
Merkel Cell Carcinoma from Molecular Pathology to Novel Therapies

 ,
,  , , and
, , and 
Abstract
:1. Introduction
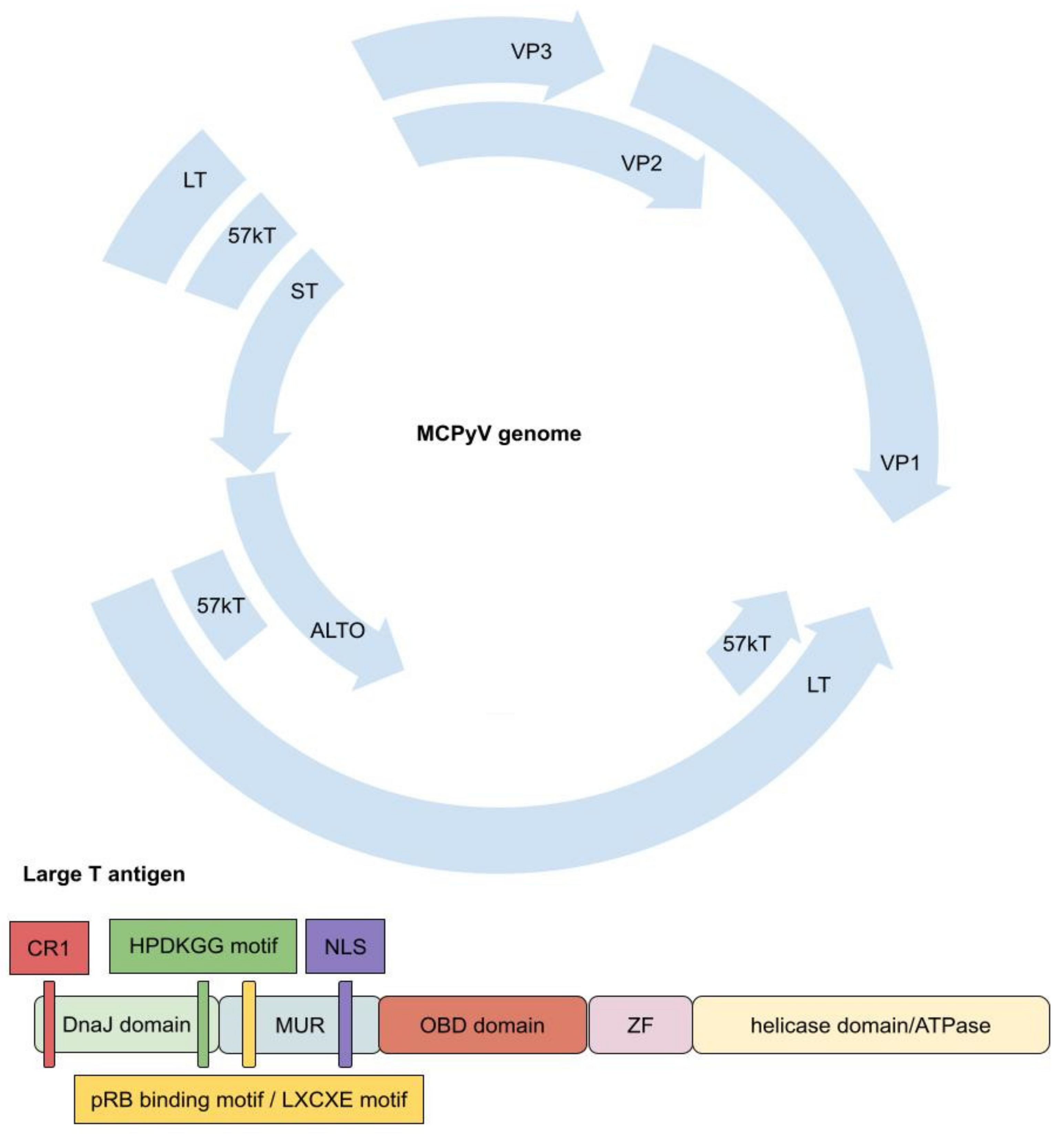
2. Merkel Cell Polyomavirus
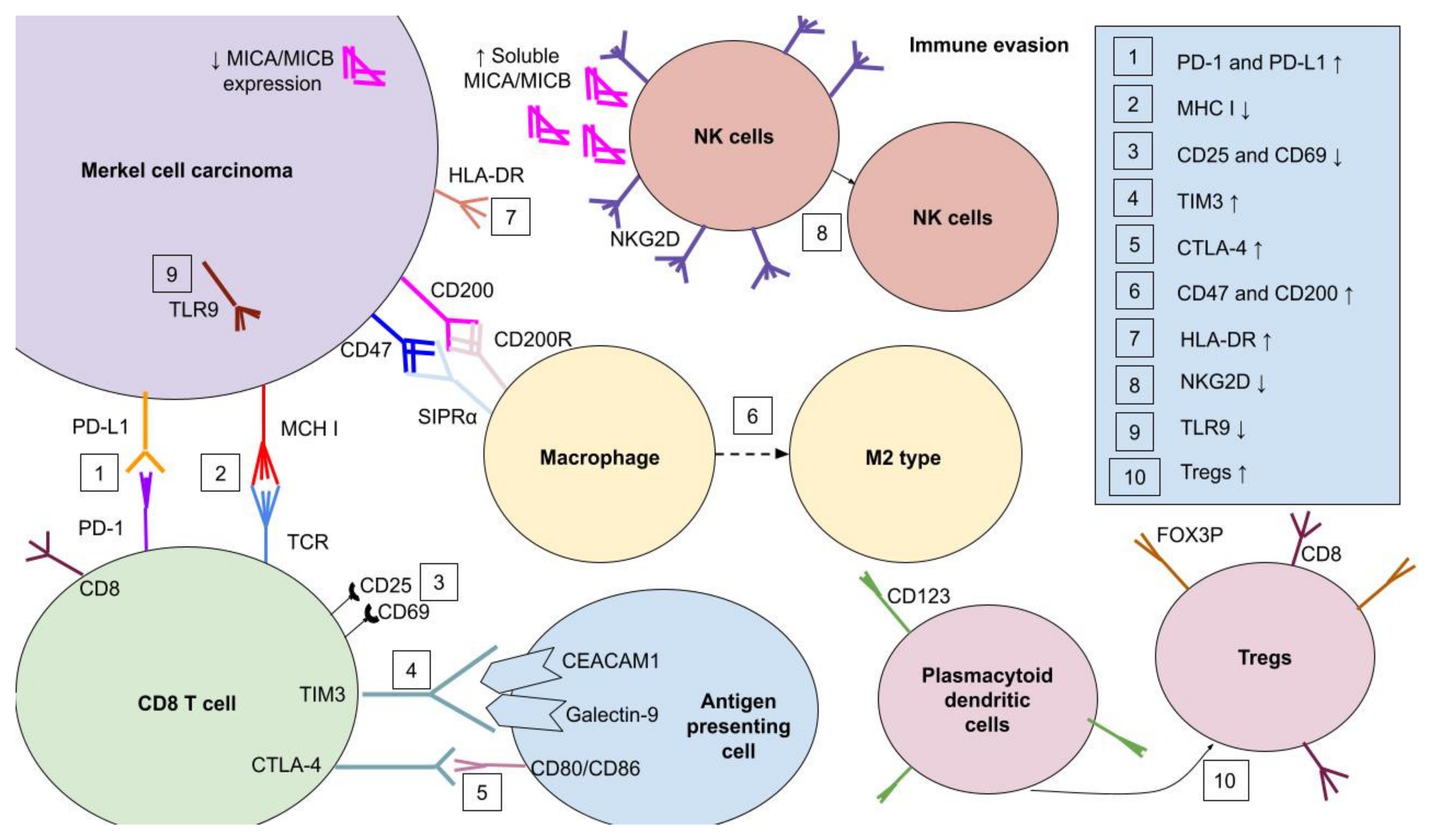
3. Immunogenicity of Merkel Cell Carcinoma
3.1. PD-1/PD-L1 Signaling in MCC
3.2. Other Immune Receptors in MCC
4. Mutational Tumor Burden
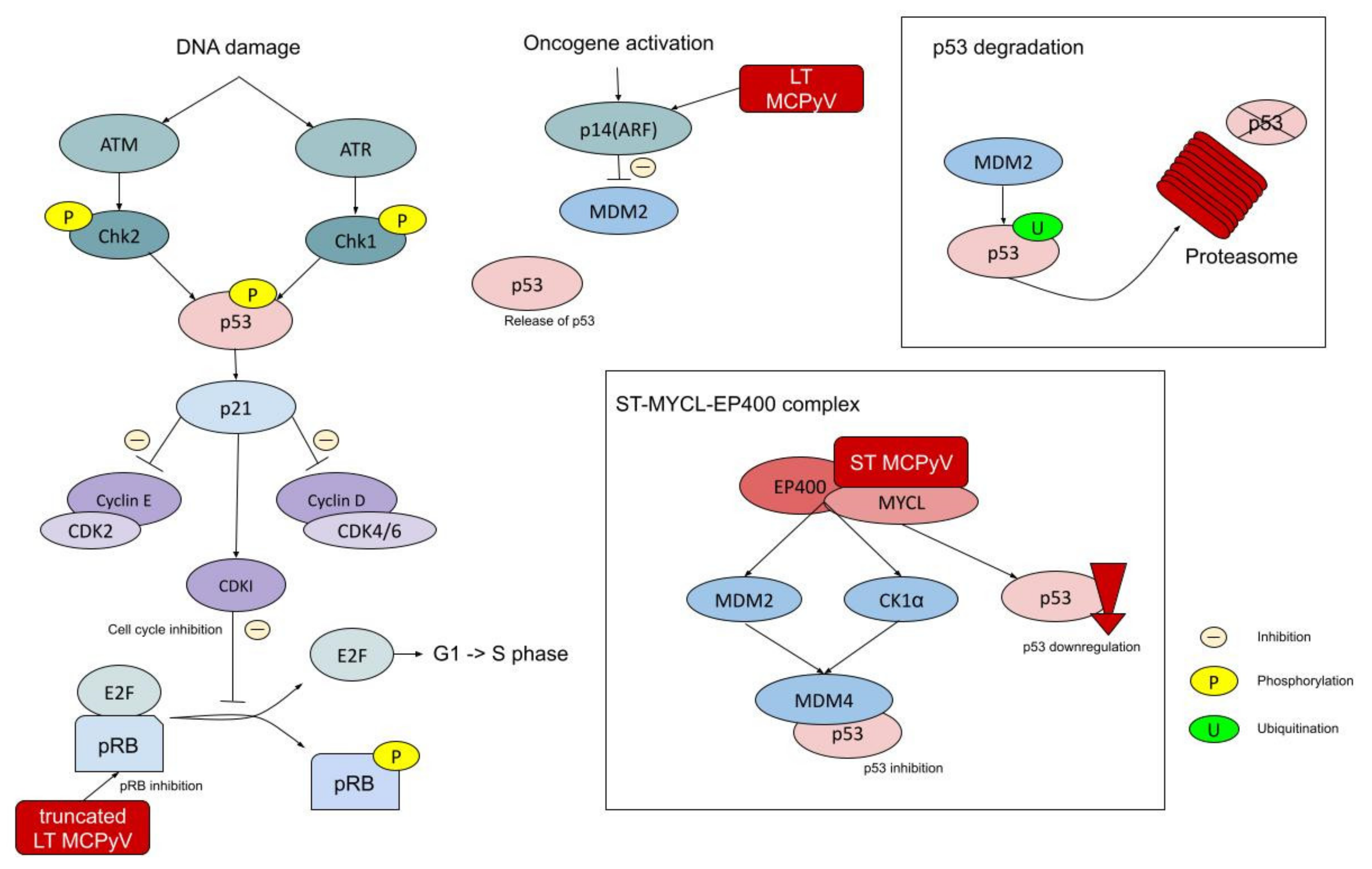
5. Deregulated Genes and Signaling Pathways
5.1. RB1 Gene
5.2. TP53 Gene
5.3. NOTCH Genes
5.4. Hedgehog Signaling Pathway
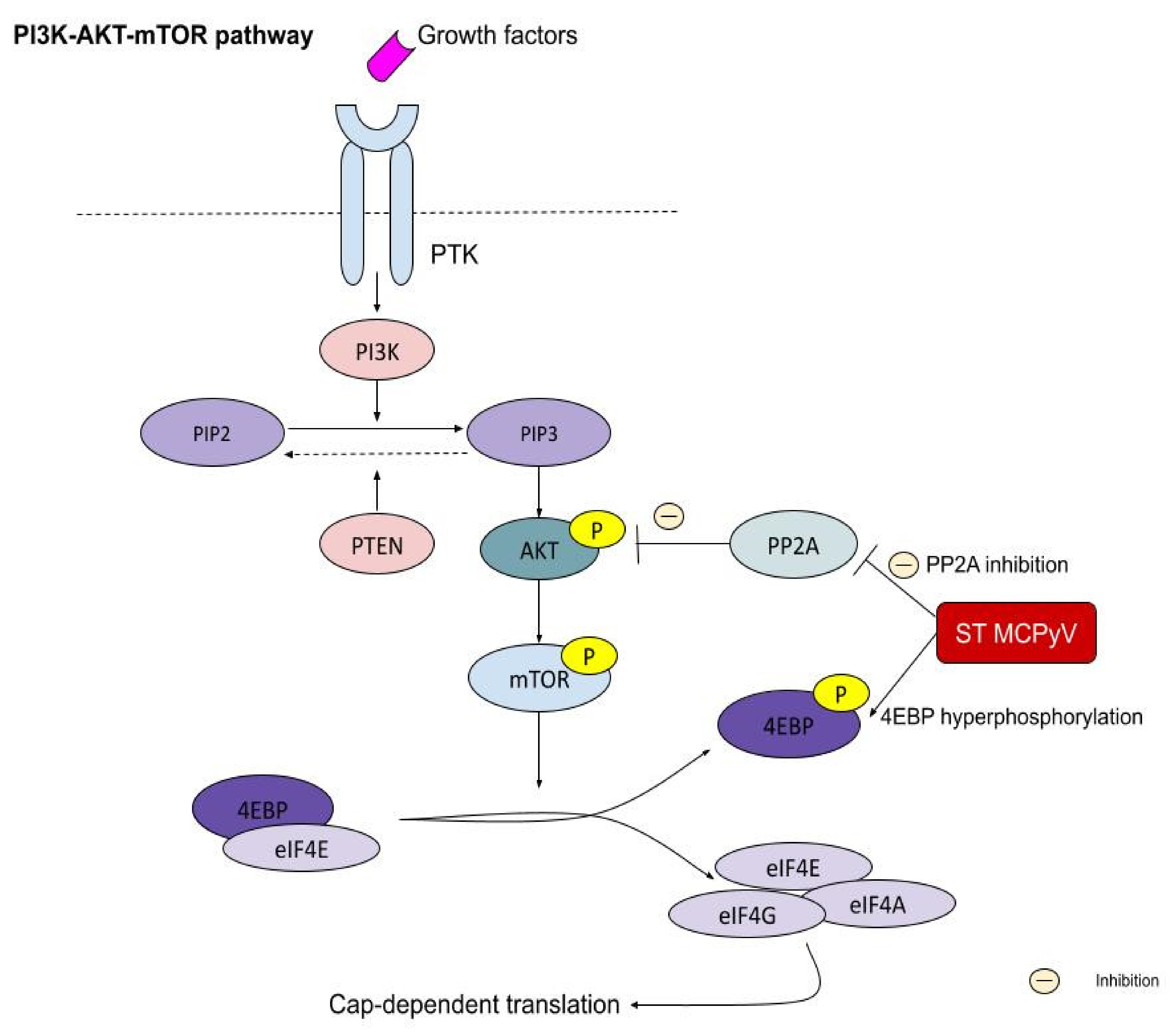
5.5. PI3K–AKT–mTOR Pathway
5.6. Chromatin Modifying Genes
5.7. Vascular Endothelial Growth Factors
5.8. Tyrosine Kinase Receptors
5.9. BCL-2 Family
5.10. Toll-Like Receptors
5.11. Chromosomal Abnormalities
5.12. MicroRNAs
5.13. Other Genes
6. MCC Treatment
6.1. General Rules for MCC Treatment and Prognosis
6.2. MCC Staging
6.3. Treatment of MCC Stage I–II
6.4. Treatment of MCC Stage III
6.5. Radiotherapy
6.6. Follow-Up after Definitive Treatment
6.7. Treatment of Local and Disseminated Recurrences
6.8. Treatment of MCC Stage IV
6.8.1. Chemotherapy
6.8.2. Immunotherapy
6.8.3. The Resistance to the Available Systemic Therapies
7. Materials and Methods
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, W.; MacDonald, M.; You, J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr. Opin. Virol. 2016, 20, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, C.M.; Haugg, A.M.; Chteinberg, E.; Rennspiess, D.; Winnepenninckx, V.; Speel, E.J.; Becker, J.C.; Kurz, A.K.; Zur Hausen, A. Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit. Rev. Oncol. Hematol. 2017, 116, 99–105. [Google Scholar] [CrossRef]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Owczarek, W. Skin carcinomas. Oncol. Clin. Pract. 2018, 14, 129–147. [Google Scholar] [CrossRef]
- Dudzisz-Śledź, M.; Zdzienicki, M.; Rutkowski, P. Merkel cell carcinoma (MCC)–neuroendocrine skin cancer. Nowotw. J. Oncol. 2019, 69, 111–116. [Google Scholar] [CrossRef]
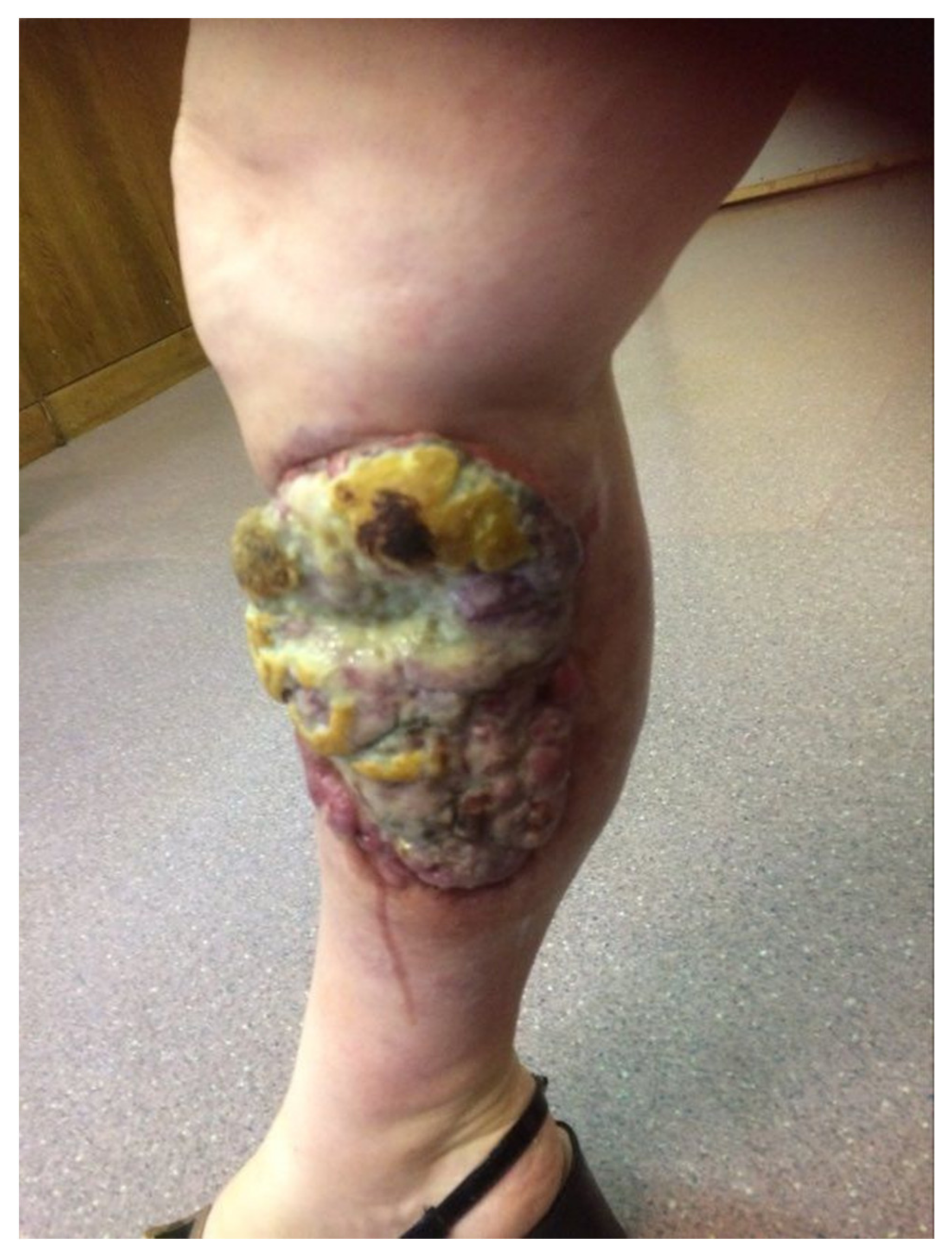
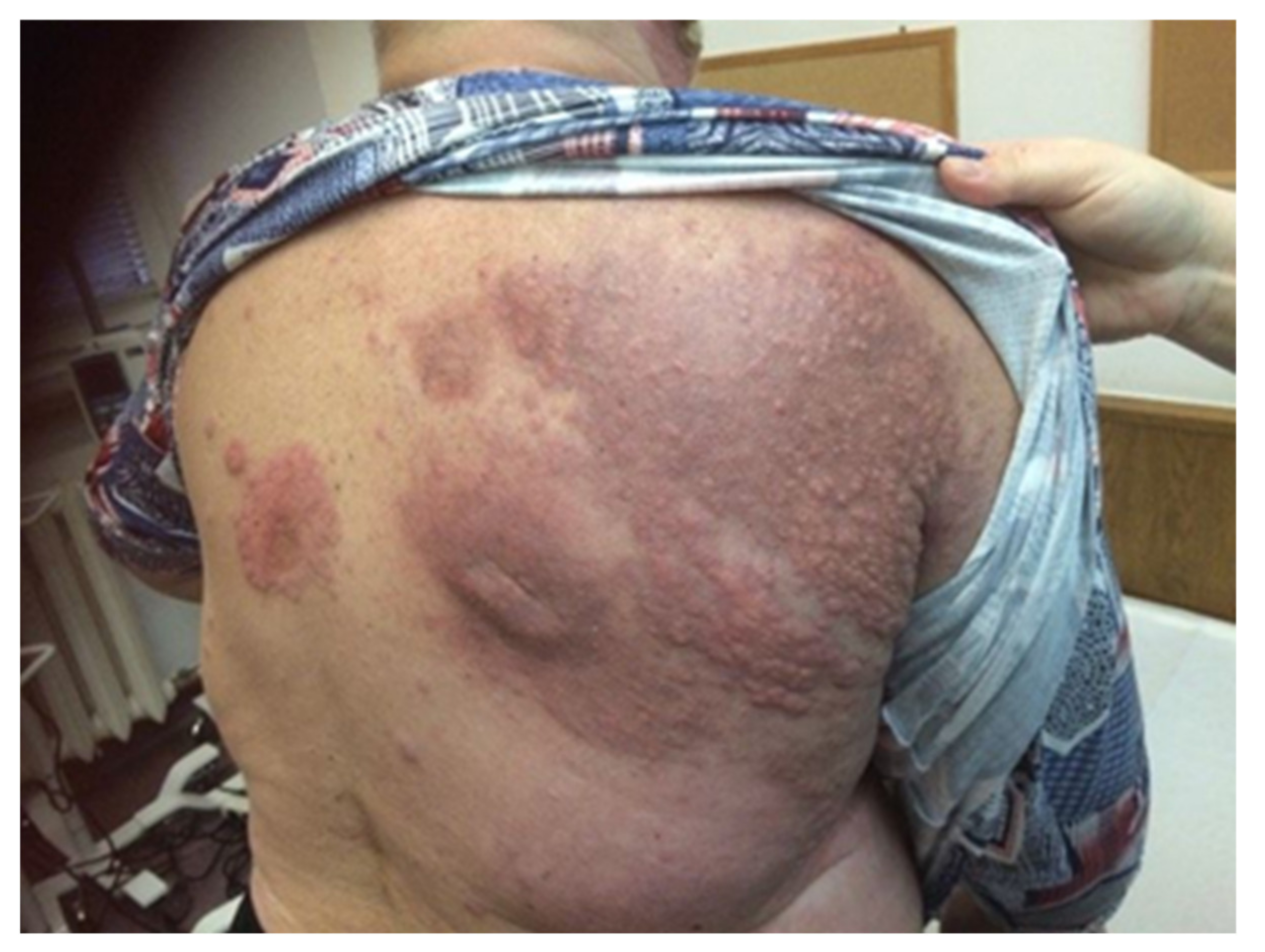
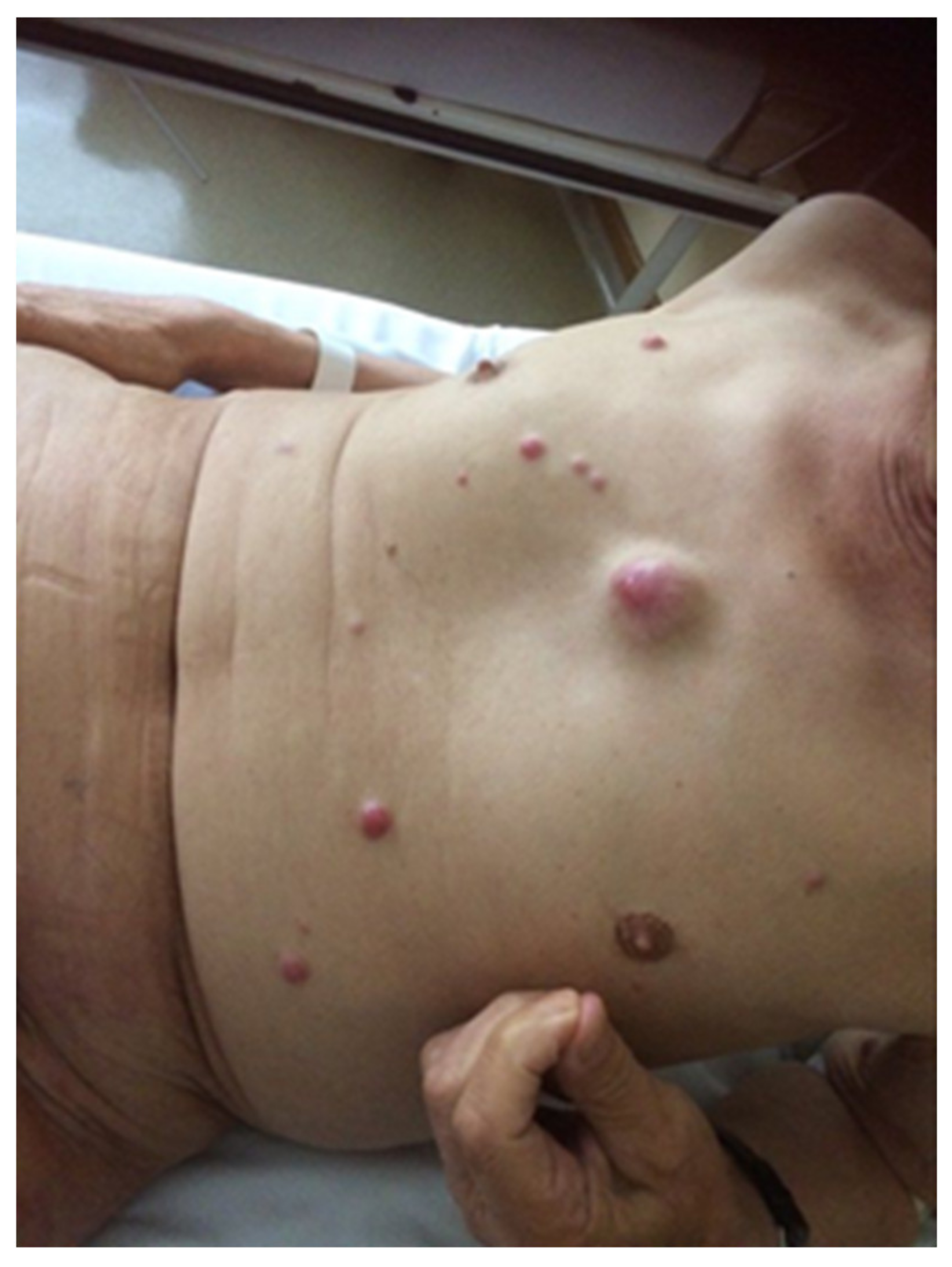
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Peñas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, T.; Ring, J.; Andres, C. Histological, immunohistological, and clinical features of merkel cell carcinoma in correlation to merkel cell polyomavirus status. J. Skin Cancer 2012, 2012, 983421. [Google Scholar] [CrossRef] [Green Version]
- Zwijnenburg, E.M.; Lubeek, S.F.K.; Werner, J.E.M.; Amir, A.L.; Weijs, W.L.J.; Takes, R.P.; Pegge, S.A.H.; van Herpen, C.M.L.; Adema, G.J.; Kaanders, J. Merkel Cell Carcinoma: New Trends. Cancers 2021, 13, 1614. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef]
- Clarke, C.A.; Robbins, H.A.; Tatalovich, Z.; Lynch, C.F.; Pawlish, K.S.; Finch, J.L.; Hernandez, B.Y.; Fraumeni, J.F., Jr.; Madeleine, M.M.; Engels, E.A. Risk of merkel cell carcinoma after solid organ transplantation. J. Natl. Cancer Inst. 2015, 107, dju382. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef] [Green Version]
- Kaae, J.; Hansen, A.V.; Biggar, R.J.; Boyd, H.A.; Moore, P.S.; Wohlfahrt, J.; Melbye, M. Merkel Cell Carcinoma: Incidence, Mortality and Risk of Other Cancers. J. Natl. Cancer Inst. 2010, 102, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 938–945. [Google Scholar] [CrossRef]
- Donizy, P.; Wroblewska, J.P.; Dias-Santagata, D.; Woznica, K.; Biecek, P.; Mochel, M.C.; Wu, C.L.; Kopczynski, J.; Pieniazek, M.; Rys, J.; et al. Merkel Cell Carcinoma of Unknown Primary: Immunohistochemical and Molecular Analyses Reveal Distinct UV-Signature/MCPyV-Negative and High Immunogenicity/MCPyV-Positive Profiles. Cancers 2021, 13, 1621. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.J.; Tanhuanco-Kho, G. Merkel cell carcinoma frequently shows histologic features of basal cell carcinoma: A study of 30 cases. J. Cutan. Pathol. 2007, 34, 612–619. [Google Scholar] [CrossRef]
- Mercuri, S.R.; Brianti, P.; Dattola, A.; Bennardo, L.; Silvestri, M.; Schipani, G.; Nisticò, S.P. CO(2) laser and photodynamic therapy: Study of efficacy in periocular BCC. Dermatol. Ther. 2018, 31, e12616. [Google Scholar] [CrossRef]
- Colunga, A.; Pulliam, T.; Nghiem, P. Merkel Cell Carcinoma in the Age of Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 2035–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keohane, S.G.; Proby, C.M.; Newlands, C.; Motley, R.J.; Nasr, I.; Mohd Mustapa, M.F.; Slater, D.N. The new 8th edition of TNM staging and its implications for skin cancer: A review by the British Association of Dermatologists and the Royal College of Pathologists, U.K. Br. J. Dermatol. 2018, 179, 824–828. [Google Scholar] [CrossRef] [PubMed]
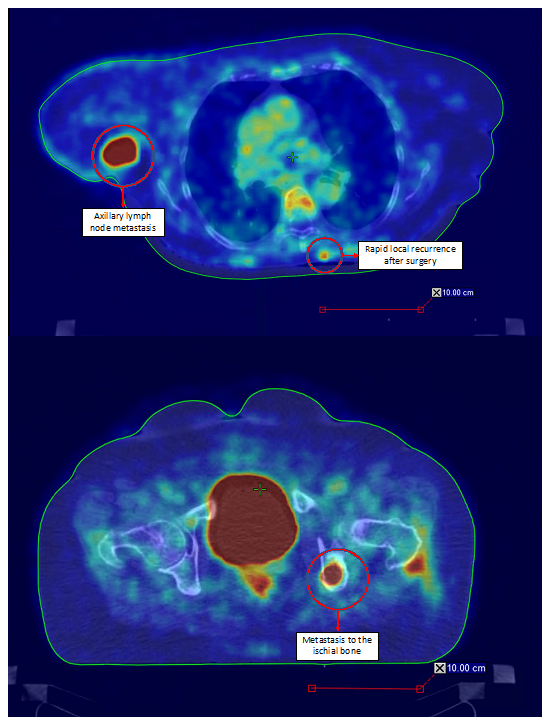
- Sachpekidis, C.; Sidiropoulou, P.; Hassel, J.C.; Drakoulis, N.; Dimitrakopoulou-Strauss, A. Positron Emission Tomography in Merkel Cell Carcinoma. Cancers 2020, 12, 2897. [Google Scholar] [CrossRef]
- Liu, J.; Larcos, G.; Howle, J.; Veness, M. Lack of clinical impact of (18) F-fluorodeoxyglucose positron emission tomography with simultaneous computed tomography for stage I and II Merkel cell carcinoma with concurrent sentinel lymph node biopsy staging: A single institutional experience from Westmead Hospital, Sydney. Australas. J. Dermatol. 2017, 58, 99–105. [Google Scholar] [CrossRef]
- Maginnis, M.S. Human Polyomaviruses (Papillomaviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 518–527. [Google Scholar]
- Rynans, S.; Dzieciątkowski, T.; Mlynarczyk, G. Polyomavirus diseases of immunosuppressed patients. Postepy Mikrobiol. 2011, 50, 191–199. [Google Scholar]
- Martel-Jantin, C.; Pedergnana, V.; Nicol, J.T.; Leblond, V.; Trégouët, D.A.; Tortevoye, P.; Plancoulaine, S.; Coursaget, P.; Touzé, A.; Abel, L.; et al. Merkel cell polyomavirus infection occurs during early childhood and is transmitted between siblings. J. Clin. Virol. 2013, 58, 288–291. [Google Scholar] [CrossRef] [Green Version]
