
Manipulation of Host Cell Organelles by Intracellular Pathogens


1
Abt. Mikrobiologie, Fachbereich Biologie/Chemie, Barbarastr 11, Universität Osnabrück, 49076 Osnabrück, Germany
2
CellNanOs–Center of Cellular Nanoanalytics Osnabrück, Universität Osnabrück, Barbarastr 11, 49076 Osnabrück, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(12), 6484; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126484
Submission received: 5 April 2021
/
Revised: 3 June 2021
/
Accepted: 4 June 2021
/
Published: 17 June 2021
(This article belongs to the Special Issue Intracellular Organelle Rearrangement Induced by Pathogenic Infections)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Pathogenic intracellular bacteria, parasites and viruses have evolved sophisticated mechanisms to manipulate mammalian host cells to serve as niches for persistence and proliferation. The intracellular lifestyles of pathogens involve the manipulation of membrane-bound organellar compartments of host cells. In this review, we described how normal structural organization and cellular functions of endosomes, endoplasmic reticulum, Golgi apparatus, mitochondria, or lipid droplets are targeted by microbial virulence mechanisms. We focus on the specific interactions of Salmonella, Legionella pneumophila, Rickettsia rickettsii, Chlamydia spp. and Mycobacterium tuberculosis representing intracellular bacterial pathogens, and of Plasmodium spp. and Toxoplasma gondii representing intracellular parasites. The replication strategies of various viruses, i.e., Influenza A virus, Poliovirus, Brome mosaic virus, Epstein-Barr Virus, Hepatitis C virus, severe acute respiratory syndrome virus (SARS), Dengue virus, Zika virus, and others are presented with focus on the specific manipulation of the organelle compartments. We compare the specific features of intracellular lifestyle and replication cycles, and highlight the communalities in mechanisms of manipulation deployed.
1. Introduction
Infectious diseases represent a global burden for human health and are still responsible for high levels of morbidity and mortality [1]. Infections are caused by pathogenic bacteria, viruses, or parasites which enter the body through distinct paths. Foodborne pathogens can colonize daily nutrition and consequently enter hosts by consumption of contaminated food or water [2]. Other causative agents are transmitted by mosquitos or ticks that serve as vectors and enter the human blood stream to initiate colonization [3]. Additionally, several pathogens are inhaled in form of infectious droplets originating from, and propagated by infected individuals due to coughing, talking, or sneezing [4].
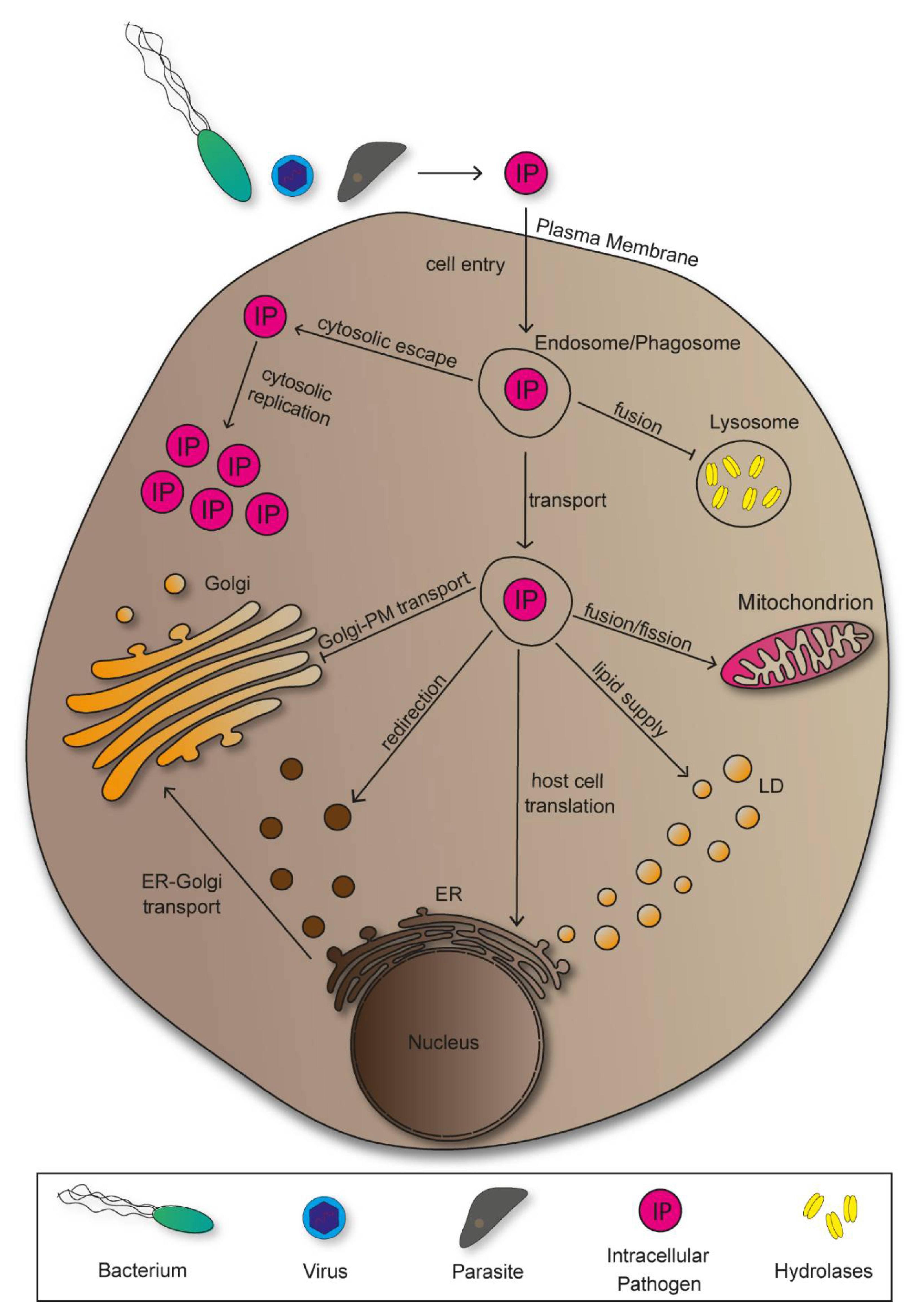
Following entry, pathogens manipulate host cells to establish an environment that provides nutrition for growth, protection against immune responses, and support spread within the host. Accordingly, various mechanisms of pathogen-induced manipulation have been discovered. A majority of bacterial pathogens deploy effector proteins for host manipulations, marking them as a central element for pathogenesis [5]. Intracellular pathogens rearrange host cell compartments into niches in which efficient proliferation occurs. To do so, however, they have to overcome certain obstacles, starting with cell entry (Figure 1).
There are various possibilities for pathogens to overcome the plasma membrane and invade host cells, both actively and passively. For active invasion, bacterial pathogens translocate effector proteins into the cytoplasm exploiting cytoskeletal elements, or deploy surface-bound invasins to induce entry in form trigger or zipper mechanisms [6]. Passive cell entry results from receptor-mediated endocytosis or phagocytosis. Following invasion, intracellular pathogens localize in endosomes or phagosomes, which they have to manipulate in order to bypass lysosomal degradation [7]. Some pathogens escape the vesicular organelles to replicate inside the host cell cytoplasm, while others take advantage of its vesicular transport along the endocytic pathways to reach target structures. Additionally, there are pathogens that remain inside compartments and redesign these into replicative-permissive environments. In order to achieve replication, viruses need to exploit host cell protein synthesis, thus making the endoplasmic reticulum (ER) a central target for viral pathogens [8]. Further pathogen-induced manipulations of the ER include the redirection of vesicles transported from ER to Golgi containing nutritional cargo. As sorting, modification, and transport of newly synthesized proteins are main Golgi functions, pathogens evolved mechanisms of manipulation to impair and take advantage of these processes to serve proliferation of pathogens [9]. There are pathogens that benefit from manipulating mitochondria to either mitochondrial fusion or fission, showing the great variety of manipulation mechanisms that evolved [10,11]. Accordingly, lipid droplets show to be exploited for lipid supply by bacteria and parasites, while viruses utilize the vesicular organelles for secretion and subsequent cellular spread [12].
2. Membrane-Bound Organelles Manipulated by Intracellular Pathogens
2.1. Endosomes and Phagosomes
Endocytosis is a process in which extracellular material is internalized by cells through formation of cargo-containing vesicular organelles, the endosomes. Subsequent transport of endosomes to cellular compartments depends on sorting mechanisms mediated by GTPases and can either lead to degradation, modification, or recycling of the content [13]. Phagosomes are intracellular vesicles created by internalization of apoptotic bodies or pathogens. The corresponding process is phagocytosis, which is crucial for cellular immunity and carried out by specialized cells, the phagocytes [14]. Degradation of pathogen-containing phagosomes requires fusion events with lysosomes, another type of cellular organelles containing several hydrolases for molecular degradation. Additionally, lysosomes take part in further degradations including endosomal content or apoptotic cells [15].
