
Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases

Abstract
:1. Introduction
2. Brief History of rSNP Discovery
3. Modern Array of Methods for Studying Individual rSNPs
4. Recent Comprehensive Examples
4.1. Allele C of rs36115365 from chr5p15.33 Multi-Cancer Risk Locus Enhances ZNF148 Binding and Telomerase Reverse Transcriptase (TERT) Expression
4.2. Allele G of rs11672691 from Chr19q13.2, Associated with Aggressive Prostate Cancer, Creates a HOXA2 Binding Site and Raises the Transcription Levels of PCAT19 and CEACAM21 Genes, Implicated in Prostate Cancer Cell Growth and Tumor Progression
4.3. Atherosclerosis Risk Variant A of rs2107595 from Chr7p21.1 Interferes with E2F3 in Putative Enhancer Region, Which Leads to HDAC9 Activation
4.4. Allele A of rs12411216 from Chr1q22 Decreases E2F4 Binding, Which Results in a Decreased GBA Expression and an Increased Cognitive Damage in Parkinson’s Disease
4.5. Allele A of rs13239597, Associated with Two Systemic Autoimmune Diseases, Enhances the Binding of EVI1, Which Promotes Formation of a Long-Range Chromatin Loop and an Increased Expression of IRF5, Located 118 kb Away
4.6. Allele T of rs17079281 Decreases Lung Cancer Risk through Creating an YY1 Binding Site to Suppress Proto-Oncogene DCBLD1 Expression
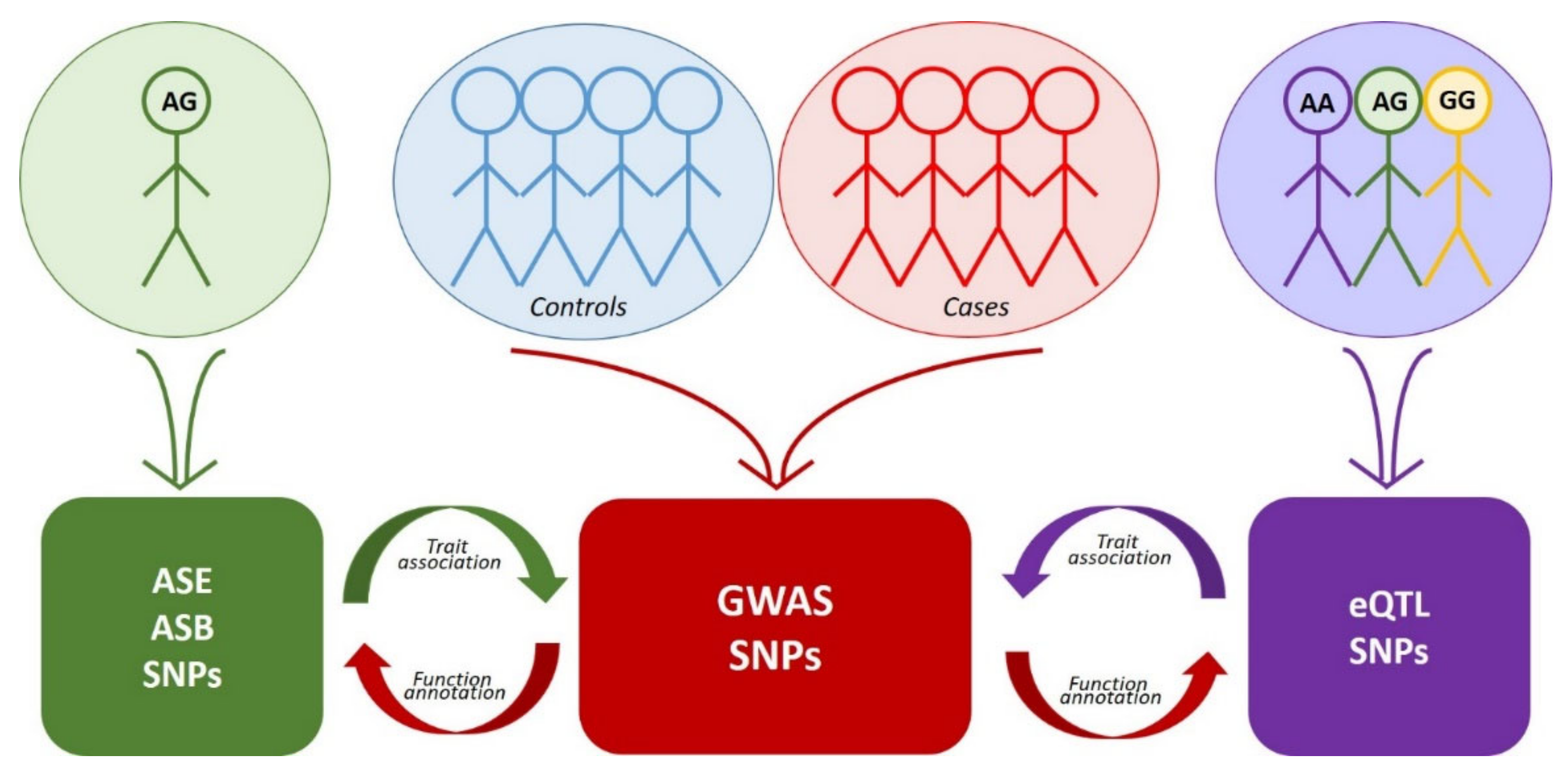
5. rSNPs on a Genome-Wide Scale
5.1. Making Molecular Sense of GWAS
5.2. eQTL Analysis
5.3. Allele-Specific Expression (ASE) Analysis
5.4. Allele-Specific Binding (ASB) Analysis
6. Conclusions
Funding
Conflicts of Interest
References
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryzgalov, L.O.; Antontseva, E.V.; Matveeva, M.Y.; Shilov, A.G.; Kashina, E.V.; Mordvinov, V.A.; Merkulova, T.I. Detection of Regulatory SNPs in Human Genome Using ChIP-seq ENCODE Data. PLoS ONE 2013, 8, e78833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farh, K.K.-H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Levo, M.; Segal, E. In pursuit of design principles of regulatory sequences. Nat. Rev. Genet. 2014, 15, 453–468. [Google Scholar] [CrossRef]
- Andersson, R. Promoter or enhancer, what’s the difference? Deconstruction of established distinctions and presentation of a unifying model. BioEssays 2015, 37, 314–323. [Google Scholar] [CrossRef]
- Erokhin, M.; Vassetzky, Y.; Georgiev, P.; Chetverina, D. Eukaryotic enhancers: Common features, regulation, and participation in diseases. Cell. Mol. Life Sci. 2015, 72, 2361–2375. [Google Scholar] [CrossRef]
- Chen, H.; Pugh, B.F. What Do Transcription Factors Interact with? J. Mol. Biol. 2021, 166883. [Google Scholar] [CrossRef]
- Tobias, I.C.; Abatti, L.E.; Moorthy, S.D.; Mullany, S.; Taylor, T.; Khader, N.; Filice, M.A.; Mitchell, J.A. Transcriptional enhancers: From prediction to functional assessment on a genome-wide scale. Genome 2021, 64, 426–448. [Google Scholar] [CrossRef]
- Singh, G.; Mullany, S.; Moorthy, S.D.; Zhang, R.; Mehdi, T.; Tian, R.; Duncan, A.G.; Moses, A.M.; Mitchell, J.A. A flexible repertoire of transcription factor binding sites and a diversity threshold determines enhancer activity in embryonic stem cells. Genome Res. 2021, 31, 564–575. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 175, 598–599. [Google Scholar] [CrossRef] [Green Version]
- Lelli, K.M.; Slattery, M.; Mann, R.S. Disentangling the Many Layers of Eukaryotic Transcriptional Regulation. Annu. Rev. Genet. 2012, 46, 43–68. [Google Scholar] [CrossRef] [Green Version]
- Merkulova, T.I.; Ananko, E.A.; Ignat’eva, E.V.; Kolchanov, N.A. Regulatory transcription codes in eukaryotic genomes. Genetika 2013, 49, 37–54. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, R.; Liu, B.; Kong, J.; Lin, H.; Yu, X.; Wang, R.; Li, L.; Gao, M.; Zhou, B.; et al. SNP rs17079281 decreases lung cancer risk through creating an YY1-binding site to suppress DCBLD1 expression. Oncogene 2020, 39, 4092–4102. [Google Scholar] [CrossRef] [Green Version]
- Padhy, B.; Hayat, B.; Nanda, G.G.; Mohanty, P.P.; Alone, D.P. Pseudoexfoliation and Alzheimer’s associated CLU risk variant, rs2279590, lies within an enhancer element and regulates CLU, EPHX2 and PTK2B gene expression. Hum. Mol. Genet. 2017, 26, 4519–4529. [Google Scholar] [CrossRef]
- Krause, M.D.; Huang, R.-T.; Wu, D.; Shentu, T.-P.; Harrison, D.L.; Whalen, M.B.; Stolze, L.K.; Di Rienzo, A.; Moskowitz, I.P.; Civelek, M.; et al. Genetic variant at coronary artery disease and ischemic stroke locus 1p32.2 regulates endothelial responses to hemodynamics. Proc. Natl. Acad. Sci. USA 2018, 115, e11349–e11358. [Google Scholar] [CrossRef] [Green Version]
- Hazelett, D.J.; Rhie, S.K.; Gaddis, M.; Yan, C.; Lakeland, D.L.; Coetzee, S.G.; Henderson, B.E.; Noushmehr, H.; Cozen, W.; Kote-Jarai, Z.; et al. Comprehensive Functional Annotation of 77 Prostate Cancer Risk Loci. PLoS Genet. 2014, 10, e1004102. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Xia, J.-H.; Sipeky, C.; Dong, X.-M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and Clinical Implications of the 19q13 Aggressive Prostate Cancer Susceptibility Locus. Cell 2018, 174, 576–589.e18. [Google Scholar] [CrossRef] [Green Version]
- Afanasyeva, M.A.; Putlyaeva, L.V.; Demin, D.E.; Kulakovskiy, I.V.; Vorontsov, I.E.; Fridman, M.V.; Makeev, V.J.; Kuprash, D.V.; Schwartz, A.M. The single nucleotide variant rs12722489 determines differential estrogen receptor binding and enhancer properties of an IL2RA intronic region. PLoS ONE 2017, 12, e0172681. [Google Scholar] [CrossRef]
- Korneev, K.V.; Sviriaeva, E.N.; Mitkin, N.A.; Gorbacheva, A.M.; Uvarova, A.N.; Ustiugova, A.S.; Polanovsky, O.L.; Kulakovskiy, I.V.; Afanasyeva, M.A.; Schwartz, A.M.; et al. Minor C allele of the SNP rs7873784 associated with rheumatoid arthritis and type-2 diabetes mellitus binds PU.1 and enhances TLR4 expression. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165626. [Google Scholar] [CrossRef]