- Faust, H.; Dillner, J. Merkel Cell Polyomavirus: Epidemiology and Clinical Features of Related Cancer. In Viruses and Human Cancer; Hudnall, S.D., Ed.; Springer: New York, NY, USA, 2014; pp. 357–367. [Google Scholar]
- Johnson, E.M. Structural evaluation of new human polyomaviruses provides clues to pathobiology. Trends Microbiol. 2010, 18, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyo, M.; Guerrero-Preston, R.; Brait, M.; Hoque, M.O.; Chuang, A.; Kim, M.S.; Sharma, R.; Liégeois, N.J.; Koch, W.M.; Califano, J.A.; et al. Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int. J. Cancer 2010, 126, 2991–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicol, J.T.; Robinot, R.; Carpentier, A.; Carandina, G.; Mazzoni, E.; Tognon, M.; Touzé, A.; Coursaget, P. Age-specific seroprevalences of merkel cell polyomavirus, human polyomaviruses 6, 7, and 9, and trichodysplasia spinulosa-associated polyomavirus. Clin. Vaccine Immunol. 2013, 20, 363–368. [Google Scholar] [CrossRef]
- Sroller, V.; Hamšíková, E.; Ludvíková, V.; Vochozková, P.; Kojzarová, M.; Fraiberk, M.; Saláková, M.; Morávková, A.; Forstová, J.; Němečková, S. Seroprevalence rates of BKV, JCV, and MCPyV polyomaviruses in the general Czech Republic population. J. Med. Virol. 2014, 86, 1560–1568. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, F.; He, Z.; Deng, Q.; Pan, Y.; Liu, Y.; Zhang, C.; Ning, T.; Guo, C.; Liang, Y.; et al. Seroprevalence of Merkel cell polyomavirus in the general rural population of Anyang, China. PLoS ONE 2014, 9, e106430. [Google Scholar] [CrossRef]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.J.; Paulson, K.G.; Wipf, G.C.; Miranda, D.; Madeleine, M.M.; Johnson, L.G.; Lemos, B.D.; Lee, S.; Warcola, A.H.; Iyer, J.G.; et al. Association of Merkel cell polyomavirus-specific antibodies with Merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 1510–1522. [Google Scholar] [CrossRef]
- Chen, T.; Hedman, L.; Mattila, P.S.; Jartti, T.; Ruuskanen, O.; Soderlund-Venermo, M.; Hedman, K. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J. Clin. Virol. 2011, 50, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, C.; Amako, Y.; Harold, A.; Toptan, T.; Chang, Y.; Shuda, M. Characterization of a Merkel Cell Polyomavirus-Positive Merkel Cell Carcinoma Cell Line CVG-1. Front. Microbiol. 2018, 9, 713. [Google Scholar] [CrossRef]
- Laude, H.C.; Jonchère, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.F.; Dupin, N.; et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar] [CrossRef]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International Workshop on Merkel Cell Carcinoma Research Working Group. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Baez, C.F.; Brandão Varella, R.; Villani, S.; Delbue, S. Human Polyomaviruses: The Battle of Large and Small Tumor Antigens. Virology 2017, 8, 1178122–17744785. [Google Scholar] [CrossRef] [PubMed]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef]
- Becker, M.; Dominguez, M.; Greune, L.; Soria-Martinez, L.; Pfleiderer, M.M.; Schowalter, R.; Buck, C.B.; Blaum, B.S.; Schmidt, M.A.; Schelhaas, M. Infectious Entry of Merkel Cell Polyomavirus. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Neu, U.; Hengel, H.; Blaum, B.S.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.W.; Imamura, A.; Ando, H.; Kiso, M.; et al. Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PLoS Pathog. 2012, 8, e1002738. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Choi, J.E.; Harms, P.W.; Chandrani, P. Merkel Cell Polyomavirus in Merkel Cell Carcinoma: Integration Sites and Involvement of the KMT2D Tumor Suppressor Gene. Viruses 2020, 12, 966. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Doolittle-Hall, J.M.; Cunningham Glasspoole, D.L.; Seaman, W.T.; Webster-Cyriaque, J. Meta-Analysis of DNA Tumor-Viral Integration Site Selection Indicates a Role for Repeats, Gene Expression and Epigenetics. Cancers 2015, 7, 2217–2235. [Google Scholar] [CrossRef]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhbari, P.; Tobin, D.; Poterlowicz, K.; Roberts, W.; Boyne, J.R. MCV-miR-M1 Targets the Host-Cell Immune Response Resulting in the Attenuation of Neutrophil Chemotaxis. J. Investig. Dermatol. 2018, 138, 2343–2354. [Google Scholar] [CrossRef] [Green Version]
- Adam, C.; Baeurle, A.; Brodsky, J.L.; Wipf, P.; Schrama, D.; Becker, J.C.; Houben, R. The HSP70 modulator MAL3-101 inhibits Merkel cell carcinoma. PLoS ONE 2014, 9, e92041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech-Sioli, M.; Siebels, S.; Radau, S.; Zahedi, R.P.; Schmidt, C.; Dobner, T.; Grundhoff, A.; Fischer, N. The Ubiquitin-Specific Protease Usp7, a Novel Merkel Cell Polyomavirus Large T-Antigen Interaction Partner, Modulates Viral DNA Replication. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Kumar, S.; Xie, H.; Shi, H.; Gao, J.; Juhlin, C.C.; Björnhagen, V.; Höög, A.; Lee, L.; Larsson, C.; Lui, W.O. Merkel cell polyomavirus oncoproteins induce microRNAs that suppress multiple autophagy genes. Int. J. Cancer 2020, 146, 1652–1666. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Wendzicki, J.A.; Shuda, Y.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen induces genome instability by E3 ubiquitin ligase targeting. Oncogene 2017, 36, 6784–6792. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, D.A.; Abdul-Sada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Afanasiev, O.; Nghiem, P. Immunobiology of Merkel cell carcinoma: Implications for immunotherapy of a polyomavirus-associated cancer. Curr. Oncol. Rep. 2011, 13, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, I.; Coati, I.; Alaibac, M. Interaction between Merkel cell carcinoma and the immune system: Pathogenetic and therapeutic implications. Mol. Clin. Oncol. 2017, 7, 729–732. [Google Scholar] [CrossRef] [Green Version]