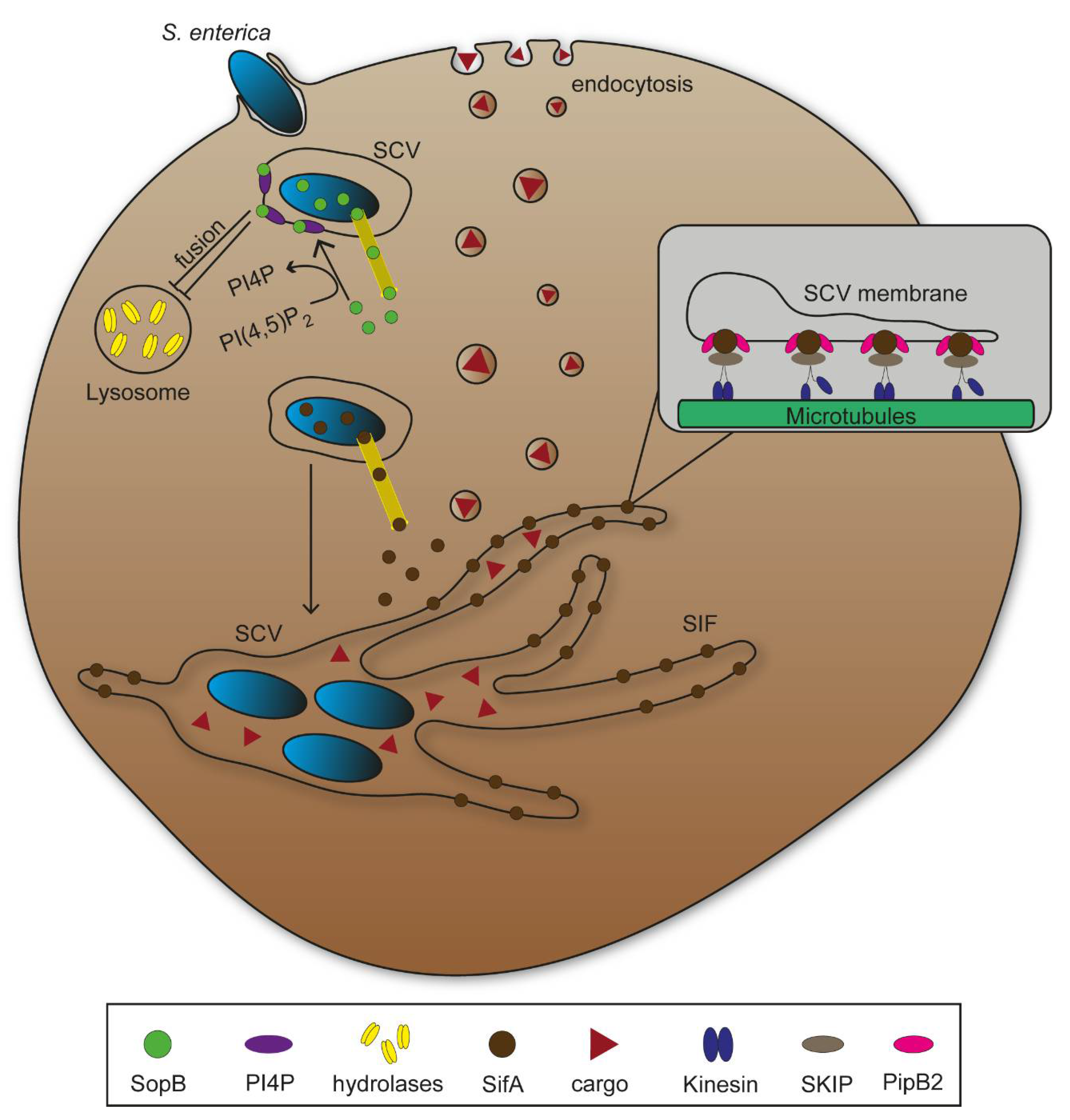
Various pathogens rearrange phagosomes into compartments that promote proliferation, or hijack endosomal transport to reach specific subcellular targets. Salmonella enterica is a facultative intracellular Gram-negative Enterobacterium causing diseases ranging from self-limiting gastroenteritis to systemic life-threatening typhoid fever [16]. Salmonella actively invades non-phagocytic epithelial cells using the Salmonella pathogenicity island 1 (SPI1)-encoded type III secretion system (T3SS) that translocates effector proteins mediating trigger invasion [17]. Salmonella can also gain access to host cells by phagocytosis. Both entry mechanisms lead to bacteria residing in vesicular compartments that are rearranged during infection into Salmonella-containing vacuoles (SCV). The SCV allows bacterial survival and replication, and further remodelling of the host cell endosomal system ensures continuous nutritional supply [18].
While phagosome fusion with lysosomes eliminate bacterial agents through enzymatic degradation, many pathogens have evolved manipulation mechanisms to bypass this process. S. enterica translocates the SPI1-T3SS effector protein SopB, a phosphoinositide phosphatase that mediates dephosphorylation of phosphatidylinositol-4,5-bisphospate (PI(4,5)P2) at the plasma membrane promoting bacterial invasion [7]. Bakowski et al. [19] showed that SopB activity reduced amount of PI(4,5)P2 as well as phosphatidylserine (PS) in SCV membranes, resulting in a decrease of the negative membrane surface charge (Figure 2). The authors concluded an impairment of fusion events between phagosomes and lysosomes on the basis of such membrane alterations. Additionally, numerous other pathogens that reside in intracellular vacuoles following invasion such as Shigella flexneri show similar membrane alterations to prevent lysosomal fusion [20,21,22]. While S. flexneri escapes from its vacuole to replicate in host cell cytosol and cause severe inflammation, S. enterica continues to inhabit the SCV [23]. SCV remodelling to provide nutritional supply requires further expression and translocation of effector proteins of the SPI2-T3SS. Several SPI2-T3SS effectors contribute to formation of an endomembrane network that evolves from the SCV through effector-mediated fusions between endosomes and the SCV. Tubular vesicles interconnect and branch throughout the cytosol, increase fusion events with endosomal vesicles, and thereby increase membrane surface and volume of the vesicular network (Figure 2). Salmonella-induced filaments (SIF) form the major portion of tubular structures, and their biogenesis critically depend on function of SifA, with contribution of additional SPI2-T3SS effector proteins [24]. SifA has an N-terminal binding domain for the SifA-Kinesin-Interacting-Protein (SKIP), which interacts with microtubule motor protein kinesin, leading to SIF extension along microtubules throughout the cell (Figure 2) [25,26,27]. Additionally, SPI2-T3SS translocated effector protein PipB2 serves as linker between kinesin and SCV [28]. Functional loss of SifA leads to decreased SCV integrity and subsequent release of bacteria into host cell cytosol [29]. Intracellular S. enterica show higher metabolic activity if SIF are formed and connected to SCV compared to mutant strains lacking SIF, underlining the importance of vesicular rearrangements for the pathogen to survive and proliferate in host cells [18].
Viral replication depends on utilization of the host cell biosynthetic machinery for replication of their genome [30]. Following cell entry through receptor-mediated endocytosis, hijacking endosomal transport therefore is pivotal to reach replication sites. There are several viral entry mechanisms including clathrin-mediated endocytosis, macropinocytosis and caveolar endocytosis that lead to pathogens residing in vesicular organelles. As viruses do not alter the endosomal composition to bypass lysosomal degradation, they have to escape or release their genome into the cytosol before fusion events [31]. Enveloped viruses such as Influenza A virus (IAV) mainly relocate their genome into the cytosol through receptor-mediated fusion with endosomal membranes. Influenza viruses are enveloped negative-strand RNA viruses causing mild respiratory diseases, mostly limited to the upper respiratory tract, and in some cases lethal pneumonia [32]. IAV particles contain the protein hemagglutinin that, after activation by organelle acidification, mediates endosomal receptor binding and subsequent membrane fusion [33,34]. In contrast, a majority of non-enveloped viruses such as the positive-strand RNA Poliovirus (PVS)—the causative agent of poliomyelitis—induce conformational changes of the capsid on the basis of receptors interacting with endosomal structures [35]. Following those changes, PVS protein VP4 is liberated into the endosome and functional PVS protein VP1 domains are uncovered to carry out their function in pore formation in the endosomal membrane to promote genome release [36]. The non-enveloped double-strand RNA Reovirus (RV) is released from the endosome as intact particle. Due to membrane-penetrating activities of Reovirus capsid protein µ1 that require protein hydrolysis by endosomal proteases for activation, whole viral particles can escape from the endosome leading to productive infections, inducing apoptosis in cultured cells in vitro, and in vivo heart tissue, and the central nervous system [37,38]. After intracellular replication, many viruses such as the enveloped double-strand DNA Epstein–Barr Virus (EBV) are packed into secretory vesicles leading to exocytosis and cellular spread after assembly [39]. EBV infections mostly appear asymptomatic, but can cause infectious mononucleosis in particular if infection occurs in adolescence or adulthood [40]. Utilization of the secretory pathway by viral pathogens constitutes an additional way for viruses to exploit host cell organelles.