- Fang, J.; Jia, J.; Makowski, M.; Xu, M.; Wang, Z.; Zhang, T.; Hoskins, J.W.; Choi, J.; Han, Y.; Zhang, M.; et al. Functional characterization of a multi-cancer risk locus on chr5p15.33 reveals regulation of TERT by ZNF148. Nat. Commun. 2017, 8, 15034. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Zhang, T.; Vu, A.; Ablain, J.; Makowski, M.M.; Colli, L.M.; Xu, M.; Hennessey, R.C.; Yin, J.; Rothschild, H.; et al. Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun. 2020, 11, 2718. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, D.; Jiang, D.; Zhang, X.; Wu, T.; Cui, J.; Qian, M.; Zhao, J.; Oesterreich, S.; Sun, W.; et al. A sequential methodology for the rapid identification and characterization of breast cancer-associated functional SNPs. Nat. Commun. 2020, 11, 3340. [Google Scholar] [CrossRef]
- Prestel, M.; Prell-Schicker, C.; Webb, T.; Malik, R.; Lindner, B.; Ziesch, N.; Rex-Haffner, M.; Röh, S.; Viturawong, T.; Lehm, M.; et al. The Atherosclerosis Risk Variant rs2107595 Mediates Allele-Specific Transcriptional Regulation of HDAC9 via E2F3 and Rb1. Stroke 2019, 50, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Trapani, D.; Goodyer-Sait, L.; Tomkova, M.; Fernandez-Rozadilla, C.; Sahnane, N.; Woolley, C.; Davis, H.; Chegwidden, L.; Kriaucionis, S.; et al. The polymorphic variant rs1800734 influences methylation acquisition and allele-specific TFAP4 binding in the MLH1 promoter leading to differential mRNA expression. Sci. Rep. 2019, 9, 13463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Huang, Y.; Zhang, P.; Han, C.; Lu, Y.; Mo, Z.; Zhang, Z.; Li, X.; Zhao, S.; Cai, F.; et al. Characterization of a pathogenic variant in GBA for Parkinson’s disease with mild cognitive impairment patients. Mol. Brain 2020, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.K.; Randolph, A.G.; Bhangale, T.; Dogra, P.; Ohlson, M.; Oshansky, C.M.; Zamora, A.E.; Shannon, J.P.; Finkelstein, D.; Dressen, A.; et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 2017, 23, 975–983. [Google Scholar] [CrossRef]
- Vasiliev, G.V.; Merkulov, V.M.; Kobzev, V.F.; Merkulova, T.I.; Ponomarenko, M.P.; Kolchanov, N.A. Point mutations within 663–666 bp of intron 6 of the human TDO2 gene, associated with a number of psychiatric disorders, damage the YY-1 transcription factor binding site. FEBS Lett. 1999, 462, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D. The human gene mutation database. Nucleic Acids Res. 1998, 26, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Deplancke, B.; Alpern, D.; Gardeux, V. The Genetics of Transcription Factor DNA Binding Variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, J.V. rSNP_Guide, a database system for analysis of transcription factor binding to target sequences: Application to SNPs and site-directed mutations. Nucleic Acids Res. 2001, 29, 312–316. [Google Scholar] [CrossRef] [Green Version]
- Bienvenu, T.; Lacronique, V.; Raymondjean, M.; Cazeneuve, C.; Hubert, D.; Kaplan, J.-C.; Beldjord, C. Three novel sequence variations in the 5? upstream region of the cystic fibrosis transmembrane conductance regulator (CFTR) gene: Two polymorphisms and one putative molecular defect. Hum. Genet. 1995, 95, 698–702. [Google Scholar] [CrossRef]
- Ludlow, L.B.; Schick, B.P.; Budarf, M.L.; Driscoll, D.A.; Zackai, E.H.; Cohen, A.; Konkle, B.A. Identification of a Mutation in a GATA Binding Site of the Platelet Glycoprotein Ibβ Promoter Resulting in the Bernard-Soulier Syndrome. J. Biol. Chem. 1996, 271, 22076–22080. [Google Scholar] [CrossRef] [Green Version]
- Comings, D.E.; Gade, R.; Muhleman, D.; Chiu, C.; Wu, S.; To, M.; Spence, M.; Dietz, G.; Winn-Deen, E.; Rosenthal, R.J.; et al. Exon and intron variants in the human tryptophan 2,3-dioxygenase gene: Potential association with Tourette syndrome, substance abuse and other disorders. Pharmacogenetics 1996, 6, 307–318. [Google Scholar] [CrossRef]
- Merkulov, V.M.; Merkulova, T.I. Nucleotide sequence of a fragment of the rat tryptophan oxygenase gene showing high affinity to glucocorticoid receptor in vitro. Biochim. Biophys. Acta Gene Struct. Expr. 1992, 1132, 100–102. [Google Scholar] [CrossRef]
- Ponomarenko, J.V.; Ponomarenko, M.P.; Frolov, A.S.; Vorobyev, D.G.; Overton, G.C.; Kolchanov, N.A. Conformational and physicochemical DNA features specific for transcription factor binding sites. Bioinformatics 1999, 15, 654–668. [Google Scholar] [CrossRef]
- Verheul, T.C.J.; van Hijfte, L.; Perenthaler, E.; Barakat, T.S. The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Knight, J.C.; Udalova, I.; Hill, A.V.S.; Greenwood, B.M.; Peshu, N.; Marsh, K.; Kwiatkowski, D. A polymorphism that affects OCT-1 binding to the TNF promoter region is associated with severe malaria. Nat. Genet. 1999, 22, 145–150. [Google Scholar] [CrossRef]
- Piedrafita, F.J.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu Element in the Myeloperoxidase Promoter Contains a Composite SP1-Thyroid Hormone-Retinoic Acid Response Element. J. Biol. Chem. 1996, 271, 14412–14420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moi, P.; Loudianos, G.; Lavinha, J.; Murru, S.; Cossu, P.; Casu, R.; Oggiano, L.; Longinotti, M.; Cao, A.; Pirastu, M. Delta-thalassemia due to a mutation in an erythroid-specific binding protein sequence 3’ to the delta-globin gene. Blood 1992, 79, 512–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingender, E. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef] [Green Version]
- Nishizaki, S.S.; Boyle, A.P. Mining the Unknown: Assigning Function to Noncoding Single Nucleotide Polymorphisms. Trends Genet. 2017, 33, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Montgomery, S.B. Identifying causal variants and genes using functional genomics in specialized cell types and contexts. Hum. Genet. 2020, 139, 95–102. [Google Scholar] [CrossRef]
- Moyerbrailean, G.A.; Richards, A.L.; Kurtz, D.; Kalita, C.A.; Davis, G.O.; Harvey, C.T.; Alazizi, A.; Watza, D.; Sorokin, Y.; Hauff, N.; et al. High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 2016, 26, 1627–1638. [Google Scholar] [CrossRef]
- Chen, Q.; Deng, X.; Hu, X.; Guan, S.; He, M.; Wang, Y.; Wei, B.; Zhang, J.; Zhao, H.; Yao, W.; et al. Breast Cancer Risk–Associated SNPs in the mTOR Promoter Form De Novo KLF5- and ZEB1-Binding Sites that Influence the Cellular Response to Paclitaxel. Mol. Cancer Res. 2019, 17, 2244–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matana, A.; Ziros, P.G.; Chartoumpekis, D.V.; Renaud, C.O.; Polašek, O.; Hayward, C.; Zemunik, T.; Sykiotis, G.P. Rare and common genetic variations in the Keap1/Nrf2 antioxidant response pathway impact thyroglobulin gene expression and circulating levels, respectively. Biochem. Pharmacol. 2020, 173, 113605. [Google Scholar] [CrossRef]