- Koljonen, V.; Kukko, H.; Pukkala, E.; Sankila, R.; Böhling, T.; Tukiainen, E.; Sihto, H.; Joensuu, H. Chronic lymphocytic leukaemia patients have a high risk of Merkel-cell polyomavirus DNA-positive Merkel-cell carcinoma. Br. J. Cancer 2009, 101, 1444–1447. [Google Scholar] [CrossRef]
- Goldstein, R.H.; DeCaprio, J.A. Merkel Cell Carcinoma in the HIV-1/AIDS Patient. Cancer Treat. Res. 2019, 177, 211–229. [Google Scholar] [CrossRef] [PubMed]
- Vandeven, N.; Nghiem, P. Merkel Cell Carcinoma Etiology, Imugenicity and Current Treatments Strategies. Ph.D. Thesis, Merkel Cell Carcinoma: Immunogenicity and the Characterization of CD4 T Cell Responses to the Merkel Cell Polyomavirus, University of Washington, Washington, DC, USA, 2016. [Google Scholar]
- Pang, C.; Sharma, D.; Sankar, T. Spontaneous regression of Merkel cell carcinoma: A case report and review of the literature. Int. J. Surg. Case. Rep. 2015, 7, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.; Roman, J.W.; Reuben, A.; Hill, N.; Hudgens, C.W.; Tetzlaff, M.; Logemann, N.F.; Brownell, I. Spontaneous regression of Merkel cell carcinoma is driven by adaptive immune activation and clonal T cell expansion. Cancer Res. 2018, 78, 4676. [Google Scholar] [CrossRef]
- Wang, L.; Toda, M.; Saito, K.; Hori, T.; Horii, T.; Shiku, H.; Kuribayashi, K.; Kato, T. Post-immune UV irradiation induces Tr1-like regulatory T cells that suppress humoral immune responses. Int. Immunol. 2008, 20, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.; Iyer, J.; Tegeder, A.; Thibodeau, R.; Schelter, J.; Koba, S.; Schrama, D.; Simonson, W.; Lemos, B.; Byrd, D.; et al. Transcriptome-Wide Studies of Merkel Cell Carcinoma and Validation of Intratumoral CD8+ Lymphocyte Invasion As an Independent Predictor of Survival. J. Clin. Oncol. 2011, 29, 1539–1546. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.J.; Church, C.D.; Dong, L.; Crispin, D.; Fitzgibbon, M.P.; Lachance, K.; Jing, L.; Shinohara, M.; Gavvovidis, I.; Willimsky, G.; et al. Tumor-Infiltrating Merkel Cell Polyomavirus-Specific T Cells Are Diverse and Associated with Improved Patient Survival. Cancer Immunol. Res. 2017, 5, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.M.; Fleming, K.E.; Hanly, J.G.; Dakin Hache, K.; Doucette, S.; Ferrara, G.; Cerroni, L. A morphological and immunophenotypic map of the immune response in Merkel cell carcinoma. Hum. Pathol. 2016, 52, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.; Lai, I.; et al. Merkel cell polyomavirus-specific CD8⁺ and CD4⁺ T-cell responses identified in Merkel cell carcinomas and blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar] [CrossRef] [Green Version]
- Lyngaa, R.; Pedersen, N.W.; Schrama, D.; Thrue, C.A.; Ibrani, D.; Met, O.; Thor Straten, P.; Nghiem, P.; Becker, J.C.; Hadrup, S.R. T-cell responses to oncogenic merkel cell polyomavirus proteins distinguish patients with merkel cell carcinoma from healthy donors. Clin. Cancer Res. 2014, 20, 1768–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowlatshahi, M.; Huang, V.; Gehad, A.E.; Jiang, Y.; Calarese, A.; Teague, J.E.; Dorosario, A.A.; Cheng, J.; Nghiem, P.; Schanbacher, C.F.; et al. Tumor-specific T cells in human Merkel cell carcinomas: A possible role for Tregs and T-cell exhaustion in reducing T-cell responses. J. Investig. Dermatol. 2013, 133, 1879–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samimi, M.; Benlalam, H.; Aumond, P.; Gaboriaud, P.; Fradin, D.; Kervarrec, T.; Florenceau, L.; Vignard, V.; Blom, A.; Touzé, A.; et al. Viral and tumor antigen-specific CD8 T-cell responses in Merkel cell carcinoma. Cell Immunol. 2019, 344, 103961. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.M.; Castonguay, M.C.; Carter, M.D.; Pasternak, S.; Ly, T.Y.; Doucette, S.; Hanly, J.G.; Saggini, A.; Cerroni, L. Global PD-L1 Signals and Tumor-Infiltrating Lymphocytes: Markers of Immunogenicity in Different Subsets of Merkel Cell Carcinoma and Potential Therapeutic Implications. Am. J. Dermatopathol. 2019, 41, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.J.; Church, C.D.; Fling, S.P.; Kulikauskas, R.; Ramchurren, N.; Shinohara, M.M.; Kluger, H.M.; Bhatia, S.; Lundgren, L.; Cheever, M.A.; et al. Merkel cell polyomavirus-specific immune responses in patients with Merkel cell carcinoma receiving anti-PD-1 therapy. J. Immunother. Cancer 2018, 6, 131. [Google Scholar] [CrossRef] [Green Version]
- Longino, N.V.; Yang, J.; Iyer, J.G.; Ibrani, D.; Chow, I.T.; Laing, K.J.; Campbell, V.L.; Paulson, K.G.; Kulikauskas, R.M.; Church, C.D.; et al. Human CD4(+) T Cells Specific for Merkel Cell Polyomavirus Localize to Merkel Cell Carcinomas and Target a Required Oncogenic Domain. Cancer Immunol. Res. 2019, 7, 1727–1739. [Google Scholar] [CrossRef]
- Behr, D.S.; Peitsch, W.K.; Hametner, C.; Lasitschka, F.; Houben, R.; Schönhaar, K.; Michel, J.; Dollt, C.; Goebeler, M.; Marx, A.; et al. Prognostic value of immune cell infiltration, tertiary lymphoid structures and PD-L1 expression in Merkel cell carcinomas. Int. J. Clin. Exp. Pathol. 2014, 7, 7610–7621. [Google Scholar] [PubMed]
- Paulson, K.G.; Carter, J.J.; Johnson, L.G.; Cahill, K.W.; Iyer, J.G.; David, S.; Becker, J.C.; Madeleine, M.M.; Nghiem, P.; Galloway, D.A. Antibodies to Merkel cell polyomavirus T-antigen oncoproteins reflect tumor burden in Merkel cell carcinoma patients. Cancer Res. 2010, 70, 8388–8397. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Lewis, C.W.; Redman, M.W.; Simonson, W.T.; Lisberg, A.; Ritter, D.; Morishima, C.; Hutchinson, K.; Mudgistratova, L.; Blom, A.; et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: A prospective validation study. Cancer 2017, 123, 1464–1474. [Google Scholar] [CrossRef] [Green Version]
- Samimi, M.; Molet, L.; Fleury, M.; Laude, H.; Carlotti, A.; Gardair, C.; Baudin, M.; Gouguet, L.; Maubec, E.; Avenel-Audran, M.; et al. Prognostic value of antibodies to Merkel cell polyomavirus T antigens and VP1 protein in patients with Merkel cell carcinoma. Br. J. Dermatol. 2016, 174, 813–822. [Google Scholar] [CrossRef]