2.2. Endoplasmic Reticulum
The endoplasmic reticulum (ER) is among the largest organelles of mammalian cells and consists of a single continuous cytoplasmic membrane that shapes a dynamic network system through its distribution into structural and functional distinct domains [41]. There are two subdomains: The nuclear envelope (NE) and the peripheral ER. Two flat membrane bilayers form the inner and outer nuclear membrane (INM/ONM) which serve as barrier between nuclear and cytoplasmic space [42,43]. Maintenance of the NE requires several interactions such as INM-associated protein binding to chromatin and lamina, linker proteins between INM and ONM for constant spacing, anchoring to cytoskeletal elements, and the positioning of nuclear pore complexes (NPCs) [41,44]. NPCs are essential transport machinery for exchange of selected material through NE and therefore play an important part in regulation of gene expression [45].
There are interconnected tubular and flat sheet membrane structures branching from the NE creating the peripheral ER. Ribosome-free tubular domains of ER membrane (smooth ER) depend on high concentrations of two membrane proteins, RTN and DP1/YOP1. In contrast, there is low abundance of both proteins in membrane sheets that might contribute to the structural curvature of edges [46,47]. Additionally, it was proposed that membrane-bound polyribosomes influence the flat sheet structure forming the rough ER [48]. Several membrane proteins in the ER mediate linkage to microtubules (MT), enabling dynamic changes, e.g., during cell division [41,49]. Further interactions between ER and various other cellular structures indicate additional functions.
Besides protein biosynthesis through translation by membrane-bound ribosomes, regulation of cellular homeostasis in stress situations through induction of the unfolded protein response (UPR), and the modification of translated proteins for further destinations are considered to be main functions of the ER there is also a domain that takes part in lipid biogenesis [50]. The required close proximity to the Golgi apparatus creates the ER-Golgi intermediate compartment (ERGIC), as the organelles interact in distributing mobilized lipids and proteins through the cell [42,48]. Furthermore, the ER serves as store for Ca2+ ions and is able to regulate Ca2+-dependent signalling pathways such as muscle contraction or apoptosis in various organs [51,52].
The ER is target for manipulations by various intracellular pathogens. Legionella pneumophila is a Gram-negative facultative intracellular bacterial pathogen causing Legionnaires’ disease [53]. This pathogen resides in the phagosome after host cell invasion, that is remodelled to an ER-like compartment, termed Legionella-containing vacuole (LCV). The LCV enabled bacterial replication, and ultimately fuses with ER membranes for constant nutritional supply. Following invasion, L. pneumophila activates expression of virulence genes encoding a type IV secretion system (T4SS) for translocation of effector proteins. By interaction with small GTPases, these effector proteins cooperatively redirect the secretory pathway between ER and Golgi apparatus, leading to fusion of ER-derived vesicles with the LCV (Figure 3 [54,55]. A similar redirection of ER vesicles for bacterial proliferation was reported for the intracellular lifestyle of Gram-negative Proteobacteria Brucella spp. and Gram-negative Chlamydiales Chlamydia trachomatis [55,56,57].
As host cell regulators for vesicle and membrane transport interact with GTPases RAB1, SAR1 and ARF1, these represent effective targets for translocated effector proteins. Once activated and recruited to the LCV through exchange of GDP to GTP by the effector protein DrrA acting as guanine nucleotide exchange factor (GEF), RAB1 recruits host-tethering factors with important roles in fusion between ER-derived vesicles and LCV (Figure 4). SAR1 is also recruited to the LCV and impacts the budding mechanism of ER-derived vesicles. Experiments interfering with SAR1 function indicated a key role in remodelling the of phagosome [58,59]. ARF1 was proposed as important factor for fusion of the LCV with ER membranes and is activated by the effector protein RalF acting as GEF. After replication in an ER-derived intracellular niche created by manipulation of endosomal pathways, L. pneumophila exits its compartment, subsequently the host cell, and spreads to adjacent cells [60].
ER is also the organelle most frequently targeted by viral particles after entry [8]. This is due to several circumstances: As one of the largest organelles and by its distribution throughout the cell, the ER can serve after viral rearrangement as structure to efficiently avoid degradation through host cell enzymes such as nucleases. Additionally, the presence of membrane-bound ribosomes for translation of mRNA is convenient for the synthesis of viral proteins. Therefore, viruses with positive-strand RNA such as brome mosaic virus (BMV) shape the peripheral ER to form single-membrane spherule vesicles by presumably modifying ER-associated proteins DP1/Yop1 and reticulons (Figure 3 and Figure 4) [41,61]. Enveloped positive-strand Hepatitis C virus (HCV) and severe acute respiratory syndrome (SARS) virus are pathogens inducing the formation of single-membrane vesicles that can develop into double-membrane vesicles with further progression of infection (Figure 3 and Figure 4). Action of non-structural viral proteins (NSP) drive membrane invaginations. Vesicles originating from ER are partly distinct in structure, but yet provide same function in protection against host cell immune factors, and space for accumulation of components essential for replication [41,62]. Another structural change results from host cell infections by adenoviruses. As this virus influences composition of NPCs through a Kinesin-1-dependent impairment to inject genetic information into the nucleus to reprogram host cells for synthesis of viral elements crucial for their spread [63]. A branched ER membrane network can be observed in cells infected by positive-strand RNA viruses, e.g., Coronaviridae. These convoluted membranes are functionally similar to most viral-induced ER rearrangements as they provide space and protection for genome replication and virus assembly [8,64,65].
Figure 4.
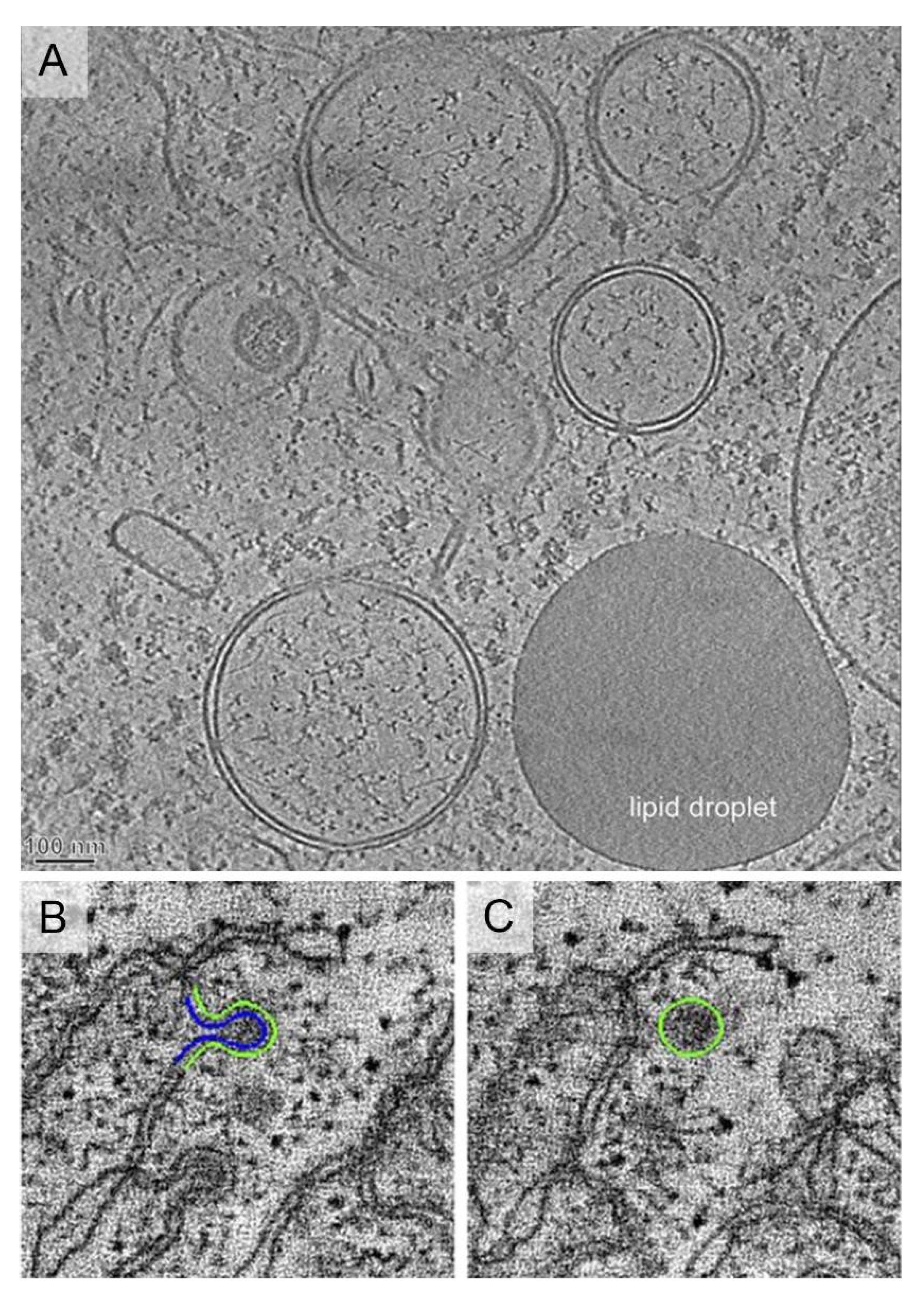
Ultrastructure of viral vesicles. (A) Endoplasmic Reticulum (ER)-derived double membrane vesicles (DMV) generated by positive-strand RNA viruses such as Hepatitis C virus (HCV), or severe acute respiratory syndrome coronavirus (SARS-CoV). Image reproduced, with permission, from [62]. Scale bar, 100 nm. (B,C) Single-membrane spherules formed by brome mosaic virus that localize inside the ER membranes. Images reproduced, with permission, from [66].
