
Recurrent Herpes Simplex Virus Type 1 (HSV-1) Infection Modulates Neuronal Aging Marks in In Vitro and In Vivo Models

 , , , , and
, , , , and 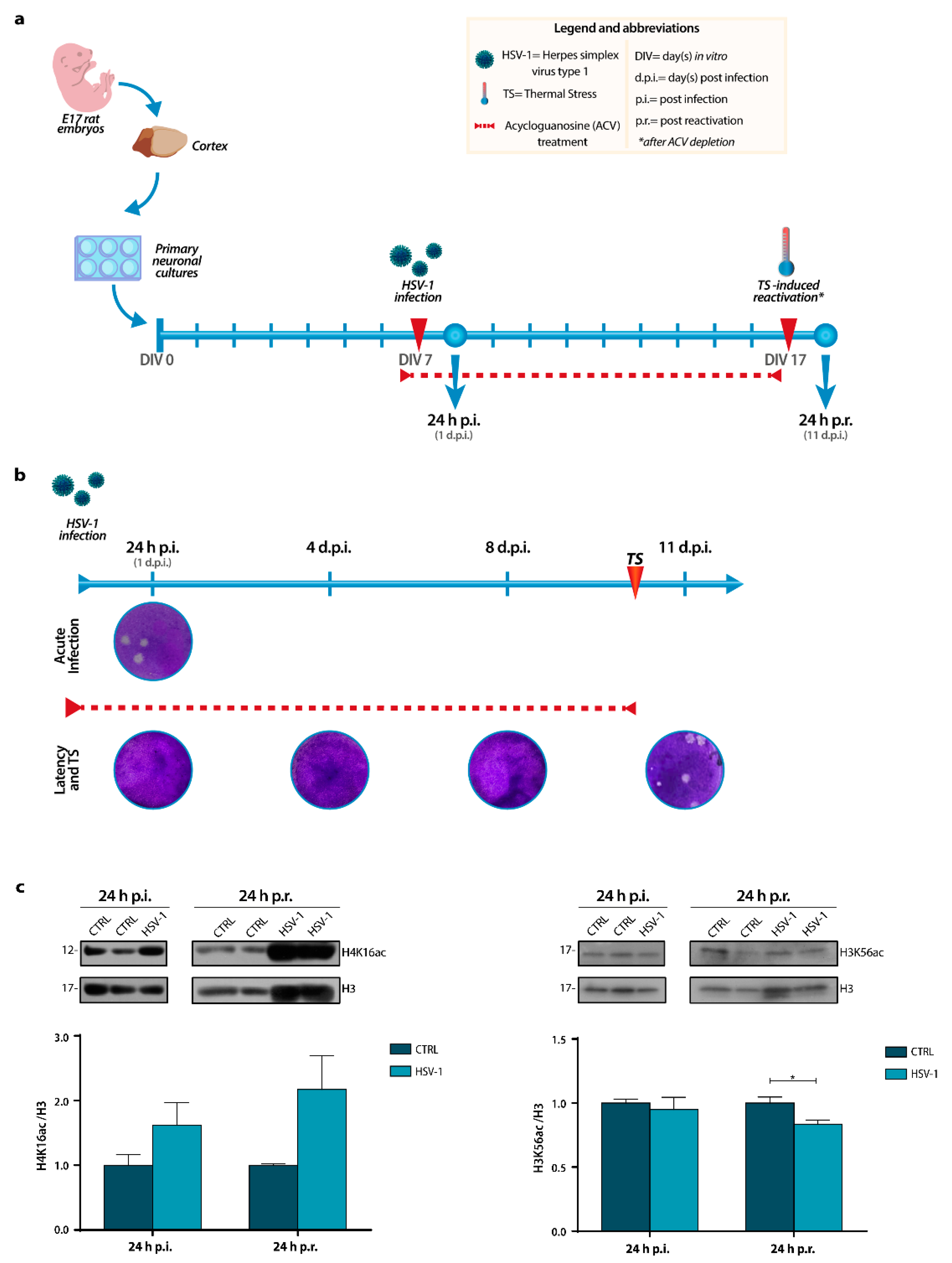{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. H3K56 and H4K16 Acetylation Levels Are Inversely Modulated by Recurrent HSV-1 Infection in Neurons
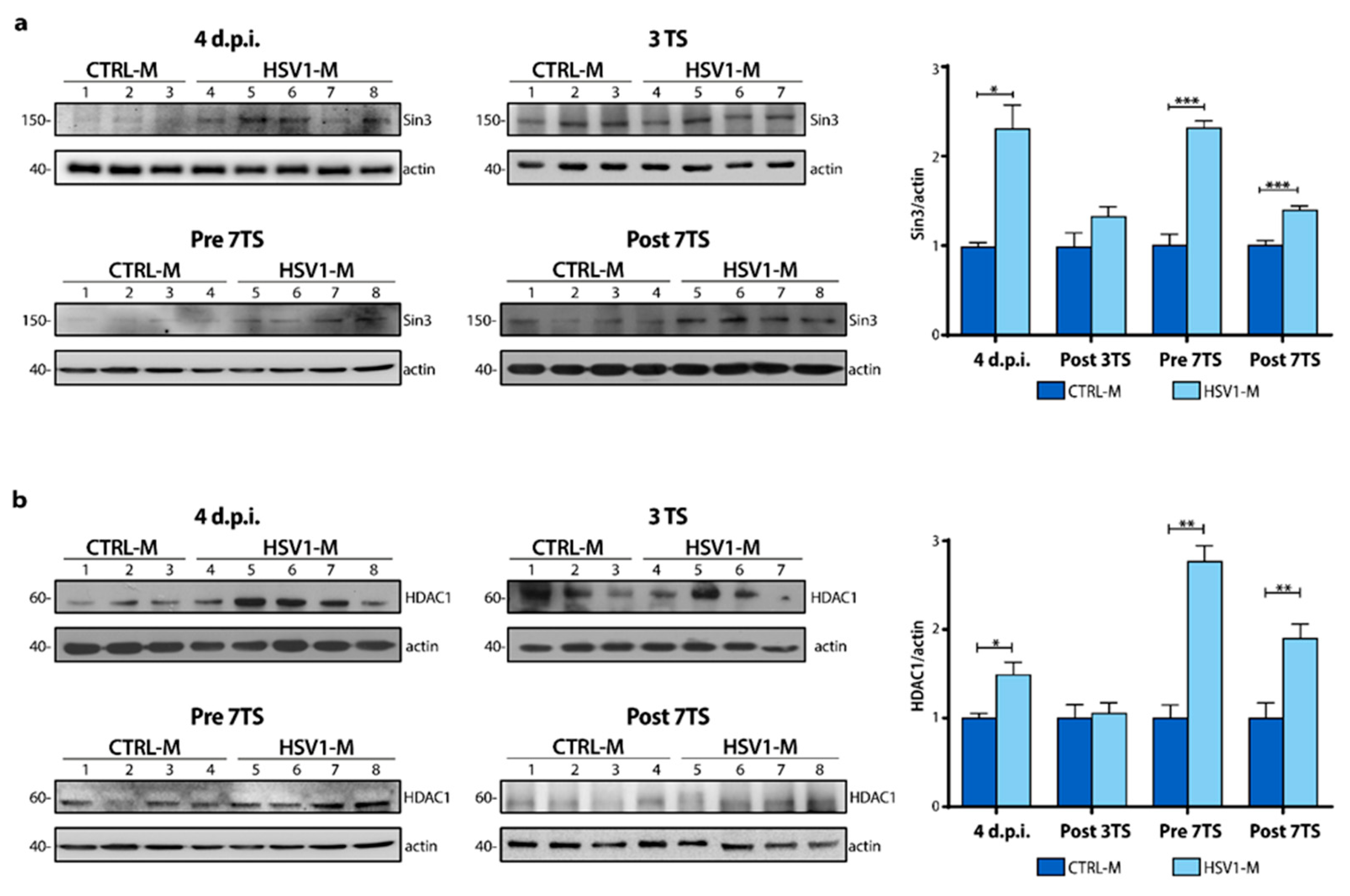
2.2. Sin3 and HDAC1 Protein Levels Increase during In Vitro Recurrent HSV-1 Infection
2.3. Recurrent HSV-1 Infection Induces HIRA Increase in 24 h p.r. Neurons
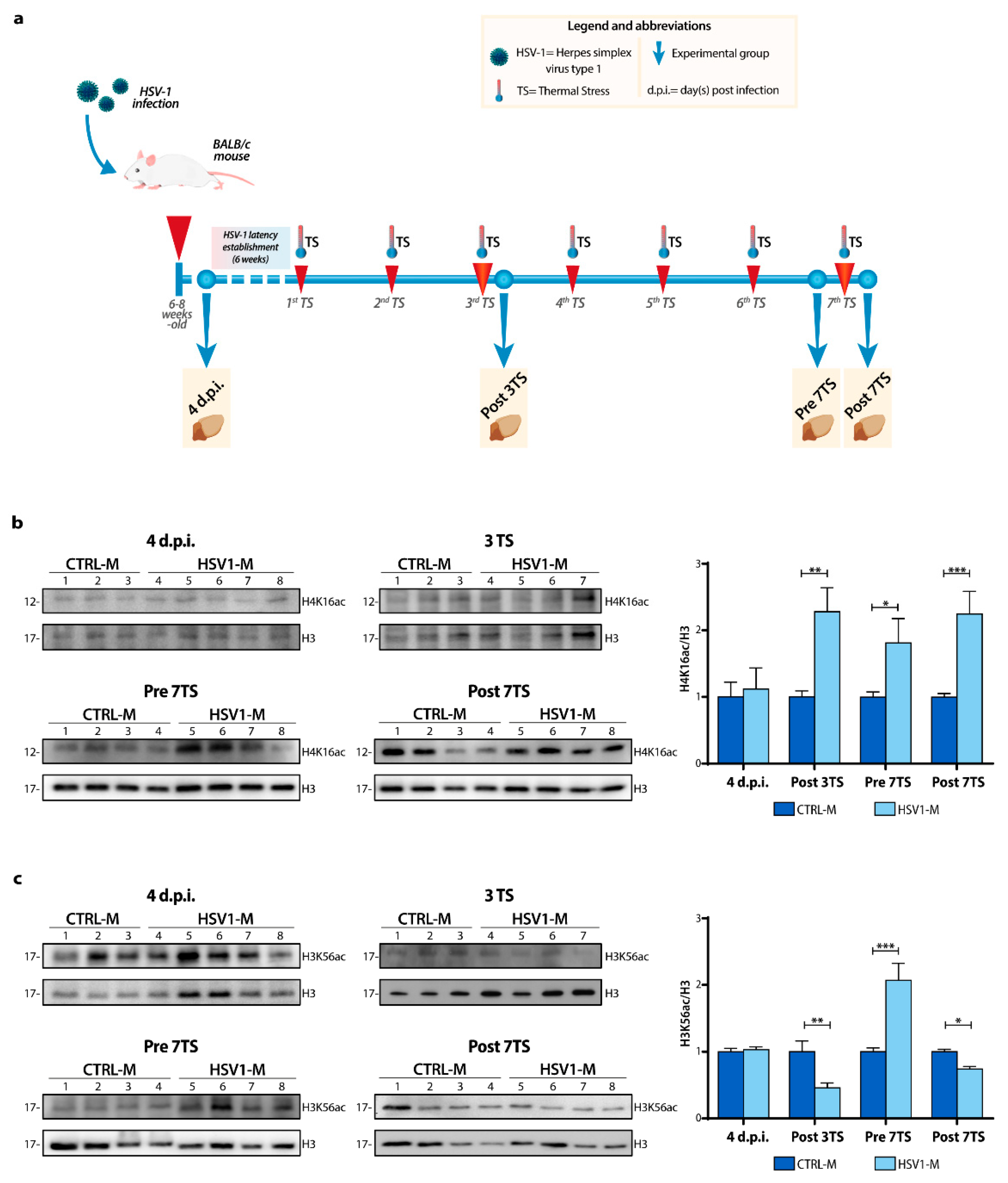
2.4. Recurrent HSV-1 Infection Modifies Histone Acetylation Profile and Sin3/HDAC1 Protein Levels in Mice
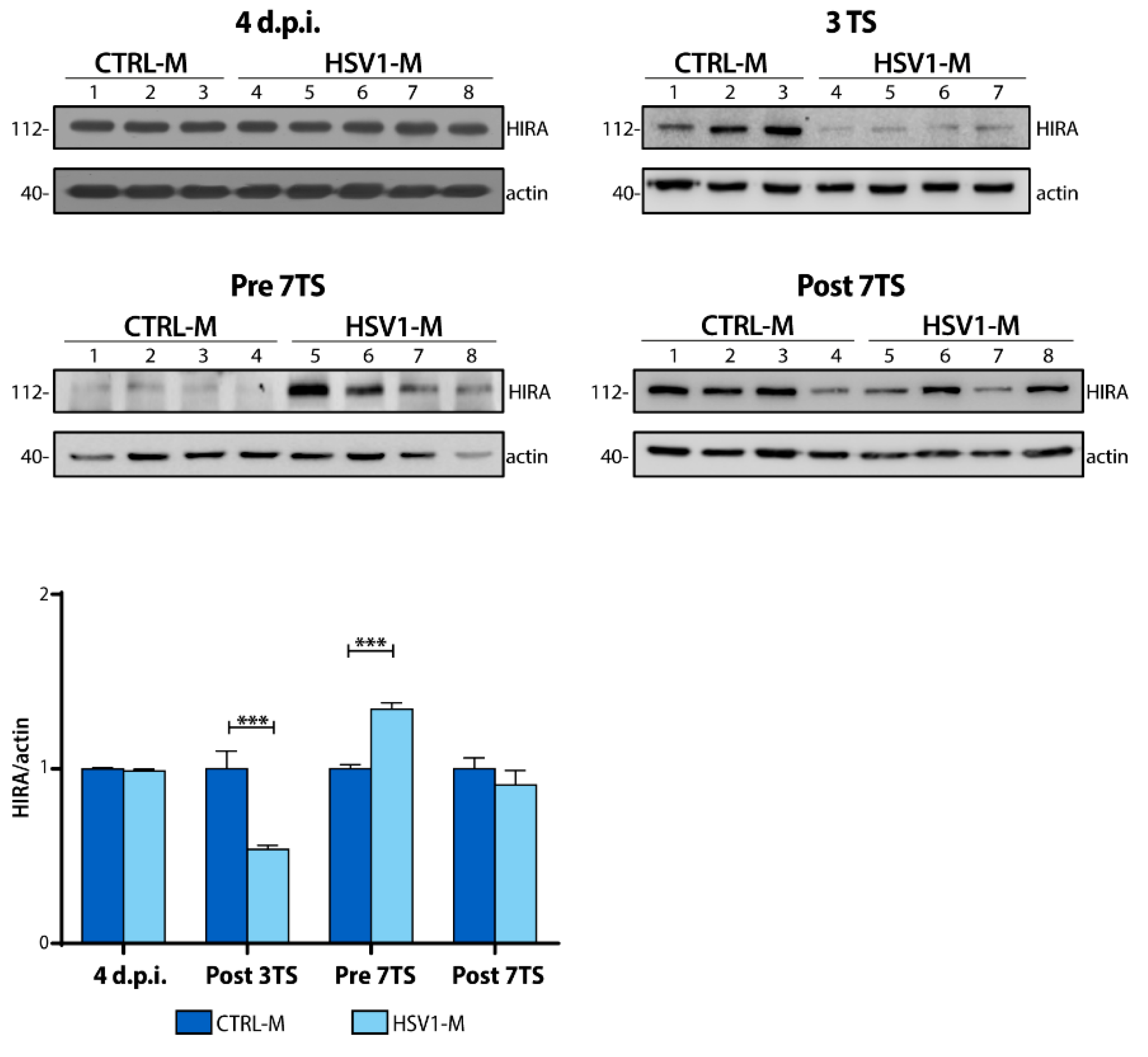
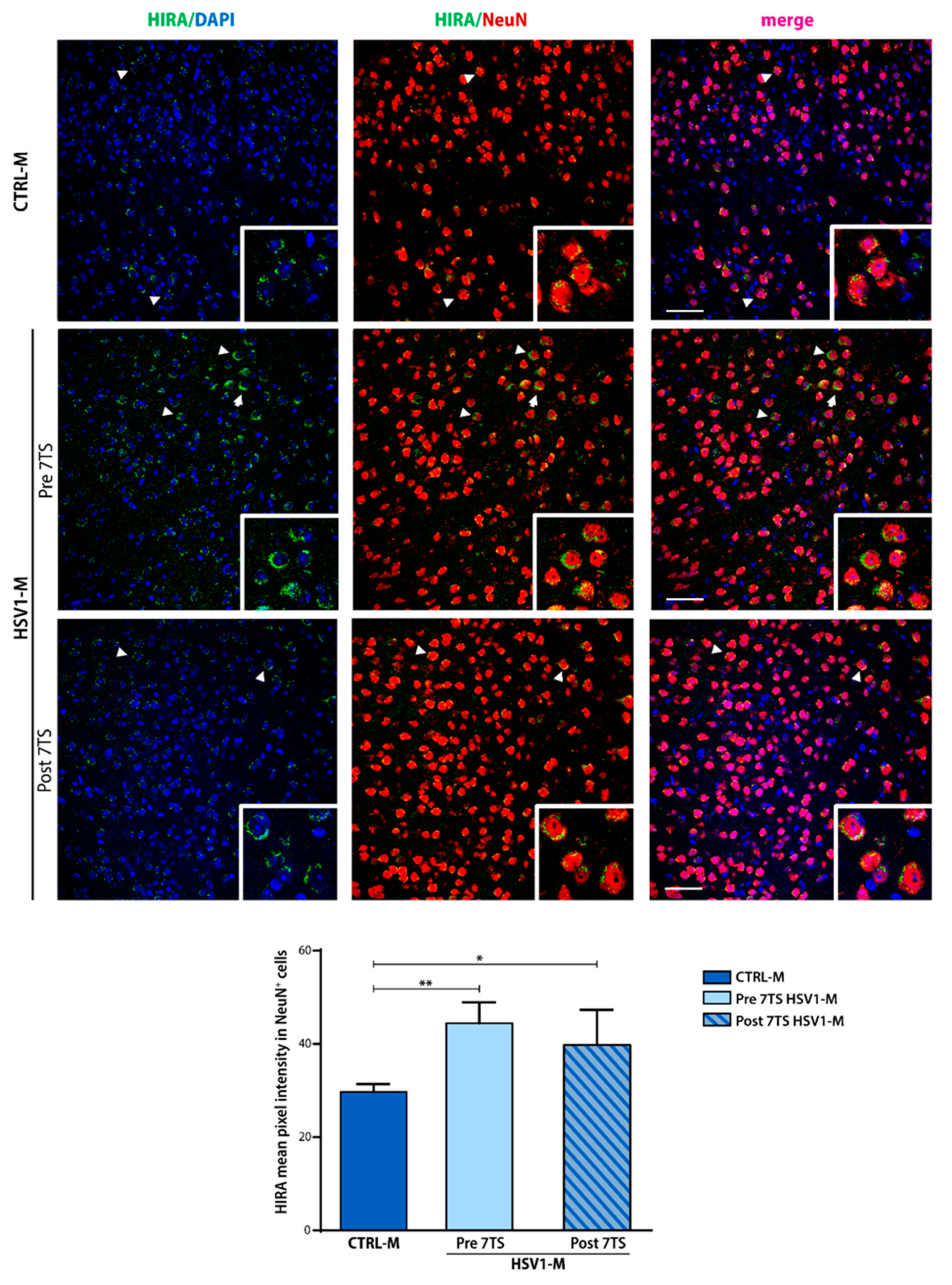
2.5. Recurrent HSV-1 Infection Induces HIRA Increase during Viral Latency in Mice
3. Discussion
4. Materials and Methods
4.1. Virus Production and Titration
4.2. Neuronal in Vitro Model of Recurrent HSV-1 Infection
4.3. Mouse Model of Recurrent HSV-1 Infection and Experimental Groups
4.4. Ethics Statement
4.5. Western Blotting
4.6. Immunofluorescence Analysis
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology, 6th ed.; Howley, D.M.K.P.M., Ed.; Lippincott Williams & Wilkins, a Wolters Kluwer Business: Alphen aan den Rijn, The Netherlands, 2014. [Google Scholar]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef] [Green Version]
- Kastrukoff, L.; Hamada, T.; Schumacher, U.; Long, C.; Doherty, P.C.; Koprowski, H. Central nervous system infection and immune response in mice inoculated into the lip with herpes simplex virus type 1. J. Neuroimmunol. 1982, 2, 295–305. [Google Scholar] [CrossRef]