- Touzé, A.; Le Bidre, E.; Laude, H.; Fleury, M.J.; Cazal, R.; Arnold, F.; Carlotti, A.; Maubec, E.; Aubin, F.; Avril, M.F.; et al. High levels of antibodies against merkel cell polyomavirus identify a subset of patients with merkel cell carcinoma with better clinical outcome. J. Clin. Oncol. 2011, 29, 1612–1619. [Google Scholar] [CrossRef]
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Paulson, K.G.; Voillet, V.; McAfee, M.S.; Hunter, D.S.; Wagener, F.D.; Perdicchio, M.; Valente, W.J.; Koelle, S.J.; Church, C.D.; Vandeven, N.; et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018, 9, 3868. [Google Scholar] [CrossRef] [Green Version]
- Afanasiev, O.K.; Yelistratova, L.; Miller, N.; Nagase, K.; Paulson, K.; Iyer, J.G.; Ibrani, D.; Koelle, D.M.; Nghiem, P. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin. Cancer Res. 2013, 19, 5351–5360. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, A.; Michel, J.; Schönhaar, K.; Goerdt, S.; Schledzewski, K. Differentiation and gene expression profile of tumor-associated macrophages. Semin. Cancer Biol. 2012, 22, 289–297. [Google Scholar] [CrossRef]
- Barros, M.H.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage polarisation: An immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef] [Green Version]
- Ritter, C.; Fan, K.; Paulson, K.G.; Nghiem, P.; Schrama, D.; Becker, J.C. Reversal of epigenetic silencing of MHC class I chain-related protein A and B improves immune recognition of Merkel cell carcinoma. Sci. Rep. 2016, 6, 21678. [Google Scholar] [CrossRef] [Green Version]
- Molfetta, R.; Quatrini, L.; Santoni, A.; Paolini, R. Regulation of NKG2D-Dependent NK Cell Functions: The Yin and the Yang of Receptor Endocytosis. Int. J. Mol. Sci. 2017, 18, 1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahl, J.; Cerwenka, A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 2017, 222, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Curr. Oncol. Rep. 2004, 6, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Hayashi, F.; Means, T.K.; Luster, A.D. Toll-like receptors stimulate human neutrophil function. Blood 2003, 102, 2660–2669. [Google Scholar] [CrossRef]
- Gelman, A.E.; LaRosa, D.F.; Zhang, J.; Walsh, P.T.; Choi, Y.; Sunyer, J.O.; Turka, L.A. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity 2006, 25, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J. Virol. 2013, 87, 13009–13019. [Google Scholar] [CrossRef] [Green Version]
- Gaiser, M.R.; Weis, C.A.; Gaiser, T.; Jiang, H.; Buder-Bakhaya, K.; Herpel, E.; Warth, A.; Xiao, Y.; Miao, L.; Brownell, I. Merkel cell carcinoma expresses the immunoregulatory ligand CD200 and induces immunosuppressive macrophages and regulatory T cells. Oncoimmunology 2018, 7, e1426517. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, M.A.; Kern, N.; Vale, R.D. CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity 2020, 53, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Vincent, J.G.; Loyo, M.; Kagohara, L.T.; Luber, B.S.; Wang, H.; Xu, H.; Nayar, S.K.; Wang, T.S.; Sidransky, D.; et al. PD-L1 expression in the Merkel cell carcinoma microenvironment: Association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol. Res. 2013, 1, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Rozali, E.N.; Hato, S.V.; Robinson, B.W.; Lake, R.A.; Lesterhuis, W.J. Programmed Death Ligand 2 in Cancer-Induced Immune Suppression. Clin. Dev. Immunol. 2012, 2012, 656340. [Google Scholar] [CrossRef]
- Kumar, P.; Marinelarena, A.; Raghunathan, D.; Ragothaman, V.K.; Saini, S.; Bhattacharya, P.; Fan, J.; Epstein, A.L.; Maker, A.V.; Prabhakar, B.S. Critical role of OX40 signaling in the TCR-independent phase of human and murine thymic Treg generation. Cell Mol. Immunol. 2019, 16, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Burgess, J.K.; Carlin, S.; Pack, R.A.; Arndt, G.M.; Au, W.W.; Johnson, P.R.; Black, J.L.; Hunt, N.H. Detection and characterization of OX40 ligand expression in human airway smooth muscle cells: A possible role in asthma? J. Allergy Clin. Immunol. 2004, 113, 683–689. [Google Scholar] [CrossRef]
- Imura, A.; Hori, T.; Imada, K.; Ishikawa, T.; Tanaka, Y.; Maeda, M.; Imamura, S.; Uchiyama, T. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J. Exp. Med. 1996, 183, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Suto, H.; Iikura, M.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast cells enhance T cell activation: Importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 2006, 176, 2238–2248. [Google Scholar] [CrossRef] [Green Version]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Sornasse, T.; Cocks, B.G.; Tanaka, Y.; Santoni, A.; Lanier, L.L. Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J. Immunol. 2004, 173, 3716–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Gough, M.J.; Weinberg, A.D. OX40 (CD134) and OX40L. Adv. Exp. Med. Biol. 2009, 647, 94–107. [Google Scholar] [CrossRef]
- Song, J.; So, T.; Cheng, M.; Tang, X.; Croft, M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity 2005, 22, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, P.R.; Song, J.; Gramaglia, I.; Killeen, N.; Croft, M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001, 15, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.M.; Maston, L.D.; Gough, M.J.; Ruby, C.E.; Redmond, W.L.; Crittenden, M.; Li, Y.; Puri, S.; Poehlein, C.H.; Morris, N.; et al. Signaling through OX40 enhances antitumor immunity. Semin. Oncol. 2010, 37, 524–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redmond, W.L.; Ruby, C.E.; Weinberg, A.D. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit. Rev. Immunol. 2009, 29, 187–201. [Google Scholar] [CrossRef]