- Levings, D.; Shaw, K.E.; Lacher, S.E. Genomic resources for dissecting the role of non-protein coding variation in gene-environment interactions. Toxicology 2020, 441, 152505. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.B.; McCarthy, M.; Ren, H.; Carrillo-Roa, T.; Shekhtman, T.; DeModena, A.; Liu, J.J.; Leckband, S.G.; Mors, O.; Rietschel, M.; et al. A functional variant in the serotonin receptor 7 gene (HTR7), rs7905446, is associated with good response to SSRIs in bipolar and unipolar depression. Mol. Psychiatry 2020, 25, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Boldes, T.; Merenbakh-Lamin, K.; Journo, S.; Shachar, E.; Lipson, D.; Yeheskel, A.; Pasmanik-Chor, M.; Rubinek, T.; Wolf, I. R269C variant of ESR1: High prevalence and differential function in a subset of pancreatic cancers. BMC Cancer 2020, 20, 531. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Zhou, J.; Narayanan, C.S.; Cui, Y.; Kumar, A. Role of C/A Polymorphism at −20 on the Expression of Human Angiotensinogen Gene. Hypertension 1999, 33, 108–115. [Google Scholar] [CrossRef]
- López Rodríguez, M.; Kaminska, D.; Lappalainen, K.; Pihlajamäki, J.; Kaikkonen, M.U.; Laakso, M. Identification and characterization of a FOXA2-regulated transcriptional enhancer at a type 2 diabetes intronic locus that controls GCKR expression in liver cells. Genome Med. 2017, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Boulling, A.; Masson, E.; Zou, W.; Paliwal, S.; Wu, H.; Issarapu, P.; Bhaskar, S.; Génin, E.; Cooper, D.N.; Li, Z.; et al. Identification of a functional enhancer variant within the chronic pancreatitis-associated SPINK1 c.101A>G (p.Asn34Ser)-containing haplotype. Hum. Mutat. 2017, 38, 1014–1024. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-X.; Peng, T.; Gao, J.; Feng, J.-G.; Wu, D.-D.; Yang, T.; Zhong, L.; Fu, W.-P.; Sun, C. Allele-specific expression identified rs2509956 as a novel long-distance cis -regulatory SNP for SCGB1A1, an important gene for multiple pulmonary diseases. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L456–L463. [Google Scholar] [CrossRef]
- Peng, T.; Zhong, L.; Gao, J.; Wan, Z.; Fu, W.-P.; Sun, C. Identification of rs11615992 as a novel regulatory SNP for human P2RX7 by allele-specific expression. Mol. Genet. Genom. 2020, 295, 23–30. [Google Scholar] [CrossRef]
- Kuang, X.; Zhou, Q.; Li, Z.; Hu, Y.; Kang, Y.; Huang, W. −254C>G SNP in the TRPC6 Gene Promoter Influences Its Expression via Interaction with the NF- κ B Subunit RELA in Steroid-Resistant Nephrotic Syndrome Children. Int. J. Genom. 2019, 2019, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; Cavalli, M.; Carlsson, B.; Skrtic, S.; Kumar, C.; Wadelius, C. rs953413 Regulates Polyunsaturated Fatty Acid Metabolism by Modulating ELOVL2 Expression. iScience 2020, 23, 100808. [Google Scholar] [CrossRef]
- Thynn, H.N.; Chen, X.-F.; Hu, W.-X.; Duan, Y.-Y.; Zhu, D.-L.; Chen, H.; Wang, N.-N.; Chen, H.-H.; Rong, Y.; Lu, B.-J.; et al. An Allele-Specific Functional SNP Associated with Two Systemic Autoimmune Diseases Modulates IRF5 Expression by Long-Range Chromatin Loop Formation. J. Investig. Dermatol. 2020, 140, 348–360.e11. [Google Scholar] [CrossRef]
- Coetzee, S.G.; Coetzee, G.A.; Hazelett, D.J. motifbreakR: An R/Bioconductor package for predicting variant effects at transcription factor binding sites: Fig. 1. Bioinformatics 2015, btv470. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Ambrosini, G.; Bucher, P. SNP2TFBS—A database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017, 45, D139–D144. [Google Scholar] [CrossRef] [Green Version]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Nishizaki, S.S.; Ng, N.; Dong, S.; Porter, R.S.; Morterud, C.; Williams, C.; Asman, C.; Switzenberg, J.A.; Boyle, A.P. Predicting the effects of SNPs on transcription factor binding affinity. Bioinformatics 2020, 36, 364–372. [Google Scholar] [CrossRef]
- Shin, S.; Hudson, R.; Harrison, C.; Craven, M.; Keleş, S. atSNP Search: A web resource for statistically evaluating influence of human genetic variation on transcription factor binding. Bioinformatics 2019, 35, 2657–2659. [Google Scholar] [CrossRef]
- Yan, J.; Qiu, Y.; Ribeiro dos Santos, A.M.; Yin, Y.; Li, Y.E.; Vinckier, N.; Nariai, N.; Benaglio, P.; Raman, A.; Li, X.; et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature 2021, 591, 147–151. [Google Scholar] [CrossRef]
- Stormo, G.D. DNA binding sites: Representation and discovery. Bioinformatics 2000, 16, 16–23. [Google Scholar] [CrossRef]
- Slattery, M.; Zhou, T.; Yang, L.; Dantas Machado, A.C.; Gordân, R.; Rohs, R. Absence of a simple code: How transcription factors read the genome. Trends Biochem. Sci. 2014, 39, 381–399. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, D.; Mahony, S. Sequence and chromatin determinants of transcription factor binding and the establishment of cell type-specific binding patterns. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194443. [Google Scholar] [CrossRef]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription factor–DNA binding: Beyond binding site motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagny, M.; Paulson, J.N.; Kuijjer, M.L.; Sonawane, A.R.; Chen, C.-Y.; Lopes-Ramos, C.M.; Glass, K.; Quackenbush, J.; Platig, J. Exploring regulation in tissues with eQTL networks. Proc. Natl. Acad. Sci. USA 2017, 114, e7841–e7850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syddall, C.M.; Reynard, L.N.; Young, D.A.; Loughlin, J. The Identification of Trans-acting Factors That Regulate the Expression of GDF5 via the Osteoarthritis Susceptibility SNP rs143383. PLoS Genet. 2013, 9, e1003557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Qin, S.; Ray, B.; Kalari, K.R.; Wang, L.; Weinshilboum, R.M. Single Nucleotide Polymorphisms (SNPs) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction. Drug Metab. Dispos. 2018, 46, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lou, J.; Cai, Y.; Rao, M.; Lu, Z.; Zhu, Y.; Zou, D.; Peng, X.; Wang, H.; Zhang, M.; et al. Risk SNP-Mediated Enhancer–Promoter Interaction Drives Colorectal Cancer through Both FADS2 and AP002754.2. Cancer Res. 2020, 80, 1804–1818. [Google Scholar] [CrossRef] [Green Version]