- Kramer, M.F.; Cook, W.J.; Roth, F.P.; Zhu, J.; Holman, H.; Knipe, D.M.; Coen, D.M. Latent herpes simplex virus infection of sensory neurons alters neuronal gene expression. J. Virol. 2003, 77, 9533–9541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious agents and neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piacentini, R.; De Chiara, G.; Li Puma, D.D.; Ripoli, C.; Marcocci, M.E.; Garaci, E.; Palamara, A.T.; Grassi, C. HSV-1 and Alzheimer’s disease: More than a hypothesis. Front. Pharmacol. 2014, 5, 97. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Lovheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.G.; Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, 593–599. [Google Scholar] [CrossRef]
- Lovheim, H.; Norman, T.; Weidung, B.; Olsson, J.; Josefsson, M.; Adolfsson, R.; Nyberg, L.; Elgh, F. Herpes Simplex Virus, APOEvarepsilon4, and Cognitive Decline in Old Age: Results from the Betula Cohort Study. J. Alzheimers Dis. 2019, 67, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Protto, V.; Tramutola, A.; Fabiani, M.; Marcocci, M.E.; Napoletani, G.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Perluigi, M.; Di Domenico, F.; et al. Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression. Microorganisms 2020, 8, 972. [Google Scholar] [CrossRef]
- Carter, C.J. Susceptibility genes are enriched in those of the herpes simplex virus 1/host interactome in psychiatric and neurological disorders. Pathog. Dis. 2013, 69, 240–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtanen, J.O.; Jacobson, S. Viruses and multiple sclerosis. CNS Neurol. Disord. Drug. Targets 2012, 11, 528–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibovitch, E.C.; Caruso, B.; Ha, S.K.; Schindler, M.K.; Lee, N.J.; Luciano, N.J.; Billioux, B.J.; Guy, J.R.; Yen, C.; Sati, P.; et