- Redmond, W.L.; Weinberg, A.D. Targeting OX40 and OX40L for the treatment of autoimmunity and cancer. Crit. Rev. Immunol. 2007, 27, 415–436. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, G.; Riccardi, C. GITR: A modulator of immune response and inflammation. Adv. Exp. Med. Biol. 2009, 647, 156–173. [Google Scholar] [CrossRef]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Schollenberger, M.D.; Dakhil, S.; Rosner, S.; Ali, O.; Sharfman, W.H.; Silk, A.W.; Bhatia, S.; Lipson, E.J. Rescue therapy for patients with anti-PD-1-refractory Merkel cell carcinoma: A multicenter, retrospective case series. J. Immunother. Cancer 2019, 7, 170. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dutta, P.; Liu, J.; Sabri, N.; Song, Y.; Li, W.X.; Li, J. Tumour cell-intrinsic CTLA4 regulates PD-L1 expression in non-small cell lung cancer. J. Cell. Mol. Med. 2019, 23, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Falà, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef] [Green Version]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, K.L.; Haaland, M.S.; Douglas-Vail, M.B.; Mujib, S.; Chew, G.M.; Ndhlovu, L.C.; Ostrowski, M.A. T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J. Immunol. 2014, 192, 782–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Uchida, C. Roles of pRB in the Regulation of Nucleosome and Chromatin Structures. Biomed. Res. Int. 2016, 2016, 5959721. [Google Scholar] [CrossRef]
- Sahi, H.; Savola, S.; Sihto, H.; Koljonen, V.; Bohling, T.; Knuutila, S. RB1 gene in Merkel cell carcinoma: Hypermethylation in all tumors and concurrent heterozygous deletions in the polyomavirus-negative subgroup. APMIS 2014, 122, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar] [CrossRef] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.H.; Hayard, N. Loss of heterozygosity of chromosome 13 in Merkel cell carcinoma. Genes Chromosom. Cancer 1997, 20, 93–97. [Google Scholar] [CrossRef]
- Cimino, P.J.; Robirds, D.H.; Tripp, S.R.; Pfeifer, J.D.; Abel, H.J.; Duncavage, E.J. Retinoblastoma gene mutations detected by whole exome sequencing of Merkel cell carcinoma. Mod. Pathol. 2014, 27, 1073–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef] [Green Version]
- Van Gele, M.; Kaghad, M.; Leonard, J.H.; Van Roy, N.; Naeyaert, J.M.; Geerts, M.L.; Van Belle, S.; Cocquyt, V.; Bridge, J.; Sciot, R.; et al. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br. J. Cancer 2000, 82, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Lassacher, A.; Heitzer, E.; Kerl, H.; Wolf, P. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J. Investig. Dermatol. 2008, 128, 1788–1796. [Google Scholar] [CrossRef] [Green Version]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef] [Green Version]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortes-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Panelos, J.; Batistatou, A.; Paglierani, M.; Zioga, A.; Maio, V.; Santi, R.; Pimpinelli, N.; De Giorgi, V.; Santucci, M.; Massi, D. Expression of Notch-1 and alteration of the E-cadherin/beta-catenin cell adhesion complex are observed in primary cutaneous neuroendocrine carcinoma (Merkel cell carcinoma). Mod. Pathol. 2009, 22, 959–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardhani, L.O.; Matsushita, M.; Kuwamoto, S.; Nonaka, D.; Nagata, K.; Kato, M.; Kitamura, Y.; Hayashi, K. Expression of Notch 3 and Jagged 1 Is Associated With Merkel Cell Polyomavirus Status and Prognosis in Merkel Cell Carcinoma. Anticancer Res. 2019, 39, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef]
- Zeng, X.; Ju, D. Hedgehog Signaling Pathway and Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huq, A.J.; Walsh, M.; Rajagopalan, B.; Finlay, M.; Trainer, A.H.; Bonnet, F.; Sevenet, N.; Winship, I.M. Mutations in SUFU and PTCH1 genes may cause different cutaneous cancer predisposition syndromes: Similar, but not the same. Fam. Cancer 2018, 17, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutierrez Garcia-Rodrigo, C.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Tilli, C.M.; Van Steensel, M.A.; Krekels, G.A.; Neumann, H.A.; Ramaekers, F.C. Molecular aetiology and pathogenesis of basal cell carcinoma. Br. J. Dermatol. 2005, 152, 1108–1124. [Google Scholar] [CrossRef] [Green Version]
- Regl, G.; Kasper, M.; Schnidar, H.; Eichberger, T.; Neill, G.W.; Ikram, M.S.; Quinn, A.G.; Philpott, M.P.; Frischauf, A.M.; Aberger, F. The zinc-finger transcription factor GLI2 antagonizes contact inhibition and differentiation of human epidermal cells. Oncogene 2004, 23, 1263–1274. [Google Scholar] [CrossRef] [Green Version]
- Kuromi, T.; Matsushita, M.; Iwasaki, T.; Nonaka, D.; Kuwamoto, S.; Nagata, K.; Kato, M.; Akizuki, G.; Kitamura, Y.; Hayashi, K. Association of expression of the hedgehog signal with Merkel cell polyomavirus infection and prognosis of Merkel cell carcinoma. Hum. Pathol. 2017, 69, 8–14. [Google Scholar] [CrossRef]
- Brunner, M.; Thurnher, D.; Pammer, J.; Heiduschka, G.; Petzelbauer, P.; Schmid, C.; Schneider, S.; Erovic, B.M. Expression of hedgehog signaling molecules in Merkel cell carcinoma. Head Neck 2010, 32, 333–340. [Google Scholar] [CrossRef]
- Van Gele, M.; Leonard, J.H.; Van Roy, N.; Cook, A.L.; De Paepe, A.; Speleman, F. Frequent allelic loss at 10q23 but low incidence of PTEN mutations in Merkel cell carcinoma. Int. J. Cancer 2001, 92, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, R.; Mustafi, S.B.; Street, M.; Dey, A.; Dwivedi, S.K. Bmi-1: At the crossroads of physiological and pathological biology. Genes Dis. 2015, 2, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Brunner, M.; Thurnher, D.; Pammer, J.; Geleff, S.; Heiduschka, G.; Reinisch, C.M.; Petzelbauer, P.; Erovic, B.M. Expression of VEGF-A/C, VEGF-R2, PDGF-alpha/beta, c-kit, EGFR, Her-2/Neu, Mcl-1 and Bmi-1 in Merkel cell carcinoma. Mod. Pathol. 2008, 21, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Veija, T.; Kero, M.; Koljonen, V.; Böhling, T. ALK and EGFR expression by immunohistochemistry are associated with Merkel cell polyomavirus status in Merkel cell carcinoma. Histopathology 2019, 74, 829–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, K.L.; Chubb, H.; Zhao, L.; Fullen, D.R.; Bichakjian, C.K.; Johnson, T.M.; Carskadon, S.; Palanisamy, N.; Harms, P.W. Increased expression of EZH2 in Merkel cell carcinoma is associated with disease progression and poorer prognosis. Hum. Pathol. 2017, 67, 78–84. [Google Scholar] [CrossRef]