Figure 4.
Ultrastructure of viral vesicles. (A) Endoplasmic Reticulum (ER)-derived double membrane vesicles (DMV) generated by positive-strand RNA viruses such as Hepatitis C virus (HCV), or severe acute respiratory syndrome coronavirus (SARS-CoV). Image reproduced, with permission, from [62]. Scale bar, 100 nm. (B,C) Single-membrane spherules formed by brome mosaic virus that localize inside the ER membranes. Images reproduced, with permission, from [66].

The parasite Toxoplasma gondii invades nucleated host cells of warm-blooded animals and can cause toxoplasmosis. Similar to a variety of bacterial pathogens, T. gondii forms parasitophorous vacuoles (PV), subcellular compartments that function as barrier against host cell immune factors, as well as niche for replication through nutrition obtained from the host cell [67,68]. In contrast to the LCV of L. pneumophila, the PV of T. gondii does not fuse with vesicles but rather forms narrow contacts with host organelles that might contribute to manipulation of cellular pathways [69]. Experiments showed associations of two organelles with the parasitophorous vacuole membrane (PVM) including the ER. Interactions of proteins secreted by T. gondii (GRA3/GRA5) residing in the PVM with the ER-anchoring protein calcium-modulating ligand (CAMLG) might induce association between PVM and organelles, though further studies have to test this hypothesis (Figure 4) [70,71]. It was also shown that the UPR was impaired during T. gondii pathogenesis through manipulation of Ca2+ efflux from ER, leading to activation of the cytoskeleton-rearranging stress sensor IRE1, promoting host cell migration and parasite dissemination [69].
2.3. Golgi Apparatus
The Golgi apparatus is a membrane-bound organelle composed of arrayed flat cisternae that stack and interact with tubular membranes to create a higher-ordered structure named Golgi ribbon [72]. Formation of Golgi ribbons depends on cytoskeletal elements as depolymerization of microtubules results in disruption of the complex structure [73,74]. There are three subdomains within the cell compartment: The cis-Golgi network (CGN) is located in close proximity to the ER and faces its membrane side. The medial-Golgi is the central part of the organelle located between the other two subdomains. The trans-Golgi faces the plasma membrane and is able to transform flat cisternae into tubular structures forming the trans-Golgi network (TGN) [75,76]. Golgi structures undergo strictly regulated morphological changes during cell cycle that reach as far as disassembly and vesicular packaging of the organelle for mitosis. Reassembly occurs shortly after cell division [77,78].
Post-translational modifications of newly synthesized proteins and of lipids originating from the ER are key roles of the Golgi apparatus. Those modifications are essential for proper transport of cargo to target membranes and functionality, and impaired functions lead to severe diseases [79,80]. The CGN is the receiving end for proteins and lipids synthesized at the ER. Transport vesicles tether and fuse with CGN membranes releasing their cargo into the organelle [81]. There are multiple enzymes throughout the Golgi compartment that perform modifications, such as glycosylation, leading to production of glycoproteins, glycolipids and further modified molecules [82]. Sorting and transport of modified molecules takes place in the TGN and requires diverse sorting signals which are present on cargo and used by coat proteins for correct packaging in vesicles. Additionally, trans-Golgi modifications are done as target membranes need to identify their cognate vesicles [83]. With sorting and transport of molecules participating in host cell immune responses, the Golgi also represents an important organelle for host defence mechanisms against intracellular pathogens. Furthermore, recent studies suggest the Golgi as pivotal element in immune signalling [84].
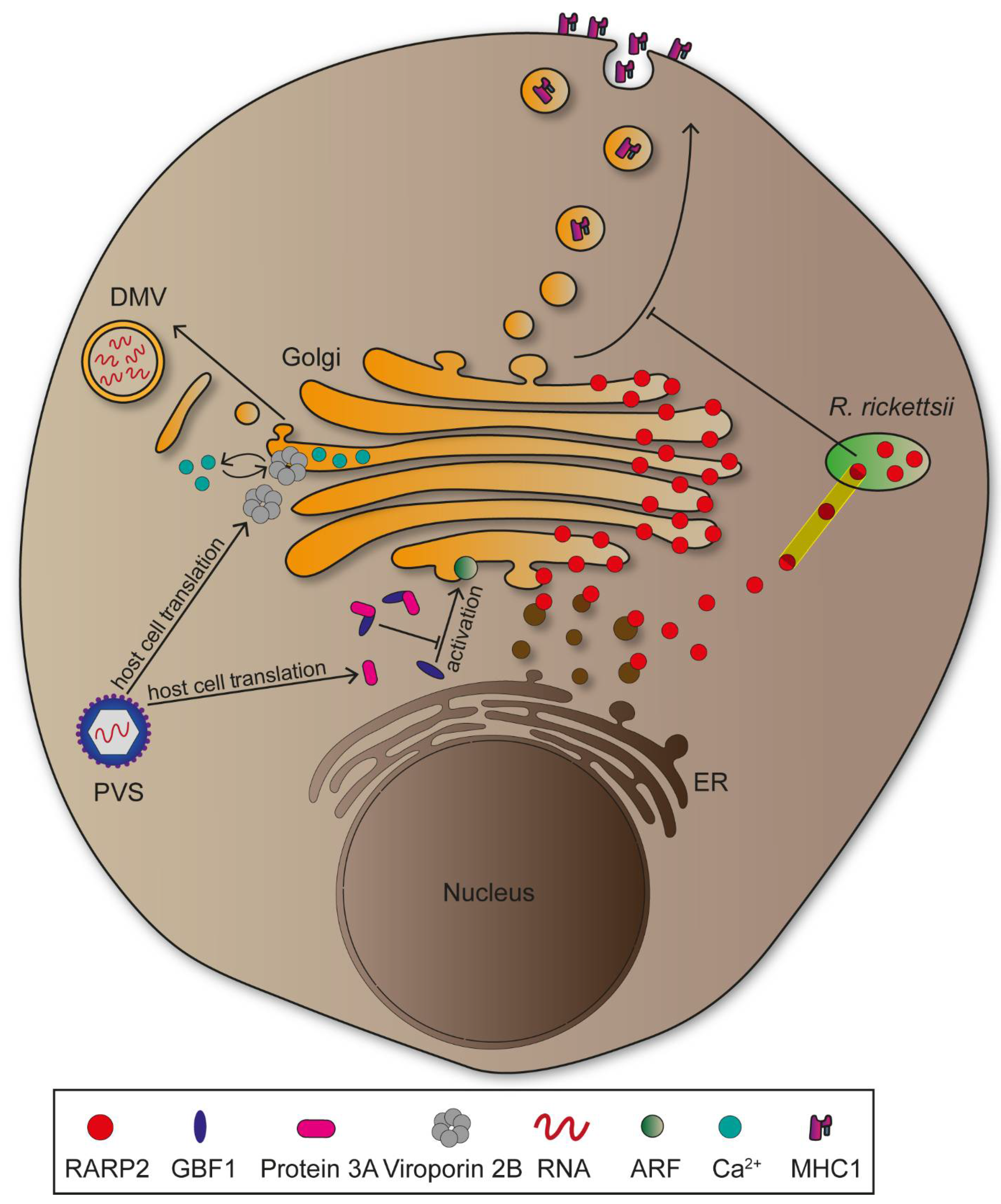
Rickettsia rickettsii is a Gram-negative obligate intracellular bacterium transmitted by ticks causing Rocky Mountain spotted fever, which can be lethal if left untreated [85]. The pathogen encodes a T4SS, as well as several effector proteins which are translocated into the cytosol subsequent to cell entry [86]. Rickettsial ankyrin repeat protein 2 (RARP2) is an effector protein mediating fragmentation of TGN, leading to attenuated vesicular transport and glycosylation defects in host cells (Figure 5) [87]. Studies suggest cysteine protease activity of RARP2 to be essential for TGN fragmentation, though possible target structures remain to be identified [9]. Two proteins are proposed to be affected by glycosylation defects during R. rickettsii infections: trans-Golgi protein TGN46 and major histocompatibility complex class 1 (MHC-I). MHC-I is transported to the plasma membrane, functions as antigen presenting complex, and this is crucial for innate and adaptive immune responses against intracellular pathogens [88,89,90]. Transport of MHC-I from TGN to plasma membrane might be impaired in R. rickettsii infections, providing protection of the pathogen against host cells innate immune responses, thus enabling pathogen proliferation (Figure 5) [9]. While the CGN is not impaired in infections by R. rickettsii, other intracellular bacteria such as Shigella disrupt the entire organelle to reduce processes maintaining integrity of the epithelial cell layer [91,92].