al. Herpesvirus trigger accelerates neuroinflammation in a nonhuman primate model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 11292–11297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukhvalova, M.S.; Mortensen, E.; Mbaye, A.; Lopez, D.; Kastrukoff, L.; Blanco, J.C.G. Herpes Simplex Virus 1 Induces Brain Inflammation and Multifocal Demyelination in the Cotton Rat Sigmodon hispidus. J. Virol. 2019, 94, e01161-19. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [Green Version]
- He, N.; Jin, W.L.; Lok, K.H.; Wang, Y.; Yin, M.; Wang, Z.J. Amyloid-beta(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013, 4, e924. [Google Scholar] [CrossRef] [Green Version]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, M.; Korsakova, E.; Allen, D.; Lee, P.; Fu, K.; Vargas, B.S.; Cinkornpumin, J.; Salas, C.; Park, J.C.; Germanguz, I.; et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep. 2018, 10, 1453–1463. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Gartner, U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J. Neural. Transm. 1998, 105, 949–960. [Google Scholar] [CrossRef]
- Arendt, T.; Rodel, L.; Gartner, U.; Holzer, M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport 1996, 7, 3047–3049. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Karlseder, J. The great unravelling: Chromatin as a modulator of the aging process. Trends Biochem. Sci. 2012, 37, 466–476. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Adams, G.E.; Chandru, A.; Cowley, S.M. Co-repressor, co-activator and general transcription factor: The many faces of the Sin3 histone deacetylase (HDAC) complex. Biochem. J. 2018, 475, 3921–3932. [Google Scholar] [CrossRef]