- Wu, X.; Bekker-Jensen, I.H.; Christensen, J.; Rasmussen, K.D.; Sidoli, S.; Qi, Y.; Kong, Y.; Wang, X.; Cui, Y.; Xiao, Z.; et al. Tumor suppressor ASXL1 is essential for the activation of INK4B expression in response to oncogene activity and anti-proliferative signals. Cell Res. 2015, 25, 1205–1218. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.; Beer, T.W.; Murray, K. Vascular density has prognostic value in Merkel cell carcinoma. Am. J. Dermatopathol. 2008, 30, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Figueras, M.T.; Puig, L.; Musulén, E.; Gilaberte, M.; Lerma, E.; Serrano, S.; Ferrándiz, C.; Ariza, A. Expression profiles associated with aggressive behavior in Merkel cell carcinoma. Mod. Pathol. 2007, 20, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Kervarrec, T.; Gaboriaud, P.; Tallet, A.; Leblond, V.; Arnold, F.; Berthon, P.; Schweinitzer, S.; Larcher, T.; Guyétant, S.; Schrama, D.; et al. VEGF-A Inhibition as a Potential Therapeutic Approach in Merkel Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 736–739. [Google Scholar] [CrossRef] [Green Version]
- Kukko, H.; Koljonen, V.; Lassus, P.; Tukiainen, E.; Haglund, C.; Böhling, T. Expression of vascular endothelial growth factor receptor-2 in Merkel cell carcinoma. Anticancer Res. 2007, 27, 2587–2589. [Google Scholar] [PubMed]
- Tay, C.M.; Ong, C.W.; Lee, V.K.; Pang, B. KIT gene mutation analysis in solid tumours: Biology, clincial applications and trends in diagnostic reporting. Pathology 2013, 45, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Kartha, R.V.; Sundram, U.N. Silent mutations in KIT and PDGFRA and coexpression of receptors with SCF and PDGFA in Merkel cell carcinoma: Implications for tyrosine kinase-based tumorigenesis. Mod. Pathol. 2008, 21, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Andea, A.A.; Patel, R.; Ponnazhagan, S.; Kumar, S.; DeVilliers, P.; Jhala, D.; Eltoum, I.E.; Siegal, G.P. Merkel cell carcinoma: Correlation of KIT expression with survival and evaluation of KIT gene mutational status. Hum. Pathol. 2010, 41, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Waltari, M.; Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. Int. J. Cancer 2011, 129, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Veija, T.; Koljonen, V.; Bohling, T.; Kero, M.; Knuutila, S.; Sarhadi, V.K. Aberrant expression of ALK and EZH2 in Merkel cell carcinoma. BMC Cancer 2017, 17, 236. [Google Scholar] [CrossRef] [Green Version]
- Aubry, A.; Galiacy, S.; Allouche, M. Targeting ALK in Cancer: Therapeutic Potential of Proapoptotic Peptides. Cancers 2019, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Plettenberg, A.; Pammer, J.; Tschachler, E. Merkel cells and Merkel cell carcinoma express the BCL-2 proto-oncogene. Exp. Dermatol. 1996, 5, 102–107. [Google Scholar] [CrossRef]
- Sahi, H.; Koljonen, V.; Kavola, H.; Haglund, C.; Tukiainen, E.; Sihto, H.; Böhling, T. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012, 461, 553–559. [Google Scholar] [CrossRef]
- Liu, W.; Krump, N.A.; Herlyn, M.; You, J. Combining DNA Damage Induction with BCL-2 Inhibition to Enhance Merkel Cell Carcinoma Cytotoxicity. Biology 2020, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, W.; Yang, C.Y.; Bai, L. MCL-1 inhibition in cancer treatment. OncoTargets Ther. 2018, 11, 7301–7314. [Google Scholar] [CrossRef] [Green Version]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Centre 2019, 43, 187. [Google Scholar] [CrossRef] [Green Version]
- Jouhi, L.; Koljonen, V.; Böhling, T.; Haglund, C.; Hagström, J. The expression of Toll-like receptors 2, 4, 5, 7 and 9 in Merkel cell carcinoma. Anticancer Res. 2015, 35, 1843–1849. [Google Scholar]
- Larramendy, M.L.; Koljonen, V.; Böhling, T.; Tukiainen, E.; Knuutila, S. Recurrent DNA copy number changes revealed by comparative genomic hybridization in primary Merkel cell carcinomas. Mod. Pathol. 2004, 17, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Härle, M.; Arens, N.; Moll, I.; Back, W.; Schulz, T.; Scherthan, H. Comparative genomic hybridization (CGH) discloses chromosomal and subchromosomal copy number changes in Merkel cell carcinomas. J. Cutan. Pathol. 1996, 23, 391–397. [Google Scholar] [CrossRef]
- Erstad, D.J.; Cusack, J.C., Jr. Mutational analysis of merkel cell carcinoma. Cancers 2014, 6, 2116–2136. [Google Scholar] [CrossRef] [Green Version]
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int. J. Cancer 2002, 99, 352–360. [Google Scholar] [CrossRef]
- Paulson, K.G.; Lemos, B.D.; Feng, B.; Jaimes, N.; Peñas, P.F.; Bi, X.; Maher, E.; Cohen, L.; Leonard, J.H.; Granter, S.R.; et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J. Investig. Dermatol. 2009, 129, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Lee, L.; Caramuta, S.; Höög, A.; Browaldh, N.; Björnhagen, V.; Larsson, C.; Lui, W.O. MicroRNA expression patterns related to merkel cell polyomavirus infection in human merkel cell carcinoma. J. Investig. Dermatol. 2014, 134, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.R.; Tomson, B.N.; Elkin, S.K.; Marchlik, E.; Carter, J.L.; Kurzrock, R. Genomic portfolio of Merkel cell carcinoma as determined by comprehensive genomic profiling: Implications for targeted therapeutics. Oncotarget 2016, 7, 23454–23467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkel Cell Carcinoma; NCCN Guidelines. Version 1.2021; NCCN Publisher: Plymouth Meeting, PA, USA, 2021.