- Merkulov, V.M.; Leberfarb, E.Y.; Merkulova, T.I. Regulatory SNPs and their widespread effects on the transcriptome. J. Biosci. 2018, 43, 1069–1075. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Berner, D.; Hoja, U.; Zenkel, M.; Ross, J.J.; Uebe, S.; Paoli, D.; Frezzotti, P.; Rautenbach, R.M.; Ziskind, A.; Williams, S.E.; et al. The protective variant rs7173049 at LOXL1 locus impacts on retinoic acid signaling pathway in pseudoexfoliation syndrome. Hum. Mol. Genet. 2019, 28, 2531–2548. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.W.; Patro, C.P.K.; Zhu, J.J.; Dampier, C.H.; Plummer, S.J.; Kuscu, C.; Adli, M.; Lau, C.; Lai, R.K.; Casey, G. A functional variant on 20q13.33 related to glioma risk alters enhancer activity and modulates expression of multiple genes. Hum. Mutat. 2021, 42, 77–88. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Zhang, Q.; Wu, J.; Liang, J.; Yu, S.; Wei, G.-H.; White, K.P.; Wang, X. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017, 18, 194. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.M.; Hadaya, J.; Trehan, A.; Zekavat, S.M.; Roselli, C.; Klarin, D.; Emdin, C.A.; Hilvering, C.R.E.; Bianchi, V.; Mueller, C.; et al. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell 2017, 170, 522–533. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.-L.; Chen, X.-F.; Hu, W.-X.; Dong, S.-S.; Lu, B.-J.; Rong, Y.; Chen, Y.-X.; Chen, H.; Thynn, H.N.; Wang, N.-N.; et al. Multiple Functional Variants at 13q14 Risk Locus for Osteoporosis Regulate RANKL Expression through Long-Range Super-Enhancer. J. Bone Miner. Res. 2018, 33, 1335–1346. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hayes, J.E.; Xu, X.; Gao, X.; Mehta, D.; Lilja, H.G.; Klein, R.J. Validation of prostate cancer risk variants rs10993994 and rs7098889 by CRISPR/Cas9 mediated genome editing. Gene 2021, 768, 145265. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Baglaenko, Y.; Arora, J.; Hannes, S.; Luo, Y.; Amariuta, T.; Teslovich, N.; Rao, D.A.; Ermann, J.; Jonsson, A.H.; et al. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat. Genet. 2020, 52, 247–253. [Google Scholar] [CrossRef]
- Klein, J.C.; Keith, A.; Rice, S.J.; Shepherd, C.; Agarwal, V.; Loughlin, J.; Shendure, J. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 2019, 10, 2434. [Google Scholar] [CrossRef] [Green Version]
- Azghandi, S.; Prell, C.; van der Laan, S.W.; Schneider, M.; Malik, R.; Berer, K.; Gerdes, N.; Pasterkamp, G.; Weber, C.; Haffner, C.; et al. Deficiency of the Stroke Relevant HDAC9 Gene Attenuates Atherosclerosis in Accord with Allele-Specific Effects at 7p21.1. Stroke 2015, 46, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Roca-Ayats, N.; Martínez-Gil, N.; Cozar, M.; Gerousi, M.; Garcia-Giralt, N.; Ovejero, D.; Mellibovsky, L.; Nogués, X.; Díez-Pérez, A.; Grinberg, D.; et al. Functional characterization of the C7ORF76 genomic region, a prominent GWAS signal for osteoporosis in 7q21.3. Bone 2019, 123, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Malecová, B.; Morris, K.V. Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. Curr. Opin. Mol. Ther. 2010, 12, 214–222. [Google Scholar]
- Butter, F.; Davison, L.; Viturawong, T.; Scheibe, M.; Vermeulen, M.; Todd, J.A.; Mann, M. Proteome-Wide Analysis of Disease-Associated SNPs That Show Allele-Specific Transcription Factor Binding. PLoS Genet. 2012, 8, e1002982. [Google Scholar] [CrossRef]
- Amin Al Olama, A.; Kote-Jarai, Z.; Schumacher, F.R.; Wiklund, F.; Berndt, S.I.; Benlloch, S.; Giles, G.G.; Severi, G.; Neal, D.E.; Hamdy, F.C.; et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum. Mol. Genet. 2013, 22, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.; Chauhan, G.; Traylor, M.; Sargurupremraj, M.; Okada, Y.; Mishra, A.; Rutten-Jacobs, L.; Giese, A.-K.; van der Laan, S.W.; Gretarsdottir, S.; et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 2018, 50, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef]
- Takeuchi, F.; Akiyama, M.; Matoba, N.; Katsuya, T.; Nakatochi, M.; Tabara, Y.; Narita, A.; Saw, W.-Y.; Moon, S.; Spracklen, C.N.; et al. Interethnic analyses of blood pressure loci in populations of East Asian and European descent. Nat. Commun. 2018, 9, 5052. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, T.J.; Ehret, G.B.; Nandakumar, P.; Ranatunga, D.; Schaefer, C.; Kwok, P.-Y.; Iribarren, C.; Chakravarti, A.; Risch, N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017, 49, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Giri, A.; Hellwege, J.N.; Keaton, J.M.; Park, J.; Qiu, C.; Warren, H.R.; Torstenson, E.S.; Kovesdy, C.P.; Sun, Y.V.; Wilson, O.D.; et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 2019, 51, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Karasu, N.; Sexton, T. 4C-Seq: Interrogating Chromatin Looping with Circular Chromosome Conformation Capture. In Capturing Chromosome Conformation; Humana: New York, NY, USA, 2021; pp. 19–34. [Google Scholar]
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Factor, S.A.; Wood-Siverio, C.; et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 2016, 31, 95–102. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, S.J.; Knol, M.J.; Chauhan, G.; Satizabal, C.L.; Smith, A.V.; Hofer, E.; Bis, J.C.; Hibar, D.P.; Hilal, S.; van den Akker, E.B.; et al. A genome-wide association study identifies genetic loci associated with specific lobar brain volumes. Commun. Biol. 2019, 2, 285. [Google Scholar] [CrossRef]
- Lan, Q.; Hsiung, C.A.; Matsuo, K.; Hong, Y.-C.; Seow, A.; Wang, Z.; Hosgood, H.D.; Chen, K.; Wang, J.-C.; Chatterjee, N.; et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet. 2012, 44, 1330–1335. [Google Scholar] [CrossRef] [Green Version]