- Bardai, F.H.; Price, V.; Zaayman, M.; Wang, L.; D’Mello, S.R. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 2012, 287, 35444–35453. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Shen, S.; Dietz, K.; He, Y.; Howell, O.; Reynolds, R.; Casaccia, P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci. 2010, 13, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, T.M.; Zurcher, N.R.; Catanese, M.C.; Tseng, C.J.; Di Biase, M.A.; Lyall, A.E.; Hightower, B.G.; Parmar, A.J.; Bhanot, A.; Wu, C.J.; et al. Neuroepigenetic signatures of age and sex in the living human brain. Nat. Commun. 2019, 10, 2945. [Google Scholar] [CrossRef] [Green Version]
- Ray-Gallet, D.; Ricketts, M.D.; Sato, Y.; Gupta, K.; Boyarchuk, E.; Senda, T.; Marmorstein, R.; Almouzni, G. Functional activity of the H3.3 histone chaperone complex HIRA requires trimerization of the HIRA subunit. Nat. Commun. 2018, 9, 3103. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, M.D.; Marmorstein, R. A Molecular Prospective for HIRA Complex Assembly and H3.3-Specific Histone Chaperone Function. J. Mol. Biol. 2017, 429, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banumathy, G.; Somaiah, N.; Zhang, R.; Tang, Y.; Hoffmann, J.; Andrake, M.; Ceulemans, H.; Schultz, D.; Marmorstein, R.; Adams, P.D. Human UBN1 is an ortholog of yeast Hpc2p and has an essential role in the HIRA/ASF1a chromatin-remodeling pathway in senescent cells. Mol. Cell Biol. 2009, 29, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, T.S.; Adams, P.D. Lessons from senescence: Chromatin maintenance in non-proliferating cells. Biochim. Biophys. Acta 2012, 1819, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, R.D.; Rechter, S.; Papior, P.; Tavalai, N.; Stamminger, T.; Orr, A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006, 80, 7995–8005. [Google Scholar] [CrossRef] [Green Version]
