADD | Integration of multiple annotations into one metric, annotation of non-coding and coding variants | [136] |
| ANNOVAR | Annotation based upon gene, region, and filter | [137] |
| Oncotator | Aggregation of annotations from genomic, cancer variants, protein, and non-cancer variant annotations | [138] |
| SnpEff | Annotation and prediction of the impact of variants on genes | [139] |
| Bioinformatics-Based Tools/Databases | Role | Refs. |
|---|---|---|
| SRA database | High-throughput sequencing data repository | [152] |
| Fast QC | Quality control check on raw sequences in WGS | [153] |
| AUGUSTUS | Gene prediction in eukaryotic genome sequencing | [154] |
| MaSuRCA | Assembly of genome | [155] |
| Prokka | Prokaryotic genome annotation in WGS | [156] |
| Cutadapt | Recognizing and eliminating adaptor sequences, poly A tails, primer, and other unrequired sequences in WGS and metagenomics | [157] |
| Ragout | Reference-assisted assembly tool in WGS | [158] |
| Gene expression omnibus (GEO) database | Repository of data related to functional genomics | [159] |
| dbSNP | Repository for single-base nucleotide substitutions in SNP discovery | [160] |
| UCSC genome browser | Collection and analysis of model organism annotations in genomics | [161] |
| PROVEAN | Estimations of effect of substitution of amino acid on biological role of protein in SNP discovery | [162] |
| Kyoto encyclopaedia of genes and genome (KEGG) | Analysis of metabolic pathway | [163] |
| SIFT | Estimation of amino acid substitution on functional role of proteins | [164] |
| Conserved domain (CD) Search | Sequence alignment | [165] |
| NCBI gene database | Genetic data repository | [166] |
| PAUP | Evaluation of phylogenetic relationship between molecular sequences by using parsimony method | [167] |
| UniProt | Stores functional data on proteins | [168] |
| PopArt | Phylogenetic evaluation with visualization of haplotype diversity network | [169] |
| Molecular evolutionary genetics analysis (MEGA) | Alignment of multiple sequences, generation and statistical evaluation of phylogenetic relationships | [170] |
| Primer 3 | Primer design in high-throughput genomics | [171] |
| PubChem | Chemical structure database for drug designing | [172] |
| Basic local alignment search tool (BLAST) | Finding similarity between sequences | [173] |
| AutoDock, Patch dock, Swiss dock, Zdock | Molecular docking tools | [142] |
| Protein databank (PDB) | 3D-protein structure database | [174] |
| Drug bank | Comprises of data on FDA approved drugs | [142] |
| Modeler | Homology of 3D protein structures | [175] |
| PyMol | Editing and visualization of molecular structure | [176] |
| GROMACS | Tool for simulation of molecular dynamics | [142] |
| NAMD | Parallel molecular dynamics code | [142] |
| Open Babel | Chemical toolbox aiding in drug designing | [177] |
| VMD | Built-in scripting and 3D graphics-based visualization program | [178] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Bhatia, S.; Al-Harrasi, A.; Zengin, G.; Babes, E.E.; Brisc, C.; Stoicescu, M.; et al. Bioinformatics Accelerates the Major Tetrad: A Real Boost for the Pharmaceutical Industry. Int. J. Mol. Sci. 2021, 22, 6184. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126184
Behl T, Kaur I, Sehgal A, Singh S, Bhatia S, Al-Harrasi A, Zengin G, Babes EE, Brisc C, Stoicescu M, et al. Bioinformatics Accelerates the Major Tetrad: A Real Boost for the Pharmaceutical Industry. International Journal of Molecular Sciences. 2021; 22(12):6184. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126184
Chicago/Turabian StyleBehl, Tapan, Ishnoor Kaur, Aayush Sehgal, Sukhbir Singh, Saurabh Bhatia, Ahmed Al-Harrasi, Gokhan Zengin, Elena Emilia Babes, Ciprian Brisc, Manuela Stoicescu, and et al. 2021. "Bioinformatics Accelerates the Major Tetrad: A Real Boost for the Pharmaceutical Industry" International Journal of Molecular Sciences 22, no. 12: 6184. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126184