- Villani, A.; Fabbrocini, G.; Costa, C.; Carmela Annunziata, M.; Scalvenzi, M. Merkel Cell Carcinoma: Therapeutic Update and Emerging Therapies. Dermatol. Ther. 2019, 9, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.A.; Camp, E.R.; Lentsch, E.J. Merkel cell carcinoma: Identification of prognostic factors unique to tumors located in the head and neck based on analysis of SEER data. Laryngoscope 2012, 122, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Steuten, L.; Garmo, V.; Phatak, H.; Sullivan, S.D.; Nghiem, P.; Ramsey, S.D. Treatment Patterns, Overall Survival, and Total Healthcare Costs of Advanced Merkel Cell Carcinoma in the USA. Appl. Health Econ. Health Policy 2019, 17, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Liang, E.; Brower, J.V.; Rice, S.R.; Buehler, D.G.; Saha, S.; Kimple, R.J. Merkel Cell Carcinoma Analysis of Outcomes: A 30-Year Experience. PLoS ONE 2015, 10, e0129476. [Google Scholar] [CrossRef]
- Wright, G.P.; Holtzman, M.P. Surgical resection improves median overall survival with marginal improvement in long-term survival when compared with definitive radiotherapy in Merkel cell carcinoma: A propensity score matched analysis of the National Cancer Database. Am. J. Surg. 2018, 215, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.L.; Healy, M.A.; Nghiem, P.; Sober, A.J.; Johnson, T.M.; Bichakjian, C.K.; Wong, S.L. Analysis of prognostic factors from 9387 Merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann. Surg. Oncol. 2016, 23, 3564–3571. [Google Scholar] [CrossRef]
- Amin, M.B.; Edge, S.B.; Greene, F.L. Merkel Cell Carcinoma. In AJCC Cancer Staging Manual, 8th ed.; Springer: Cham, Switzerland, 2017; pp. 549–561. [Google Scholar]
- Bichakjian, C.K.; Nghiem, P.; Johnson, T.; Wright, C.L.; Robert, J. Merkel Cell Carcinoma. In AJCC Cancer Staging Manual, 8th ed.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Gupta, S.G.; Wang, L.C.; Peñas, P.F.; Gellenthin, M.; Lee, S.J.; Nghiem, P. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The Dana-Farber experience and meta-analysis of the literature. Arch. Dermatol. 2006, 142, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.J.; Bowne, W.B.; Jaques, D.P.; Brennan, M.F.; Busam, K.; Coit, D.G. Merkel cell carcinoma: Prognosis and treatment of patients from a single institution. J. Clin. Oncol. 2005, 23, 2300–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, T.; Carr, M.; Zager, J.S.; Naghavi, A.; Smith, F.O.; Cruse, C.W.; Messina, J.L.; Russell, J.; Rao, N.G.; Fulp, W.; et al. Radiation Therapy is Associated with Improved Outcomes in Merkel Cell Carcinoma. Ann. Surg. Oncol. 2016, 23, 3572–3578. [Google Scholar] [CrossRef] [PubMed]
- Garneski, K.M.; Nghiem, P. Merkel cell carcinoma adjuvant therapy: Current data support radiation but not chemotherapy. J. Am. Acad. Dermatol. 2007, 57, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, M.G.; Rischin, D.; Porter, I.; Walpole, E.; Harvey, J.; Hamilton, C.; Keller, J.; Tripcony, L. Does chemotherapy improve survival in high-risk stage I and II Merkel cell carcinoma of the skin? Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 114–119. [Google Scholar] [CrossRef]
- Poulsen, M.; Rischin, D.; Walpole, E.; Harvey, J.; Mackintosh, J.; Ainslie, J.; Hamilton, C.; Keller, J.; Tripcony, L. High-risk Merkel cell carcinoma of the skin treated with synchronous carboplatin/etoposide and radiation: A Trans-Tasman Radiation Oncology Group Study--TROG 96:07. J. Clin. Oncol. 2003, 21, 4371–4376. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Bhatia, S.; Kudchadkar, R.R.; Amin, A.; Sharfman, W.H.; Lebbe, C.; Delord, J.-P.; Shinohara, M.M.; Baxi, S.S.; Chung, C.H.; et al. Nivolumab (Nivo) as neoadjuvant therapy in patients with resectable Merkel cell carcinoma (MCC) in CheckMate 358. J. Clin. Oncol. 2018, 36, 9505. [Google Scholar] [CrossRef]
- Bhatia, S.; Storer, B.E.; Iyer, J.G.; Moshiri, A.; Parvathaneni, U.; Byrd, D.; Sober, A.J.; Sondak, V.K.; Gershenwald, J.E.; Nghiem, P. Adjuvant Radiation Therapy and Chemotherapy in Merkel Cell Carcinoma: Survival Analyses of 6908 Cases From the National Cancer Data Base. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Harrington, C.; Kwan, W. Radiotherapy and Conservative Surgery in the Locoregional Management of Merkel Cell Carcinoma: The British Columbia Cancer Agency Experience. Ann. Surg. Oncol. 2016, 23, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Roman, S.A.; Sosa, J.A.; Judson, B.L. The role of adjuvant therapy in the management of head and neck merkel cell carcinoma: An analysis of 4815 patients. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Han, A.Y.; Patel, P.B.; Anderson, M.; Diaz, M.F.P.; Chin, R.; St John, M.A. Adjuvant radiation therapy improves patient survival in early-stage merkel cell carcinoma: A 15-year single-institution study. Laryngoscope 2018, 128, 1862–1866. [Google Scholar] [CrossRef] [PubMed]
- Hruby, G.; Scolyer, R.A.; Thompson, J.F. The important role of radiation treatment in the management of Merkel cell carcinoma. Br. J. Dermatol. 2013, 169, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Foote, M.; Harvey, J.; Porceddu, S.; Dickie, G.; Hewitt, S.; Colquist, S.; Zarate, D.; Poulsen, M. Effect of radiotherapy dose and volume on relapse in Merkel cell cancer of the skin. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 677–684. [Google Scholar] [CrossRef]
- Medina-Franco, H.; Urist, M.M.; Fiveash, J.; Heslin, M.J.; Bland, K.I.; Beenken, S.W. Multimodality treatment of Merkel cell carcinoma: Case series and literature review of 1024 cases. Ann. Surg. Oncol. 2001, 8, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Tai, P.T.; Yu, E.; Winquist, E.; Hammond, A.; Stitt, L.; Tonita, J.; Gilchrist, J. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: Case series and review of 204 cases. J. Clin. Oncol. 2000, 18, 2493–2499. [Google Scholar] [CrossRef]
- Desch, L.; Kunstfeld, R. Merkel cell carcinoma: Chemotherapy and emerging new therapeutic options. J. Skin Cancer 2013, 2013, 327150. [Google Scholar] [CrossRef] [Green Version]
- Vargo, J.A.; Ghareeb, E.R.; Balasubramani, G.K.; Beriwal, S. RE: Adjuvant Radiation Therapy and Chemotherapy in Merkel Cell Carcinoma: Survival Analyses of 6908 Cases From the National Cancer Data Base. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Naseri, S.; Steiniche, T.; Ladekarl, M.; Bonnelykke-Behrndtz, M.L.; Holmich, L.R.; Langer, S.W.; Venzo, A.; Tabaksblat, E.; Klausen, S.; Skaarup Larsen, M.; et al. Management Recommendations for Merkel Cell Carcinoma-A Danish Perspective. Cancers 2020, 12, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbé, C.; Lewis, K.D.; et al. Two-year efficacy and safety update from JAVELIN Merkel 200 part A: A registrational study of avelumab in metastatic Merkel cell carcinoma progressed on chemotherapy. J. Clin. Oncol. 2018, 36, 9507. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Milella, M.; Brownell, I.; et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥ 1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbé, C.; Lewis, K.D.; et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: Long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Russell, J.; Hassel, J.C.; Lebbe, C.; Chmielowski, B.; Rabinowits, G.; Terheyden, P.; Brownell, I.; Zwiener, I.; Bajars, M.; et al. First-line (1L) avelumab treatment in patients (pts) with metastatic Merkel cell carcinoma (mMCC): Preliminary data from an ongoing study. J. Clin. Oncol. 2017, 35, 9530. [Google Scholar] [CrossRef]