- Rai, T.S.; Glass, M.; Cole, J.J.; Rather, M.I.; Marsden, M.; Neilson, M.; Brock, C.; Humphreys, I.R.; Everett, R.D.; Adams, P.D. Histone chaperone HIRA deposits histone H3.3 onto foreign viral DNA and contributes to anti-viral intrinsic immunity. Nucleic Acids Res. 2017, 45, 11673–11683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jiang, C.; Trudeau, S.J.; Narita, Y.; Zhao, B.; Teng, M.; Guo, R.; Gewurz, B.E. Histone Loaders CAF1 and HIRA Restrict Epstein-Barr Virus B-Cell Lytic Reactivation. mBio 2020, 11, e01063-20. [Google Scholar] [CrossRef]
- McFarlane, S.; Orr, A.; Roberts, A.P.E.; Conn, K.L.; Iliev, V.; Loney, C.; da Silva Filipe, A.; Smollett, K.; Gu, Q.; Robertson, N.; et al. The histone chaperone HIRA promotes the induction of host innate immune defences in response to HSV-1 infection. PLoS Pathog. 2019, 15, e1007667. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Levine, A.J. HIV-1 Infection Accelerates Age According to the Epigenetic Clock. J. Infect. Dis. 2015, 212, 1563–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naggie, S. Hepatitis C Virus, Inflammation, and Cellular Aging: Turning Back Time. Top. Antivir. Med. 2017, 25, 3–6. [Google Scholar] [PubMed]
- Naggie, S.; Swiderska-Syn, M.; Choi, S.; Lusk, S.; Lan, A.; Ferrari, G.; Syn, W.K.; Guy, C.D.; Diehl, A.M. Markers of Tissue Repair and Cellular Aging Are Increased in the Liver Tissue of Patients With HIV Infection Regardless of Presence of HCV Coinfection. Open Forum Infect. Dis. 2018, 5, ofy138. [Google Scholar] [CrossRef] [PubMed]