Disruption of the Golgi apparatus is not exclusively induced by bacterial pathogens, since numerous viruses cause similar effects. Positive-stranded RNA viruses such as Poliovirus (PVS) need to associate with, and rearrange host cell membranes for genome replication [93]. PVS is an Enterovirus of the Picornaviridae and causative agent of Poliomyelitis, a severe infectious disease that ranges from mild symptoms to heavy paralysis [94]. Following invasion, the PVS genome is translated into non-structural proteins that differ in function and create an intracellular niche for viral replication. Protein 2B is a viroporin involved in Golgi disruption, as its expression was followed by membrane fragmentation [95]. These membranes are consecutively rearranged into double-membrane vesicles which provide protection against host immune factors, as mentioned above (Figure 5) [96]. The viroporin might support Golgi fragmentation through integration into membranes where it influences Ca2+ signalling and promotes permeabilization [97]. Additionally, PVS is able to hinder vesicular transport between the ER and the Golgi, thus inhibiting the secretory pathway. This is due to protein 3A interacting and recruiting GBF1, a GEF for small GTPases ADP ribosylation factor (ARF), to viral vesicles (Figure 5) [98]. Delocalization of GBF1 might lead to lower activation of GTPases that are essential for maintaining the secretory pathway, ultimately leading to reduced translocation of immunity-related molecules such as MHC-I or interleukins [96]. Similar to PVS, HCV induces Golgi disruption to form double-membrane vesicles due to manipulation of GBF1. However, the GEF is not delocalized, but rather phosphorylated mediated by human immunity-related GTPase M (IRGM), that along with GBF1 and ARF, promotes Golgi fragmentation [99]. Taken together, GBF1 appears central for viral replication mechanisms and therefore represents a key target for further investigations [100].
Plasmodium spp. parasites cause malaria, leading to 228 million cases globally with 405,000 deaths in 2018 (WHO Malaria report). The parasites are transmitted by mosquitos and possess a complex life cycle with transition between sexual and asexual stages [101]. Humans are infected by sporozoites of the parasite that rapidly enter hepatocytes where they proliferate extensively [102]. Nutritional supply of PV is required so that Plasmodium spp. can effectively proliferate, suggesting the existence of host cell manipulation mechanisms. P. berghei showed associations of the PVM with Golgi membranes that were maintained throughout the proliferation stage in hepatocytes and followed by Golgi rearrangements to expand membrane interactions which are proposed to improve the parasites nutritional supply. The small GTPase RAB11 is crucial for the organelle’s morphological changes during P. berghei infections, as functional mutations diminished this effect [103]. RAB11 takes part in the translocation of trans-Golgi vesicles to cellular target membranes and therefore represents an important element of the secretory pathway [104]. T. gondii as well utilizes small GTPases of the Rab family for nutritional supply, indicating common mechanisms of manipulation of secretory pathway elements by intracellular pathogens to promote proliferation [105,106].
2.4. Mitochondria
The mitochondrion is a dynamic organelle likely originating from an endosymbiotic α-proteobacterium [107]. Basis for this endosymbiotic theory are similarities in key characteristics of mitochondria and Gram-negative bacteria, e. g. presence of two membranes. The outer membrane separates cell compartments from the cytoplasm and creates an intermembrane space through its distance to the inner membrane. The inner membrane forms structural dynamic cristae due to invaginations and encloses a sub-compartment called the mitochondrial matrix [108,109]. Another indicator for an evolutionary adaptation from an α-proteobacterium is presence of mitochondrial DNA (mtDNA) as circular genome, ribosomes and t-RNAs for autonomous protein translation and protein complexes that might be homologous to those of bacteria [110,111].
Over time, mitochondria acquired several important cellular functions such as synthesis of energy source adenosine triphosphate (ATP) through oxidative phosphorylation, being crucial as disruption or impairment can lead to severe diseases and death in higher eukaryotes [112,113]. As central coordination element for Ca2+-dependent pathways inducing intrinsic apoptosis, mitochondria play a key role in regulating programmed cell death. Contact sides to the Ca2+-storing ER for material exchange are therefore essential [108,114]. Furthermore, mitochondria release pro-apoptotic proteins such as cytochrome C into the cytosol, following activation of the intrinsic pathway supporting its importance in apoptosis [115,116]. Mitochondria also engage in initiation of immune signalling and take part in innate immune response by producing reactive oxygen species (ROS) that restrict pathogen proliferation and protect the cell from damage [117,118]. Like the ER, mitochondria have a response mechanism to lower mitochondrial stress created through accumulation of unfolded proteins in the matrix that contributes to the regulation of cellular homeostasis in stress situations overall [119]. As a highly dynamic organelle, mitochondria are able to undergo fission and fusion events depending on changing conditions in the cell. Mitochondrial fission is a necessity before cellular division, while fusion events take place when there is a low nutritional supply as it might contribute to maintain energy levels [120,121]. The multifunctional characteristic of the mitochondrion makes it a valuable target for intracellular pathogens. Studies have identified various manipulation mechanisms influencing mitochondria and support understanding pathogenesis of infectious diseases.
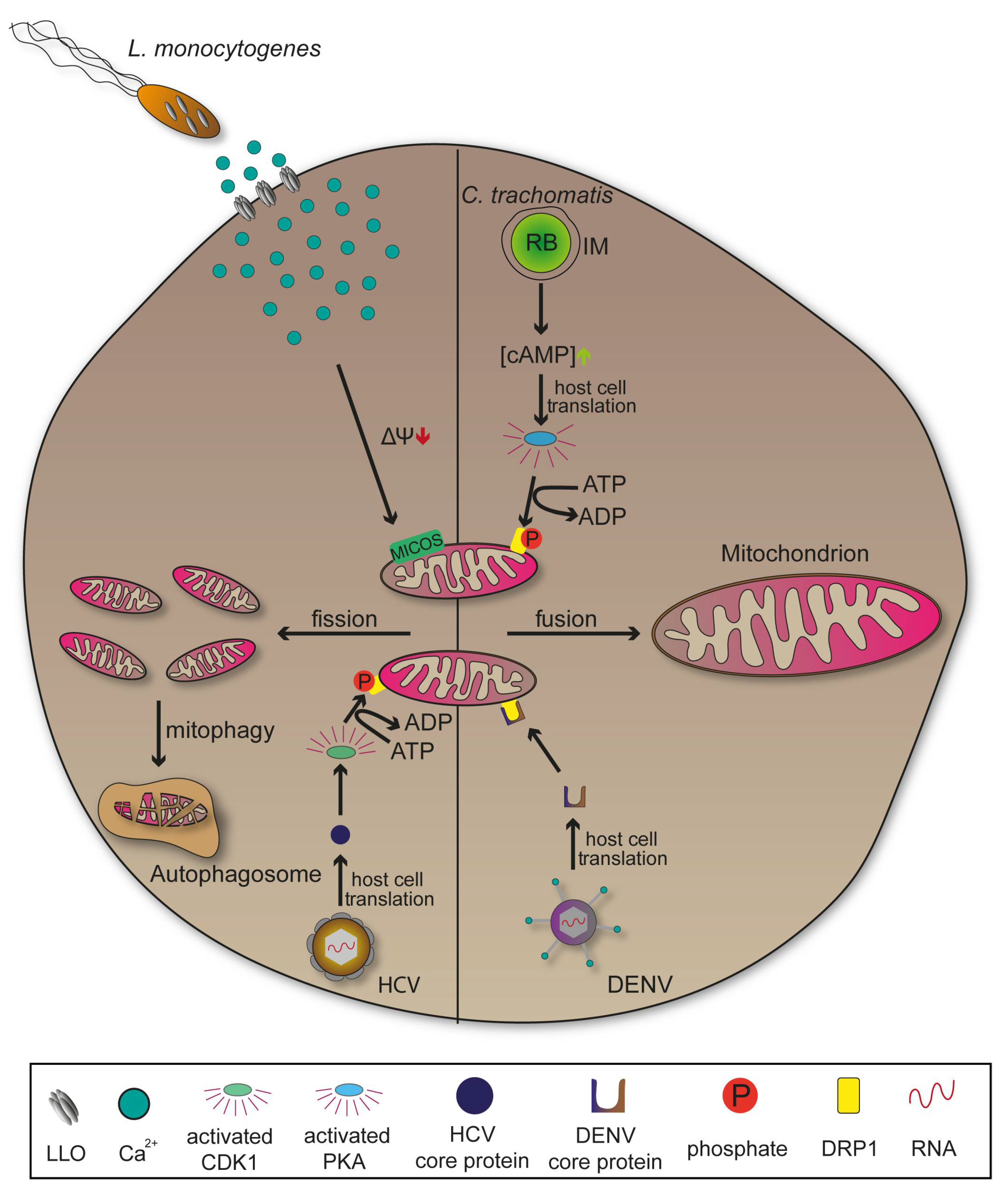
Chlamydia trachomatis is an obligate intracellular Gram-negative bacterium responsible for the blinding disease trachoma, as well as for sexually-transmitted diseases of the urogenital tract. It has a reduced genome that lacks important metabolic pathways, explaining the need for hosts to provide nutritional supply [122,123]. C. trachomatis differentiates from the infectious form elementary body (EB) to intracellular proliferating form reticulate body (RB). RBs are surrounded by an inclusion membrane (IM) whose associated membrane proteins mediate vesicle recruitment important to ensure continuous nutritional supply [57,124]. The pathogen is able to induce mitochondrial fusion and elongation following invasion. This process requires a phosphorylation of fission promoting dynamin-related protein 1 (DRP1) at serine residue 637 by cyclic AMP-dependent protein kinase (PKA) leading to expansion of the organelle and a higher production of ATP that favours bacterial replication (Figure 6) [10]. Later stages of infections with C. trachomatis showed a reversion of elongated mitochondria which might be due to a metabolic shift towards the rapid ATP yielding aerobic glycolysis or Warburg metabolism [125].