- McKay, J.D.; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; Li, Y.; et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 2017, 49, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T. Functional genomics bridges the gap between quantitative genetics and molecular biology. Genome Res. 2015, 25, 1427–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Zhang, L.; Cai, M.; Li, H.; Xu, H.; Yang, H.; Zhao, Z.; Rhie, S.K.; Farnham, P.J.; Shi, J.; et al. The prostate cancer risk variant rs55958994 regulates multiple gene expression through extreme long-range chromatin interaction to control tumor progression. Sci. Adv. 2019, 5, eaaw6710. [Google Scholar] [CrossRef] [Green Version]
- Ulirsch, J.C.; Nandakumar, S.K.; Wang, L.; Giani, F.C.; Zhang, X.; Rogov, P.; Melnikov, A.; McDonel, P.; Do, R.; Mikkelsen, T.S.; et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 2016, 165, 1530–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalita, C.A.; Brown, C.D.; Freiman, A.; Isherwood, J.; Wen, X.; Pique-Regi, R.; Luca, F. High-throughput characterization of genetic effects on DNA–protein binding and gene transcription. Genome Res. 2018, 28, 1701–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwardhan, R.P.; Lee, C.; Litvin, O.; Young, D.L.; Pe’er, D.; Shendure, J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009, 27, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Xia, J.-H.; Zhu, J.; Gao, P.; Tian, Y.-J.; Du, M.; Guo, Y.-C.; Suleman, S.; Zhang, Q.; Kohli, M.; et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018, 9, 2022. [Google Scholar] [CrossRef] [Green Version]
- Kolchanov, N.A.; Merkulova, T.I.; Ignatieva, E.V.; Ananko, E.A.; Oshchepkov, D.Y.; Levitsky, V.G.; Vasiliev, G.V.; Klimova, N.V.; Merkulov, V.M.; Charles Hodgman, T. Combined experimental and computational approaches to study the regulatory elements in eukaryotic genes. Brief. Bioinform. 2007, 8, 266–274. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Li, X.; Liu, J.; Huo, Y.; Wang, J.; Liu, Z.; Li, M.; Luo, X.-J. Regulatory mechanisms of major depressive disorder risk variants. Mol. Psychiatry 2020, 25, 1926–1945. [Google Scholar] [CrossRef]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Yao, S.; Tang, J.; Liu, S.; Chen, J.; Deng, D.; Zeng, C. Integrative analysis of super enhancer SNPs for type 2 diabetes. PLoS ONE 2018, 13, e0192105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Qiu, C.; Huang, D.; Zhang, Y.; Yu, S.; Zeng, C. Integrative functional analysis of super enhancer SNPs for coronary artery disease. J. Hum. Genet. 2018, 63, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Du, Y.; Qu, S.; Wang, J. rVarBase: An updated database for regulatory features of human variants. Nucleic Acids Res. 2016, 44, D888–D893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Peng, P.-C.; Coetzee, S.G.; Tyrer, J.; Reyes, A.L.P.; Corona, R.I.; Davis, B.; Chen, S.; Dezem, F.; Seo, J.-H.; et al. Ovarian Cancer Risk Variants Are Enriched in Histotype-Specific Enhancers and Disrupt Transcription Factor Binding Sites. Am. J. Hum. Genet. 2020, 107, 622–635. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Segrè, A.V.; van de Bunt, M.; Wen, X.; Xi, H.S.; Hormozdiari, F.; Ongen, H.; Konkashbaev, A.; Derks, E.M.; Aguet, F.; et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 2018, 50, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Barbeira, A.N.; Bonazzola, R.; Gamazon, E.R.; Liang, Y.; Park, Y.; Kim-Hellmuth, S.; Wang, G.; Jiang, Z.; Zhou, D.; Hormozdiari, F.; et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021, 22, 49. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Ray, J.P.; de Boer, C.G.; Fulco, C.P.; Lareau, C.A.; Kanai, M.; Ulirsch, J.C.; Tewhey, R.; Ludwig, L.S.; Reilly, S.K.; Bergman, D.T.; et al. Prioritizing disease and trait causal variants at the TNFAIP3 locus using functional and genomic features. Nat. Commun. 2020, 11, 1237. [Google Scholar] [CrossRef]
- Zeng, B.; Lloyd-Jones, L.R.; Montgomery, G.W.; Metspalu, A.; Esko, T.; Franke, L.; Vosa, U.; Claringbould, A.; Brigham, K.L.; Quyyumi, A.A.; et al. Comprehensive Multiple eQTL Detection and Its Application to GWAS Interpretation. Genetics 2019, 212, 905–918. [Google Scholar] [CrossRef]
- Zhao, J.; Cheng, F.; Jia, P.; Cox, N.; Denny, J.C.; Zhao, Z. An integrative functional genomics framework for effective identification of novel regulatory variants in genome–phenome studies. Genome Med. 2018, 10, 7. [Google Scholar] [CrossRef]
- Gerring, Z.F.; Vargas, A.M.; Gamazon, E.R.; Derks, E.M. An integrative systems-based analysis of substance use: eQTL-informed gene-based tests, gene networks, and biological mechanisms. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2020. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Hu, J.; Xue, C.; Zhang, H.; Susztak, K.; Reilly, M.P.; Xiao, R.; Li, M. ASEP: Gene-based detection of allele-specific expression across individuals in a population by RNA sequencing. PLoS Genet. 2020, 16, e1008786. [Google Scholar] [CrossRef]
- Kim-Hellmuth, S.; Bechheim, M.; Pütz, B.; Mohammadi, P.; Nédélec, Y.; Giangreco, N.; Becker, J.; Kaiser, V.; Fricker, N.; Beier, E.; et al. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun. 2017, 8, 266. [Google Scholar] [CrossRef] [Green Version]
- Werling, D.M.; Pochareddy, S.; Choi, J.; An, J.-Y.; Sheppard, B.; Peng, M.; Li, Z.; Dastmalchi, C.; Santpere, G.; Sousa, A.M.M.; et al. Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep. 2020, 31, 107489. [Google Scholar] [CrossRef]
- Göring, H.H.H.; Curran, J.E.; Johnson, M.P.; Dyer, T.D.; Charlesworth, J.; Cole, S.A.; Jowett, J.B.M.; Abraham, L.J.; Rainwater, D.L.; Comuzzie, A.G.; et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet. 2007, 39, 1208–1216. [Google Scholar] [CrossRef]
- Westra, H.-J.; Franke, L. From genome to function by studying eQTLs. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Umans, B.D.; Battle, A.; Gilad, Y. Where Are the Disease-Associated eQTLs? Trends Genet. 2021, 37, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Hormozdiari, F.; Jew, B.; Castel, S.E.; Lappalainen, T.; Ernst, J.; Sul, J.H.; Eskin, E. Leveraging allelic imbalance to refine fine-mapping for eQTL studies. PLoS Genet. 2019, 15, e1008481. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Zwinderman, A.H.; Cox, N.J.; Denys, D.; Derks, E.M. Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nat. Genet. 2019, 51, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Gerring, Z.F.; Lupton, M.K.; Edey, D.; Gamazon, E.R.; Derks, E.M. An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimers Res. Ther. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef] [Green Version]