- Stowe, R.P.; Peek, M.K.; Cutchin, M.P.; Goodwin, J.S. Reactivation of herpes simplex virus type 1 is associated with cytomegalovirus and age. J. Med. Virol. 2012, 84, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Kim, J.Y.; Camarena, V.; Roehm, P.C.; Chao, M.V.; Wilson, A.C.; Mohr, I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 2012, 3823. [Google Scholar] [CrossRef]
- Feser, J.; Tyler, J. Chromatin structure as a mediator of aging. FEBS Lett. 2011, 585, 2041–2048. [Google Scholar] [CrossRef] [Green Version]
- Cohen, C.; Corpet, A.; Roubille, S.; Maroui, M.A.; Poccardi, N.; Rousseau, A.; Kleijwegt, C.; Binda, O.; Texier, P.; Sawtell, N.; et al. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/Histone H3.3/H3.3 Chaperone Axis. PLoS Pathog. 2018, 14, e1007313. [Google Scholar] [CrossRef] [Green Version]
- Michishita, E.; McCord, R.A.; Boxer, L.D.; Barber, M.F.; Hong, T.; Gozani, O.; Chua, K.F. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 2009, 8, 2664–2666. [Google Scholar] [CrossRef] [Green Version]
- Feser, J.; Truong, D.; Das, C.; Carson, J.J.; Kieft, J.; Harkness, T.; Tyler, J.K. Elevated histone expression promotes life span extension. Mol. Cell 2010, 39, 724–735. [Google Scholar] [CrossRef] [Green Version]
- Kulej, K.; Avgousti, D.C.; Sidoli, S.; Herrmann, C.; Della Fera, A.N.; Kim, E.T.; Garcia, B.A.; Weitzman, M.D. Time-resolved Global and Chromatin Proteomics during Herpes Simplex Virus Type 1 (HSV-1) Infection. Mol. Cell Proteom. 