- Bullement, A.; D’Angelo, S.P.; Amin, A.; Stapelkamp, C.; Willis, A.; Lilley, C.; Hatswell, A.; Bharmal, M. Predicting overall survival in patients (pts) with treatment-naive metastatic Merkel cell carcinoma (mMCC) treated with avelumab. J. Clin. Oncol. 2018, 36, e21620. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Russell, J.; Lebbé, C.; Chmielowski, B.; Gambichler, T.; Grob, J.J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I.; et al. Efficacy and Safety of First-line Avelumab Treatment in Patients With Stage IV Metastatic Merkel Cell Carcinoma: A Preplanned Interim Analysis of a Clinical Trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.; Lebbe, C.; Mortier, L.; Brohl, A.; Fazio, N.; Grob, J.-J.; Prinzi, N.; Hanna, G.; Hassel, J.; Kiecker, F. First-Line Avelumab Treatment in Patients with Metastatic Merkel Cell Carcinoma: Primary Analysis After>= 15 Months of Follow-Up from Javelin Merkel 200, a Registrational Phase 2 Trial. Proc. J. Immunother. Cancer 2020. [Google Scholar] [CrossRef]
- Walker, J.W.; Lebbé, C.; Grignani, G.; Nathan, P.; Dirix, L.; Fenig, E.; Ascierto, P.A.; Sandhu, S.; Munhoz, R.; Benincasa, E.; et al. Efficacy and safety of avelumab treatment in patients with metastatic Merkel cell carcinoma: Experience from a global expanded access program. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Topalian, S.; Bhatia, S.; Hollebecque, A.; Awada, A.; Boer, J.D.; Kudchadkar, R.R.; Gonçalves, A.; Delord, J.; Martens, U.; Picazo, J.L.; et al. Abstract CT074: Non-comparative, open-label, multiple cohort, phase 1/2 study to evaluate nivolumab (NIVO) in patients with virus-associated tumors (CheckMate 358): Efficacy and safety in Merkel cell carcinoma (MCC). Cancer Res. 2017, 77. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
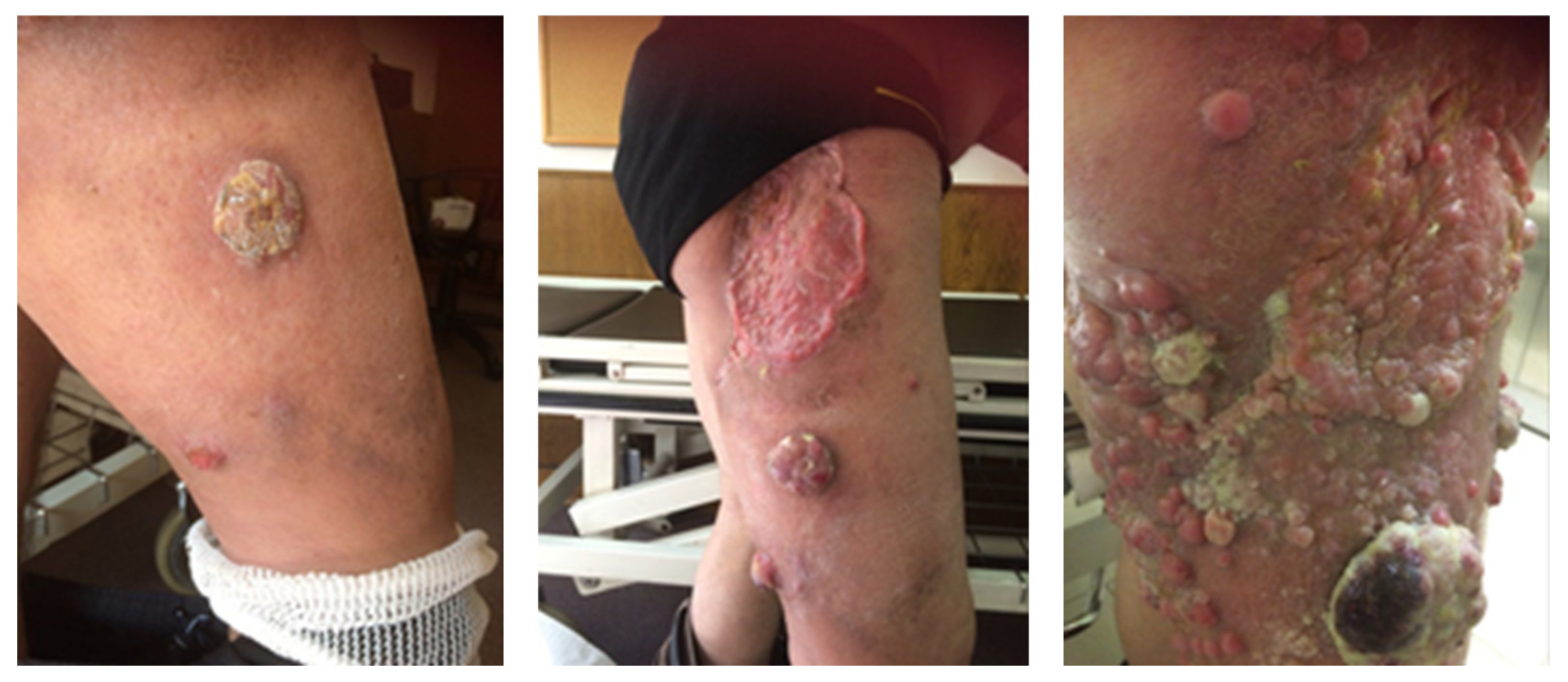{kind=link}
| Setting | Indication | Dose Range |
|---|---|---|
| Adjuvant | Negative resection margins, high-risk | 50–56 Gy |
| Microscopically positive resection margins | 56–60 Gy | |
| Macroscopically positive resection margins | 60–66 Gy | |
| Positive sentinel lymph node biopsy with no evidence of clinically palpable or radiologically suspicious for nodal metastases without lymphadenectomy | 50–56 Gy | |
| Multiple involved lymph nodes and/or extracapsular extension after lymphadenectomy | 56–60 Gy | |
| Definitive | Unresectable or refused surgery or at high risk of postsurgical morbidity | 60–66 Gy |
| Clinically palpable or radiologically evident nodal metastases without lymphadenectomy | 60–66 Gy |
| Molecule | Target | Study Name | Study Number | Number of Patients | Treatment Line | ORR | mPFS | mOS |
|---|---|---|---|---|---|---|---|---|
| avelumab | PD-L1 | JAVELIN Merkel 200 | NCT02155647 | 116 | 1 | 39.7% | 4.1 months | 20.3 months |
| avelumab | PD-L1 | JAVELIN Merkel 200 | NCT02155647 | 88 | >1 | 33% | 2.7 months | 12.6 months |
| pembrolizumab | PD-1 | Keynote-017 | NCT02267603 | 50 | 1 | 56% | 16.8 months | NR |
| nivolumab | PD-1 | CheckMate-358 | NCT02488759 | 14 | 1 | 71% | NR | NR |
| nivolumab | PD-1 | CheckMate-358 | NCT02488759 | 8 | >1 | 63% | NR | NR |
| Clinical Trial | Agent/Interventions | Phase | Study Population | Status |
|---|---|---|---|---|
| NCT04792073 | Avelumab Comprehensive Ablative Radiation Therapy | Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT03599713 | Retifanlimab | Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT04393753 | Domatinostat in combination with avelumab | Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT03787602 | KRT-232 | Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT04261855 | Avelumab External Beam Radiation Therapy (EBRT) Lutetium-177 (177Lu)-DOTATATE | Phase 1 Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT03853317 | Avelumab N-803 haNK™ | Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT03071406 | Nivolumab Ipilimumab Stereotactic Body Radiation Therapy (SBRT) | Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT04590781 | XmAb18087 XmAb18087 ± Pembrolizumab | Phase 2 | Merkel Cell Carcinoma Small Cell Lung Cancer | Not yet recruiting |
| NCT03271372 | Avelumab | Phase 3 | Merkel Cell Carcinoma | Recruiting |
| NCT04160065 | IFx-Hu2.0 | Phase 1 | Merkel Cell Carcinoma Cutaneous Squamous Cell Carcinoma | Recruiting |
| NCT04291885 | Avelumab Placebo | Phase 2 | Merkel Cell Carcinoma Neuroendocrine Tumors Carcinoma Neuroendocrine Skin | Recruiting |
| NCT03684785 | Cavrotolimod Pembrolizumab Cemiplimab | Phase 1 Phase 2 | Merkel Cell Carcinoma Cutaneous Squamous Cell Carcinoma and other Solid Tumors | Recruiting |
| NCT03712605 | Best Practice Pembrolizumab Radiation Therapy | Phase 3 | Merkel Cell Carcinoma | Recruiting |
| NCT03901573 | NT-I7 Atezolizumab | Phase 1 Phase 2 | Melanoma Merkel Cell Carcinoma Cutaneous Squamous Cell Carcinoma | Recruiting |
| NCT02978625 | Nivolumab Talimogene Laherparepvec | Phase 2 | Cutaneous Squamous Cell Carcinoma Merkel Cell Carcinoma Other Rare Skin Tumors | Recruiting |
| NCT03458117 | Talimogene Laherparepvec (T-VEC) | Phase 1 | Non-melanoma Skin Cancer | Recruiting |
| NCT03747484 | Autologous MCPyV-specific HLA-A02-restricted TCR-transduced CD4+ and CD8+ T-cells FH-MCVA2TCR Avelumab Pembrolizumab Fludarabine Cyclophosphamide | Phase 1 Phase 2 | Merkel Cell Carcinoma | Recruiting |
| NCT04725331 | BT-001 Pembrolizumab | Phase 1 Phase 2 | Solid Tumor, Adult Metastatic Cancer Soft Tissue Sarcoma Merkel Cell Carcinoma Melanoma Triple Negative Breast Cancer Non Small Cell Lung Cancer | Recruiting |
| NCT04246671 | TAEK-VAC-HerBy | Phase 1 Phase 2 | Breast Cancer Gastric Cancer Chordoma Lung Cancer Ovarian Cancer Prostate Cancer Colorectal Cancer P ancreatic Cancer Hepatocellular Cancer Merkel Cell Carcinoma Small-cell Lung Cancer | Recruiting |
| NCT03935893 | Tumor Infiltrating Lymphocytes (TIL) Fludarabine + Cyclophosphamide combination | Phase 2 | Merkel Cell Carcinoma Advanced Solid Cancers | Recruiting |
| NCT04116320 | Device: Echopulse Imiquimod Standard of Care PD-1 Therapy | Phase 1 | Merkel Cell Carcinoma Advanced Solid Cancers | Recruiting |
| NCT04272034 | INCB099318 | Phase 1 | Merkel Cell Carcinoma Advanced Solid Cancers | Not yet recruiting |
| NCT04242199 | INCB099280 | Phase 1 | Merkel Cell Carcinoma Advanced Solid Cancers | Recruiting |
| NCT04260802 | OC-001 OC-001 in Combination | Phase 1 Phase 2 | Merkel Cell Carcinoma Advanced or Metastatic Cancers | Recruiting |
| NCT03841110 | FT500 Nivolumab Pembrolizumab Atezolizumab Cyclophosphamide Fludarabine IL-2 | Phase 1 | Merkel Cell Carcinoma Advanced Solid Tumors | Recruiting |
| NCT02643303 | Durvalumab Tremelimumab Poly ICLC | Phase 1 Phase 2 | Merkel Cell Carcinoma Advanced, Measurable, Biopsy-accessible Cancers | Recruiting |
| NCT03212404 | CK-301 (cosibelimab) | Phase 1 | Merkel Cell Carcinoma Advanced Cancers | Recruiting |
| NCT04551885 | FT516 Avelumab Cyclophosphamide Fludarabine Drug: IL-2 | Phase 1 | Advanced Solid Tumors | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachyra, K.; Dudzisz-Śledź, M.; Bylina, E.; Szumera-Ciećkiewicz, A.; Spałek, M.J.; Bartnik, E.; Rutkowski, P.; Czarnecka, A.M. Merkel Cell Carcinoma from Molecular Pathology to Novel Therapies. Int. J. Mol. Sci. 2021, 22, 6305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126305
Stachyra K, Dudzisz-Śledź M, Bylina E, Szumera-Ciećkiewicz A, Spałek MJ, Bartnik E, Rutkowski P, Czarnecka AM. Merkel Cell Carcinoma from Molecular Pathology to Novel Therapies. International Journal of Molecular Sciences. 2021; 22(12):6305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126305
Chicago/Turabian StyleStachyra, Karolina, Monika Dudzisz-Śledź, Elżbieta Bylina, Anna Szumera-Ciećkiewicz, Mateusz J. Spałek, Ewa Bartnik, Piotr Rutkowski, and Anna M. Czarnecka. 2021. "Merkel Cell Carcinoma from Molecular Pathology to Novel Therapies" International Journal of Molecular Sciences 22, no. 12: 6305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126305