In contrast, several other pathogens such as Listeria monocytogenes or Shigella flexneri promote mitochondrial fission following invasion to undermine immune signalling, highlighting the diversity of manipulation strategies that have evolved (Figure 6) [11]. Pores formed by Listeriolysin (LLO) mediate release of L. monocytogenes from the PCV. However, LLO also interacts with the plasma membrane, induces Ca2+ influx that result in mitochondrial fragmentation [126]. Interactions with apoptotic pathways are an additional characteristic of infections with C. trachomatis. The bacterium secretes a protease that degrades pro-apoptotic proteins, thus inhibiting mitochondrial release of cytochrome C and the intrinsic apoptosis pathway. Cellular disintegration is prevented which provides the structural stability necessary for host-dependent pathogens [127,128]. Yet, there are intracellular bacteria like M. tuberculosis inducing the opposite effect, leading to host cell disruption for an extensive spread [129].
DRP1 is also target for viral pathogenesis revealing manipulation mechanisms affecting mitochondria similar to those of bacteria. In Dengue virus (DENV) infections, mitochondrial elongation through secretion of DRP1 activity-suppressing protein NS4B is used to increase contacts between the organelle and viral membranes. This might support the arrangement of convoluted membranes, ultimately promoting viral replication (Figure 6). DENV is a positive-strand RNA virus of the Flaviviridae, causing dengue fever. Additionally, DENV-induced innate immune response was attenuated when elongation of mitochondria occurred [130,131]. Comparable influence on mitochondrial morphology can be observed in viral infections with SARS-Coronaviruses (SARS-CoV) or Zika virus (ZIKV) [130,132]. In contrast to DENV, infections with HCV lead to mitochondrial fission and mitophagy due to a viral protein that activates cyclin-dependent kinase 1 (CDK1). CDK1 subsequently phosphorylates DRP1 at the serine residue 616 promoting its translocation to the mitochondria where it is responsible for fission events and consecutive mitophagy (Figure 6). This might promote viral persistence by restraining mitochondria-mediated apoptosis [133,134]. Many viruses, analogous to C. trachomatis, manipulate cellular metabolic pathways so that glucose is used to rapidly produce ATP by aerobic glycolysis instead of the prolonged oxidative phosphorylation presenting a method for immediate supply of energy [135].
In addition to ER, mitochondria are further organelles associated with the PVM in T. gondii-infected cells [70]. The parasites possess a mitochondrial association factor 1 (MAF1) locus that encodes for several proteins linked to host cell mitochondria association and immune evasion with MAF1b protein as central mediator [136]. Primary reason for association of T. gondii with host cell organelles is presumably the need for nutritional supply, allowing the PV to extend [67]. Pernas et al. [137] showed that mitochondrial morphology is indirectly affected during T. gondii infection on the basis of a competing counter-mechanism promoting mitochondrial fusion and attenuating uptake of fatty acids by the parasite.
2.5. Lipid Droplets
Lipid droplets (LDs) are highly dynamic, lipid-containing organelles that distribute throughout the whole cell performing various functions. LDs form at the ER bilayer where local enzymes synthesize non-charged, neutral lipids such as triglycerides or cholesterol esters whose accumulation between the leaflets of ER membrane leads to lens-like protrusions [138,139]. As consequence of organelle growth through continuous lipid synthesis, LDs bud from the ER membrane into the cytosol. This process requires the activity of fat storage-inducing transmembrane proteins (FIT) which, if functionally disabled, show defects in LD budding [140,141]. Cytosolic LDs structurally distinguish into a hydrophobic core including ER-synthesized neutral lipids, and a phospholipid monolayer that separates cargo from the cytosol [142]. Parallel to cellular distribution, LDs are able to dynamically expand due to lipid-synthesizing enzymes associated with the monolayer. Additionally, various other membrane proteins are located at LDs to mediate transport to, and interactions with organelles such as mitochondria, Golgi, lysosomes, or ER. These interactions are crucial for lipid transfer between both compartments and provide the possibility to counteract starvation-induced energy deprivation of cells as catabolic pathways can break down LD-derived lipids for energy supply [143,144]. Cell growth also requires LD-derived energy supply, as well as further lipids for membrane extensions [145]. LDs represent an important cellular compartment against lipotoxicity since they are dynamic organelles that can not only synthesize and deliver neutral lipids, but also mediate uptake. Likewise, LDs are proposed to temporally store hydrophobic proteins that might accumulate and induce stress-related signalling pathways, thus reducing intracellular stress [142,146]. There are several diseases related to malfunction of lipid storage such as the neutral fat storage disease or obesity, and diverse intracellular pathogens take advantage of LDs to survive and replicate inside host cells [147,148,149].