2017, 16, S92–S107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contrepois, K.; Thuret, J.Y.; Courbeyrette, R.; Fenaille, F.; Mann, C. Deacetylation of H4-K16Ac and heterochromatin assembly in senescence. Epigenetics Chromatin 2012, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Panossian, L.; Fenik, P.; Zhu, Y.; Zhan, G.; McBurney, M.W.; Veasey, S. SIRT1 regulation of wakefulness and senescence-like phenotype in wake neurons. J. Neurosci. 2011, 31, 4025–4036. [Google Scholar] [CrossRef]
- Martin, C.; Leyton, L.; Arancibia, Y.; Cuevas, A.; Zambrano, A.; Concha, M.I.; Otth, C. Modulation of the AMPK/Sirt1 axis during neuronal infection by herpes simplex virus type 1. J. Alzheimers Dis. 2014, 42, 301–312. [Google Scholar] [CrossRef]
- De Chiara, G.; Racaniello, M.; Mollinari, C.; Marcocci, M.E.; Aversa, G.; Cardinale, A.; Giovanetti, A.; Garaci, E.; Palamara, A.T.; Merlo, D. Herpes Simplex Virus-Type1 (HSV-1) Impairs DNA Repair in Cortical Neurons. Front. Aging Neurosci. 2016, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Rielland, M.; Cantor, D.J.; Graveline, R.; Hajdu, C.; Mara, L.; Diaz Bde, D.; Miller, G.; David, G. Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression. J. Clin. Investig. 2014, 124, 2125–2135. [Google Scholar] [CrossRef]
- Rai, T.S.; Cole, J.J.; Nelson, D.M.; Dikovskaya, D.; Faller, W.J.; Vizioli, M.G.; Hewitt, R.N.; Anannya, O.; McBryan, T.; Manoharan, I.; et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014, 28, 2712–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell-Doherty, R.D.; Abbott, A.R.N.; Nelson, L.A.; Bertke, A.S. Amyloid-beta and p-Tau Anti-Threat Response to Herpes Simplex Virus 1 Infection in Primary Adult Murine Hippocampal Neurons. J. Virol. 2020, 94, e01874-19. [Google Scholar] [CrossRef] [PubMed]