- Fadason, T.; Ekblad, C.; Ingram, J.R.; Schierding, W.S.; O’Sullivan, J.M. Physical Interactions and Expression Quantitative Traits Loci Identify Regulatory Connections for Obesity and Type 2 Diabetes Associated SNPs. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, A.E.; Hoeppner, D.J.; Saito, T.; Blanpain, L.; Ukaigwe, J.; Burke, E.E.; Collado-Torres, L.; Tao, R.; Tajinda, K.; Maynard, K.R.; et al. Profiling gene expression in the human dentate gyrus granule cell layer reveals insights into schizophrenia and its genetic risk. Nat. Neurosci. 2020, 23, 510–519. [Google Scholar] [CrossRef]
- Morrow, J.D.; Cho, M.H.; Platig, J.; Zhou, X.; DeMeo, D.L.; Qiu, W.; Celli, B.; Marchetti, N.; Criner, G.J.; Bueno, R.; et al. Ensemble genomic analysis in human lung tissue identifies novel genes for chronic obstructive pulmonary disease. Hum. Genom. 2018, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, H.E.; Hannon, E.; Hill, M.J.; Toste, C.C.; Robertson, M.J.; Morgan, J.E.; McLaughlin, G.; Lewis, C.M.; Schalkwyk, L.C.; Hall, L.S.; et al. Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 2018, 19, 194. [Google Scholar] [CrossRef]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610. [Google Scholar] [CrossRef]
- Zhang, T.; Choi, J.; Kovacs, M.A.; Shi, J.; Xu, M.; Goldstein, A.M.; Trower, A.J.; Bishop, D.T.; Iles, M.M.; Duffy, D.L.; et al. Cell-type–specific eQTL of primary melanocytes facilitates identification of melanoma susceptibility genes. Genome Res. 2018, 28, 1621–1635. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Korbolina, E.E.; Bryzgalov, L.O.; Ustrokhanova, D.Z.; Postovalov, S.N.; Poverin, D.V.; Damarov, I.S.; Merkulova, T.I. A panel of rSNPs demonstrating allelic asymmetry in both ChIP-seq and RNA-seq data and the search for their phenotypic outcomes through analysis of DEGs. Int. J. Mol. Sci. 2021, (in press). [Google Scholar]
- Saha, A.; Kim, Y.; Gewirtz, A.D.H.; Jo, B.; Gao, C.; McDowell, I.C.; Engelhardt, B.E.; Battle, A. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 2017, 27, 1843–1858. [Google Scholar] [CrossRef] [Green Version]
- van der Wijst, M.; de Vries, D.; Groot, H.; Trynka, G.; Hon, C.; Bonder, M.; Stegle, O.; Nawijn, M.; Idaghdour, Y.; van der Harst, P.; et al. The single-cell eQTLGen consortium. Elife 2020, 9. [Google Scholar] [CrossRef]
- Tewhey, R.; Kotliar, D.; Park, D.S.; Liu, B.; Winnicki, S.; Reilly, S.K.; Andersen, K.G.; Mikkelsen, T.S.; Lander, E.S.; Schaffner, S.F.; et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 2016, 165, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Richard, A.C.; Peters, J.E.; Lee, J.C.; Vahedi, G.; Schäffer, A.A.; Siegel, R.M.; Lyons, P.A.; Smith, K.G.C. Targeted genomic analysis reveals widespread autoimmune disease association with regulatory variants in the TNF superfamily cytokine signalling network. Genome Med. 2016, 8, 76. [Google Scholar] [CrossRef] [Green Version]
- Beer, M.A. Predicting enhancer activity and variant impact using gkm-SVM. Hum. Mutat. 2017, 38, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Castel, S.E.; Aguet, F.; Mohammadi, P.; Ardlie, K.G.; Lappalainen, T. A vast resource of allelic expression data spanning human tissues. Genome Biol. 2020, 21, 234. [Google Scholar] [CrossRef]
- Kang, E.Y.; Martin, L.J.; Mangul, S.; Isvilanonda, W.; Zou, J.; Ben-David, E.; Han, B.; Lusis, A.J.; Shifman, S.; Eskin, E. Discovering Single Nucleotide Polymorphisms Regulating Human Gene Expression Using Allele Specific Expression from RNA-seq Data. Genetics 2016, 204, 1057–1064. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, C.; Shen, S.; Chen, X.; Szlachta, K.; Edmonson, M.N.; Shao, Y.; Ma, X.; Hyle, J.; Wright, S.; et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat. Genet. 2020, 52, 811–818. [Google Scholar] [CrossRef]
- Harvey, C.T.; Moyerbrailean, G.A.; Davis, G.O.; Wen, X.; Luca, F.; Pique-Regi, R. QuASAR: Quantitative allele-specific analysis of reads. Bioinformatics 2015, 31, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Edsgärd, D.; Iglesias, M.J.; Reilly, S.-J.; Hamsten, A.; Tornvall, P.; Odeberg, J.; Emanuelsson, O. GeneiASE: Detection of condition-dependent and static allele-specific expression from RNA-seq data without haplotype information. Sci. Rep. 2016, 6, 21134. [Google Scholar] [CrossRef]
- Maurano, M.T.; Haugen, E.; Sandstrom, R.; Vierstra, J.; Shafer, A.; Kaul, R.; Stamatoyannopoulos, J.A. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015, 47, 1393–1401. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, M.; Pan, G.; Nord, H.; Wallerman, O.; Wallén Arzt, E.; Berggren, O.; Elvers, I.; Eloranta, M.-L.; Rönnblom, L.; Lindblad Toh, K.; et al. Allele-specific transcription factor binding to common and rare variants associated with disease and gene expression. Hum. Genet. 2016, 135, 485–497. [Google Scholar] [CrossRef]
- Matys, V. TRANSFAC(R) and its module TRANSCompel(R): Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [Green Version]
- Portales-Casamar, E.; Thongjuea, S.; Kwon, A.T.; Arenillas, D.; Zhao, X.; Valen, E.; Yusuf, D.; Lenhard, B.; Wasserman, W.W.; Sandelin, A. JASPAR 2010: The greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010, 38, D105–D110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newburger, D.E.; Bulyk, M.L. UniPROBE: An online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009, 37, D77–D82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-Binding Specificities of Human Transcription Factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; ‘t Hoen, P.A.C.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, M.; Pan, G.; Nord, H.; Wallén Arzt, E.; Wallerman, O.; Wadelius, C. Allele-specific transcription factor binding in liver and cervix cells unveils many likely drivers of GWAS signals. Genomics 2016, 107, 248–254. [Google Scholar] [CrossRef]