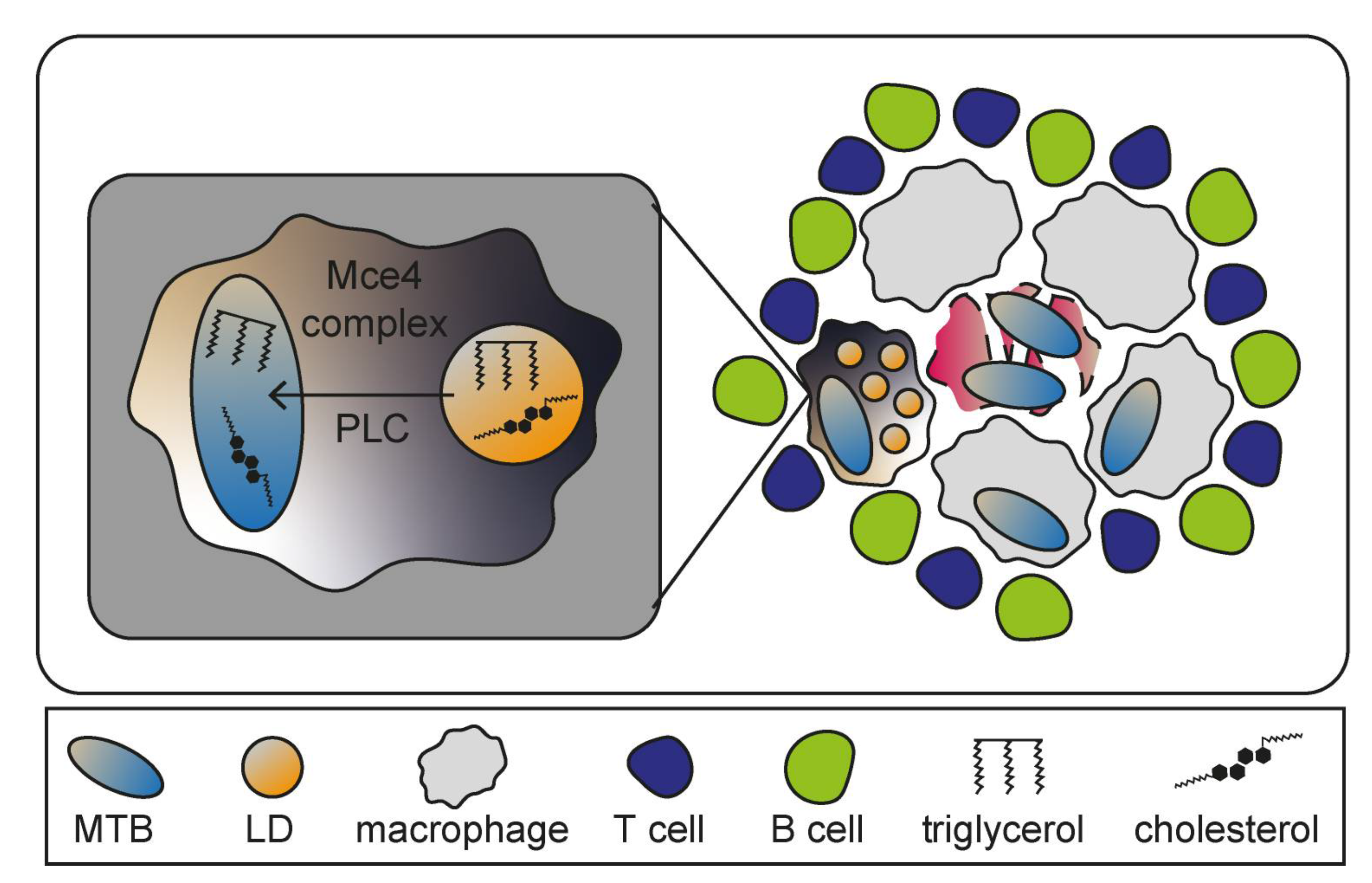
Mycobacterium tuberculosis (MTB) is a facultative intracellular pathogen and causative agent of the pulmonary disease tuberculosis (TB) which, if untreated, is often lethal. The bacteria mainly enter host cells through phagocytosis by alveolar macrophages. MTB is able to bypass lysosomal degradation and continuously resides in host cells without inducing strong immune responses, preventing the establishment of clinical symptoms [150,151]. Granuloma formation is the hallmark of TB pathogenesis and can be defined as an accumulation of various immune cells including differentiated and infected macrophages that create the inner core, as well as surrounding T- and B-lymphocytes (Figure 7) [152]. Granulomas can lead to severe tissue damage due to macrophages undergoing MTB-induced necrosis that might promote bacterial replication as necrosis liberates components utilized by MTB [153,154]. Although granulomas are temporally restricting bacterial spread, they also support MTB as protective niche [155]. One specific type of granuloma-associated macrophage acts as key element for TB pathogenesis: the foamy macrophage [156]. Those immune cells are implied to be generated through accumulations of LDs, whose biosynthesis is upregulated as result of peroxisome proliferator-activated receptor γ (PPARγ) expression and activation during MTB infections [157]. Several bacterial genes encode lipid-processing enzymes such as the lipid import system (Mce4) or phospholipase C (PLC), suggesting exploitation of LDs in foamy macrophages by MTB to ensure constant nutritional supply for persistence (Figure 8) [158].
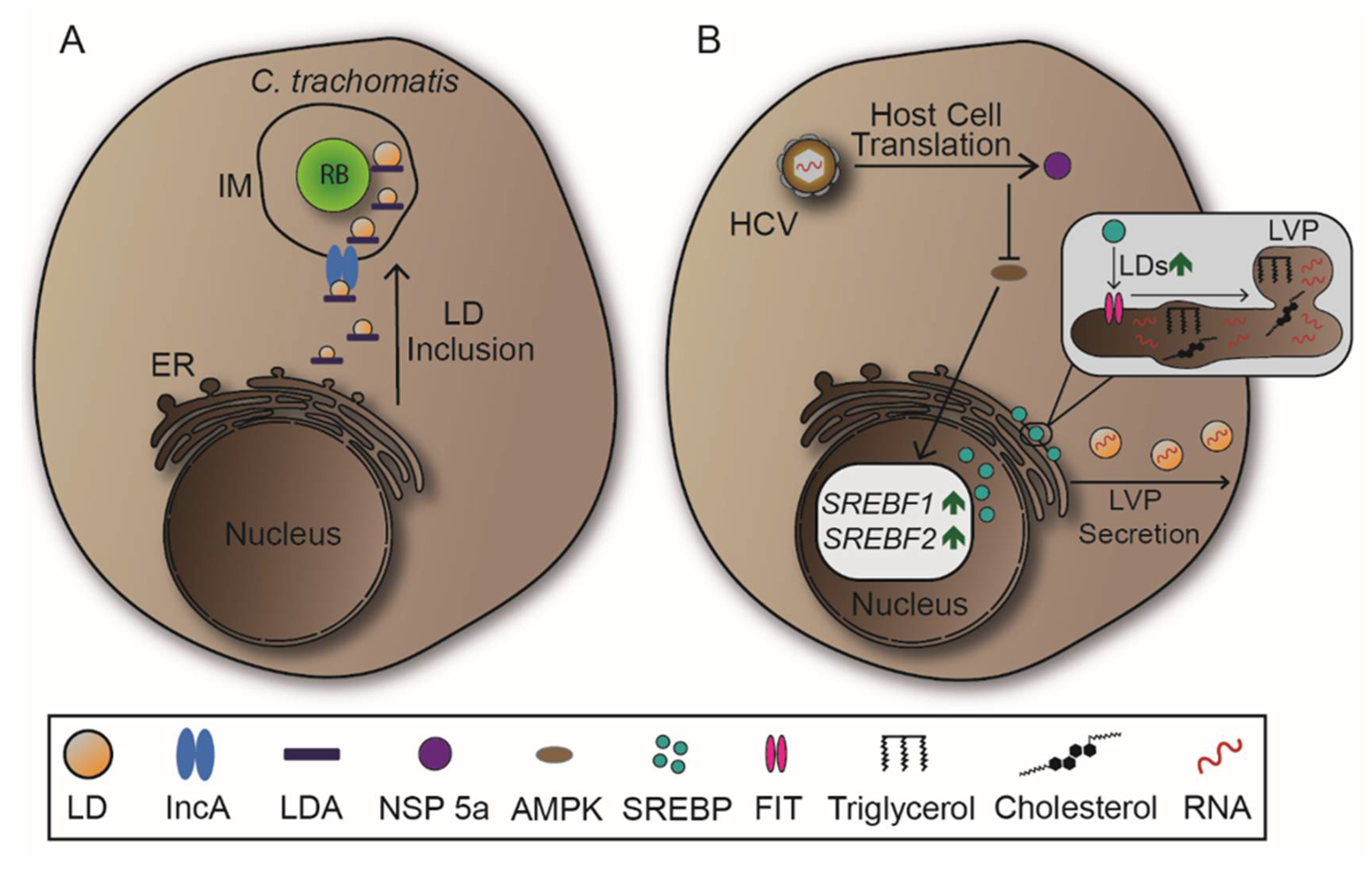
LDs are also manipulated by C. trachomatis. The bacteria redirect LDs to the IM and subsequently internalize the organelles, likely providing RBs with abundant lipid supply (Figure 8A) [12,159]. Inclusion membrane protein A (IncA), which is required for IM fusion events, is connected with LDs during C. trachomatis infections suggesting a pivotal role in LD recruitment [124,160]. There are LD-associated proteins (LDA) expressed by C. trachomatis, but their impact on LD manipulation has to be further investigated to understand their functionality [12,151].
The ER-associated transcription factor sterol regulator element-binding protein (SREBP) is essential for signalling pathways that induce lipid synthesis previous to LD biogenesis [161]. Following cell entry, HCV activates SREBP through various molecular mechanisms, resulting in increased lipid and LD synthesis. This benefits HCV as it is able to assemble replicative RNA in lipid-coated particles, or lipoviroparticles (LVPs) (Figure 8B) [162,163]. On the molecular level, HCV encodes NSPs which manipulate lipid homeostasis due to distinct interactions. NSP 5a shows to inhibit the phosphorylation and therefore the regulation activity of adenosine monophosphate-activated protein kinase (AMPK) leading to higher expressions of SREBP [164]. Furthermore, NSP 4b and core protein induce oxidative stress, resulting in the activation of phosphatidylinositol 3-kinase (PI3K), also increasing SREBP expression during HCV infection [163,165]. In DENV infection, association of DENV core protein with LDs is critical since mutations decrease viral replication [166]. These associations might provide a possible way of viral spread, as LDs can be secreted in an autophagy-mediated process. To support autophagy-mediated secretion of LDs, DENV upregulates LD biogenesis through activation of subtilisin kexin isozyme-1/site-1 protease (SKI-1/S1P), which cleaves a precursor of SREBP previous to its nuclear translocation [167,168]. In sum, SREBP is one of the main targets in viral infections that mandatorily require manipulation of lipid synthesis and LDs.
The PV harbouring T. gondii associates with host cell organelles for possible pathway manipulations and nutritional acquirements. The parasite might also recruit to PV and subsequently form contacts with LDs. Nolan et al. [169] showed growth reduction of T. gondii in mutant host cells depleted of LDs, as well as access to LD content by the parasite. Additionally, the authors confirmed the uptake of LDs into the PV, where lipids can be translocated into LDs of T. gondii, resulting in organelle expansion [169].