- Killington, R.A. Growth, assay and purification of Herpes viruses. In Virology a Practical Approach; Mahy, B.W.J., Ed.; IRL Press: Oxford, UK, 1991; pp. 207–236. [Google Scholar]
- Fabiani, M.; Limongi, D.; Palamara, A.T.; De Chiara, G.; Marcocci, M.E. A Novel Method to Titrate Herpes Simplex Virus-1 (HSV-1) Using Laser-Based Scanning of Near-Infrared Fluorophores Conjugated Antibodies. Front. Microbiol. 2017, 8, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brillinger, D. The Collected Works of John W. Tukey; Wadsworth Advanced Books & Software: Monterey, CA, USA, 1984. [Google Scholar]
- Grubbs, F.E. Procedures for Detecting Outlying Observations in Samples. Technometrics 1969, 11, 1–21. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napoletani, G.; Protto, V.; Marcocci, M.E.; Nencioni, L.; Palamara, A.T.; De Chiara, G. Recurrent Herpes Simplex Virus Type 1 (HSV-1) Infection Modulates Neuronal Aging Marks in In Vitro and In Vivo Models. Int. J. Mol. Sci. 2021, 22, 6279. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126279
Napoletani G, Protto V, Marcocci ME, Nencioni L, Palamara AT, De Chiara G. Recurrent Herpes Simplex Virus Type 1 (HSV-1) Infection Modulates Neuronal Aging Marks in In Vitro and In Vivo Models. International Journal of Molecular Sciences. 2021; 22(12):6279. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126279
Chicago/Turabian StyleNapoletani, Giorgia, Virginia Protto, Maria Elena Marcocci, Lucia Nencioni, Anna Teresa Palamara, and Giovanna De Chiara. 2021. "Recurrent Herpes Simplex Virus Type 1 (HSV-1) Infection Modulates Neuronal Aging Marks in In Vitro and In Vivo Models" International Journal of Molecular Sciences 22, no. 12: 6279. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126279