- Marinov, G.K.; Shipony, Z. Interrogating the Accessible Chromatin Landscape of Eukaryote Genomes Using ATAC-seq. In Deep Sequencing Data Analysis; Humana: New York, NY, USA, 2021; pp. 183–226. [Google Scholar] [CrossRef]
- Xu, S.; Feng, W.; Lu, Z.; Yu, C.Y.; Shao, W.; Nakshatri, H.; Reiter, J.L.; Gao, H.; Chu, X.; Wang, Y.; et al. regSNPs-ASB: A Computational Framework for Identifying Allele-Specific Transcription Factor Binding From ATAC-seq Data. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Benaglio, P.; D’Antonio-Chronowska, A.; Ma, W.; Yang, F.; Young Greenwald, W.W.; Donovan, M.K.R.; DeBoever, C.; Li, H.; Drees, F.; Singhal, S.; et al. Allele-specific NKX2-5 binding underlies multiple genetic associations with human electrocardiographic traits. Nat. Genet. 2019, 51, 1506–1517. [Google Scholar] [CrossRef]
- van Setten, J.; Brody, J.A.; Jamshidi, Y.; Swenson, B.R.; Butler, A.M.; Campbell, H.; Del Greco, F.M.; Evans, D.S.; Gibson, Q.; Gudbjartsson, D.F.; et al. PR interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity. Nat. Commun. 2018, 9, 2904. [Google Scholar] [CrossRef] [Green Version]
- D’Oliveira Albanus, R.; Kyono, Y.; Hensley, J.; Varshney, A.; Orchard, P.; Kitzman, J.O.; Parker, S.C.J. Chromatin information content landscapes inform transcription factor and DNA interactions. Nat. Commun. 2021, 12, 1307. [Google Scholar] [CrossRef]
- Li, M.; Huang, H.; Li, L.; He, C.; Zhu, L.; Guo, H.; Wang, L.; Liu, J.; Wu, S.; Liu, J.; et al. Core transcription regulatory circuitry orchestrates corneal epithelial homeostasis. Nat. Commun. 2021, 12, 420. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, B.; Aguilera-Jimenez, E.; Chu, V.S.; Zhou, J.; Wu, Z.; Francis, J.M.; Yang, X.; Choi, P.S.; Bailey, S.D.; et al. Chromatin Looping Shapes KLF5-Dependent Transcriptional Programs in Human Epithelial Cancers. Cancer Res. 2020, 80, 5464–5477. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Y.; McGreal, R.; Cohen-Tayar, Y.; Rockowitz, S.; Wilczek, C.; Ashery-Padan, R.; Shechter, D.; Zheng, D.; Cvekl, A. Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers. Epigenet. Chromatin 2016, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Korbolina, E.E.; Brusentsov, I.I.; Bryzgalov, L.O.; Leberfarb, E.Y.; Degtyareva, A.O.; Merkulova, T.I. Novel approach to functional SNPs discovery from genome-wide data reveals promising variants for colon cancer risk. Hum. Mutat. 2018, 39, 851–859. [Google Scholar] [CrossRef]
- Bryzgalov, L.O.; Korbolina, E.E.; Brusentsov, I.I.; Leberfarb, E.Y.; Bondar, N.P.; Merkulova, T.I. Novel functional variants at the GWAS-implicated loci might confer risk to major depressive disorder, bipolar affective disorder and schizophrenia. BMC Neurosci. 2018, 19, 22. [Google Scholar] [CrossRef] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Leberfarb, E.Y.; Degtyareva, A.O.; Brusentsov, I.I.; Maximov, V.N.; Voevoda, M.I.; Autenshlus, A.I.; Morozov, D.V.; Sokolov, A.V.; Merkulova, T.I. Potential regulatory SNPs in the ATXN7L3B and KRT15 genes are associated with gender-specific colorectal cancer risk. Per. Med. 2020, 17, 43–54. [Google Scholar] [CrossRef]
- Cavalli, M.; Baltzer, N.; Umer, H.M.; Grau, J.; Lemnian, I.; Pan, G.; Wallerman, O.; Spalinskas, R.; Sahlén, P.; Grosse, I.; et al. Allele specific chromatin signals, 3D interactions, and motif predictions for immune and B cell related diseases. Sci. Rep. 2019, 9, 2695. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Chevalier, J.; Mazrooei, P.; Lupien, M.; Staels, B.; Lefebvre, P.; Eeckhoute, J. Organizing combinatorial transcription factor recruitment at cis -regulatory modules. Transcription 2018, 9, 233–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstein, M.B.; Kundaje, A.; Hariharan, M.; Landt, S.G.; Yan, K.-K.; Cheng, C.; Mu, X.J.; Khurana, E.; Rozowsky, J.; Alexander, R.; et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91–100. [Google Scholar] [CrossRef]
- Lan, X.; Farnham, P.J.; Jin, V.X. Uncovering Transcription Factor Modules Using One- and Three-dimensional Analyses. J. Biol. Chem. 2012, 287, 30914–30921. [Google Scholar] [CrossRef] [Green Version]
- Gan, K.A.; Carrasco Pro, S.; Sewell, J.A.; Fuxman Bass, J.I. Identification of Single Nucleotide Non-coding Driver Mutations in Cancer. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Carrasco Pro, S.; Bulekova, K.; Gregor, B.; Labadorf, A.; Fuxman Bass, J.I. Prediction of genome-wide effects of single nucleotide variants on transcription factor binding. Sci. Rep. 2020, 10, 17632. [Google Scholar] [CrossRef]
- Badis, G.; Berger, M.F.; Philippakis, A.A.; Talukder, S.; Gehrke, A.R.; Jaeger, S.A.; Chan, E.T.; Metzler, G.; Vedenko, A.; Chen, X.; et al. Diversity and Complexity in DNA Recognition by Transcription Factors. Science 2009, 324, 1720–1723. [Google Scholar] [CrossRef] [Green Version]
- Nagy, G.; Nagy, L. Motif grammar: The basis of the language of gene expression. Comput. Struct. Biotechnol. J. 2020, 18, 2026–2032. [Google Scholar] [CrossRef]
- Crocker, J.; Preger-Ben Noon, E.; Stern, D.L. The Soft Touch: Low-affinity transcription factor binding sites in development and evolution. Curr. Top. Dev. Biol. 2016, 117, 455–469. [Google Scholar] [CrossRef]
- Levitsky, V.G.; Kulakovskiy, I.V.; Ershov, N.I.; Oshchepkov, D.; Makeev, V.J.; Hodgman, T.C.; Merkulova, T.I. Application of experimentally verified transcription factor binding sites models for computational analysis of ChIP-Seq data. BMC Genom. 2014, 15, 80. [Google Scholar] [CrossRef] [Green Version]
- Levitsky, V.G.; Oshchepkov, D.Y.; Klimova, N.V.; Ignatieva, E.; Vasiliev, G.V.; Merkulov, V.M.; Merkulova, T.I. Hidden heterogeneity of transcription factor binding sites: A case study of SF-1. Comput. Biol. Chem. 2016, 64, 19–32. [Google Scholar] [CrossRef]
- Osz, J.; McEwen, A.G.; Bourguet, M.; Przybilla, F.; Peluso-Iltis, C.; Poussin-Courmontagne, P.; Mély, Y.; Cianférani, S.; Jeffries, C.M.; Svergun, D.I.; et al. Structural basis for DNA recognition and allosteric control of the retinoic acid receptors RAR–RXR. Nucleic Acids Res. 2020, 48, 9969–9985. [Google Scholar] [CrossRef]
- Yin, M.; Wang, J.; Wang, M.; Li, X.; Zhang, M.; Wu, Q.; Wang, Y. Molecular mechanism of directional CTCF recognition of a diverse range of genomic sites. Cell Res. 2017, 27, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Afek, A.; Cohen, H.; Barber-Zucker, S.; Gordân, R.; Lukatsky, D.B. Nonconsensus Protein Binding to Repetitive DNA Sequence Elements Significantly Affects Eukaryotic Genomes. PLoS Comput. Biol. 2015, 11, e1004429. [Google Scholar] [CrossRef] [Green Version]
- Teif, V.B. Soft Power of Nonconsensus Protein-DNA Binding. Biophys. J. 2020, 118, 1797–1798. [Google Scholar] [CrossRef]
- Zheng, A.; Lamkin, M.; Zhao, H.; Wu, C.; Su, H.; Gymrek, M. Deep neural networks identify sequence context features predictive of transcription factor binding. Nat. Mach. Intell. 2021, 3, 172–180. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Q.; Shen, Z.; He, Y.; Chen, Z.-H.; Li, J.; Huang, D.-S. Predicting transcription factor binding sites using DNA shape features based on shared hybrid deep learning architecture. Mol. Ther. Nucleic Acids 2021, 24, 154–163. [Google Scholar] [CrossRef]
- Wada, K.; Wada, Y.; Ikemura, T. Mb-level CpG and TFBS islands visualized by AI and their roles in the nuclear organization of the human genome. Genes Genet. Syst. 2020, 95, 29–41. [Google Scholar] [CrossRef]
- Pei, G.; Hu, R.; Dai, Y.; Manuel, A.M.; Zhao, Z.; Jia, P. Predicting regulatory variants using a dense epigenomic mapped CNN model elucidated the molecular basis of trait-tissue associations. Nucleic Acids Res. 2021, 49, 53–66. [Google Scholar] [CrossRef]
- Jing, F.; Zhang, S.-W.; Cao, Z.; Zhang, S. An Integrative Framework for Combining Sequence and Epigenomic Data to Predict Transcription Factor Binding Sites Using Deep Learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 355–364. [Google Scholar] [CrossRef]
- Chen, C.; Hou, J.; Shi, X.; Yang, H.; Birchler, J.A.; Cheng, J. DeepGRN: Prediction of transcription factor binding site across cell-types using attention-based deep neural networks. BMC Bioinform. 2021, 22, 38. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Aim | Method | Advantages | Shortcomings | Comments |
|---|---|---|---|---|
| Registration of the fact of an effect of nucleotide substitution on TF binding | EMSA with nuclear extract (cross-competition assay when necessary) | Simple procedure | In vitro; tissue-specific effects | Testing of several cell lines is desirable |
| Identification of TF the binding site of which is disrupted by a nucleotide substitution | EMSA with purified TF or specific antibody | Unambiguous result | In vitro; requires prior knowledge about TFBS, purified TF, specific antibody | Prescreening in competition assay with unlabeled oligonucleotides may be helpful |
| Confirmation of TF binding in vivo | ChIP-PCR | In vivo | Requires prior knowledge about TFBS and specific antibody | |
| Identification of TF the binding site of which is disrupted by a nucleotide substitution | ChIP-AS-qPCR | In vivo; unambiguous result | Requires prior knowledge about TFBS and specific antibody | Copy number variation must be taken into account when using cell lines |
| Identification of TF the binding site of which is disrupted by a nucleotide substitution | Pull-down assay followed by mass spectrometry analysis | Requires no prior knowledge about TFBS | In vitro | Confirmation by EMSA with purified TF or specific antibody is necessary in some cases |
| Registration of the fact of an effect of nucleotide substitution on the activity of regulatory element | Reporter assays | Simple procedure | Out of genome context | Testing of several cell lines is desirable |
| Registration of the fact of an effect of nucleotide substitution on the activity of regulatory element | CRISPR/Cas9-mediated single nucleotide editing | In genome context | Testing of several cell lines is desirable |
| ID | Location | Risk Allele | TFs with ASB | Genes with ASE | Risk Disease According to GWAS | Ref |
|---|---|---|---|---|---|---|
| rs36115365 | chr5p15.33 intergenic region, putative enhancer | C | ZNF148 (EMSA+AB, EMSA+ purified ZNF148) | TERT (ASE, siRNA-mediated knockdown of ZNF148) | Increased pancreatic and testicular cancer risk but a decreased lung cancer and melanoma risk | [23] |
| rs11672691 | Chr19q13.2 Intron 2 of lncRNA PCAT19 | G | HOXA2 (ChIP-AS-qPCR) | PCAT19 CEACAM21 (ASE, HOXA2 knockdown CRISPR/Cas9 | Aggressive prostate cancer | [20] |
| rs2107595 | Chr7p21 noncoding DNA 3’ to the HDAC, DHSs | A | E2F3 (ChIP-PCR) | HDAC9 (ASE) | Atherosclerosis, coronary artery disease, stroke | [26] |
| rs12411216 | Chr1q22 DHSs | A | E2F4 (EMSA+AB) | GBA (ASE, CRISPR/Cas9) | Parkinson’s disease, cognitive damage | [28] |
| rs13239597 | Chr7q32.1 TNPO3 promoter | A | EVI1 (ChIP-AS-qPCR) | IRF5 (ASE, shRNA-mediated knockdown of EVI1) | Systemic lupus erythematosus and systemic sclerosis | [59] |
| rs17079281 | Chr6q22.2 DCBLD1 promoter | C | YY1 (ChIP-qPCR) | DCBLD1 (ASE, CRISPR/Cas9) | Lung cancer | [16] |
| Approach | GWAS | eQTL Analysis | ASE | ASB | |
|---|---|---|---|---|---|
| 1 | Initial association with trait | + | − | − | − |
| 2 | Initial association with function | − | + | + | + |
| 3 | Causal or in LD | Both | + | ++ | +++ |
| 4 | Number of participants | Tens and hundreds of thousands (large cohorts) | Hundreds (modestly sized cohorts) | Few | Few |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degtyareva, A.O.; Antontseva, E.V.; Merkulova, T.I. Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. Int. J. Mol. Sci. 2021, 22, 6454. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126454
Degtyareva AO, Antontseva EV, Merkulova TI. Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases. International Journal of Molecular Sciences. 2021; 22(12):6454. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126454
Chicago/Turabian StyleDegtyareva, Arina O., Elena V. Antontseva, and Tatiana I. Merkulova. 2021. "Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases" International Journal of Molecular Sciences 22, no. 12: 6454. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126454