3. Conclusions
Mammalian organelles are targets of manipulations by intracellular pathogens. Each intracellular lifestyle features distinct strategies, ranging from simple destruction of an PCV to gain access to host cytosol, to sophisticated redirection of vesicular transport and fusion, resulting in novel compartments that serve as niches for pathogen persistence and proliferation. Recent studies revealed insight into the molecular mechanisms of manipulation, indicated common traits, but also raise questions regarding pathogenesis of diseases caused by many important pathogens. Regardless of similarities and differences, these manipulation mechanisms ultimately serve pathogen survival, replication, and spread. Comparing the mechanisms of manipulation between diverse intracellular pathogens targeting the same organelle could help to understand and therapeutically target recurring virulence patterns in intracellular lifestyles.
Author Contributions
M.K., F.S. and M.H. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Work in our group was supported by grants of DFG and BMBF.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thakur, A.; Mikkelsen, H.; Jungersen, G. Intracellular pathogens: Host immunity and microbial persistence strategies. J. Immunol. Res. 2019, 2019, 1356540. [Google Scholar] [CrossRef]
- Priyanka, B.; Patil, R.K.; Dwarakanath, S. A review on detection methods used for foodborne pathogens. Indian J. Med. Res. 2016, 144, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Cabezas Cruz, A.; Valdés, J.J.; de la Fuente, J. Control of vector-borne infectious diseases by human immunity against alpha-Gal. Expert Rev. Vaccines 2016, 15, 953–955. [Google Scholar] [CrossRef] [Green Version]
- Dbouk, T.; Drikakis, D. On respiratory droplets and face masks. Phys Fluids 2020, 32, 063303. [Google Scholar] [CrossRef]
- Galan, J.E. Common themes in the design and function of bacterial effectors. Cell Host Microbe 2009, 5, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velge, P.; Wiedemann, A.; Rosselin, M.; Abed, N.; Boumart, Z.; Chausse, A.M.; Grepinet, O.; Namdari, F.; Roche, S.M.; Rossignol, A.; et al. Multiplicity of Salmonella entry mechanisms, a new paradigm for Salmonella pathogenesis. Microbiologyopen 2012, 1, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.D.; Hueffer, K.; Wenk, M.R.; Galan, J.E. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 2004, 304, 1805–1807. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aistleitner, K.; Clark, T.; Dooley, C.; Hackstadt, T. Selective fragmentation of the trans-Golgi apparatus by Rickettsia rickettsii. PLoS Pathog. 2020, 16, e1008582. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Itoh, R.; Shimizu, A.; Walenna, N.F.; Chou, B.; Ishii, K.; Soejima, T.; Fujikane, A.; Hiromatsu, K. Chlamydia trachomatis targets mitochondrial dynamics to promote intracellular survival and proliferation. Cell Microbiol. 2019, 21, e12962. [Google Scholar] [CrossRef] [Green Version]
- Tiku, V.; Tan, M.W.; Dikic, I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Kumar, Y.; Cocchiaro, J.; Valdivia, R.H. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr. Biol. 2006, 16, 1646–1651. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A fundamental process in immunity. Biomed. Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [Green Version]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Jajere, S.M. A review of Salmonella enterica with particular focus on the pathogenicity and virulence factors, host specificity and antimicrobial resistance including multidrug resistance. Vet World 2019, 12, 504–521. [Google Scholar] [CrossRef] [Green Version]
- Lorkowski, M.; Felipe-Lopez, A.; Danzer, C.A.; Hansmeier, N.; Hensel, M. Salmonella enterica invasion of polarized epithelial cells is a highly cooperative effort. Infect. Immun. 2014, 82, 2657–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liss, V.; Swart, A.L.; Kehl, A.; Hermanns, N.; Zhang, Y.; Chikkaballi, D.; Bohles, N.; Deiwick, J.; Hensel, M. Salmonella enterica remodels the host cell endosomal system for efficient intravacuolar nutrition. Cell Host Microbe 2017, 21, 390–402. [Google Scholar] [CrossRef] [Green Version]
- Bakowski, M.A.; Braun, V.; Lam, G.Y.; Yeung, T.; Heo, W.D.; Meyer, T.; Finlay, B.B.; Grinstein, S.; Brumell, J.H. The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe 2010, 7, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaughnessy, L.M.; Hoppe, A.D.; Christensen, K.A.; Swanson, J.A. Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cell Microbiol. 2006, 8, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, T.; Heit, B.; Dubuisson, J.F.; Fairn, G.D.; Chiu, B.; Inman, R.; Kapus, A.; Swanson, M.; Grinstein, S. Contribution of phosphatidylserine to membrane surface charge and protein targeting during phagosome maturation. J. Cell Biol. 2009, 185, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Creasey, E.A.; Isberg, R.R. Maintenance of vacuole integrity by bacterial pathogens. Curr. Opin. Microbiol. 2014, 17, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, M.B.; Estrada-Garcia, T. Shigella: A Highly Virulent and Elusive Pathogen. Curr. Trop. Med. Rep. 2014, 1, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Knuff, K.; Finlay, B.B. What the SIF Is Happening-The Role of Intracellular Salmonella-Induced Filaments. Front. Cell Infect. Microbiol. 2017, 7, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, A.; Boucrot, E.; Drevensek, S.; Daire, V.; Gorvel, J.P.; Pous, C.; Holden, D.W.; Meresse, S. SKIP, the host target of the Salmonella virulence factor SifA, promotes kinesin-1-dependent vacuolar membrane exchanges. Traffic 2010, 11, 899–911. [Google Scholar] [CrossRef]
- Ohlson, M.B.; Huang, Z.; Alto, N.M.; Blanc, M.P.; Dixon, J.E.; Chai, J.; Miller, S.I. Structure and function of Salmonella SifA indicate that its interactions with SKIP, SseJ, and RhoA family GTPases induce endosomal tubulation. Cell Host Microbe 2008, 4, 434–446. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Wall, D.M.; Castillo, A.; McCormick, B.A. Caspase-3 cleavage of Salmonella type III secreted effector protein SifA is required for localization of functional domains and bacterial dissemination. Gut Microbes 2019, 10, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Henry, T.; Couillault, C.; Rockenfeller, P.; Boucrot, E.; Dumont, A.; Schroeder, N.; Hermant, A.; Knodler, L.A.; Lecine, P.; Steele-Mortimer, O.; et al. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. USA 2006, 103, 13497–13502. [Google Scholar] [CrossRef] [Green Version]
- Beuzon, C.R.; Meresse, S.; Unsworth, K.E.; Ruiz-Albert, J.; Garvis, S.; Waterman, S.R.; Ryder, T.A.; Boucrot, E.; Holden, D.W. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 2000, 19, 3235–3249. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Helenius, A. Virus entry at a glance. J. Cell Sci. 2013, 126, 1289–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 226–249. [Google Scholar] [CrossRef] [Green Version]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Staring, J.; Raaben, M.; Brummelkamp, T.R. Viral escape from endosomes and host detection at a glance. J Cell Sci. 2018, 131, jcs216259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butan, C.; Filman, D.J.; Hogle, J.M. Cryo-electron microscopy reconstruction shows poliovirus 135S particles poised for membrane interaction and RNA release. J. Virol. 2014, 88, 1758–1770. [Google Scholar] [CrossRef] [Green Version]
- Mainou, B.A.; Dermody, T.S. Transport to late endosomes is required for efficient reovirus infection. J. Virol. 2012, 86, 8346–8358. [Google Scholar] [CrossRef] [Green Version]
- Forrest, J.C.; Dermody, T.S. Reovirus receptors and pathogenesis. J. Virol. 2003, 77, 9109–9115. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A. Epstein-Barr Virus Exploits the Secretory Pathway to Release Virions. Microorganisms 2020, 8, 729. [Google Scholar] [CrossRef]
- Weiss, L.M.; O’Malley, D. Benign lymphadenopathies. Mod. Pathol. 2013, 26, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rout, M.P.; Aitchison, J.D. The nuclear pore complex as a transport machine. J. Biol. Chem. 2001, 276, 16593–16596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, M.A.; Hetzer, M.W. The role of the nuclear envelope in cellular organization. Cell. Mol. Life Sci. 2006, 63, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Terasaki, M.; Shemesh, T.; Kasthuri, N.; Klemm, R.W.; Schalek, R.; Hayworth, K.J.; Hand, A.R.; Yankova, M.; Huber, G.; Lichtman, J.W.; et al. Stacked endoplasmic reticulum sheets are connected by helicoidal membrane motifs. Cell 2013, 154, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Shemesh, T.; Prinz, W.A.; Palazzo, A.F.; Kozlov, M.M.; Rapoport, T.A. Mechanisms determining the morphology of the peripheral ER. Cell 2010, 143, 774–788. [Google Scholar] [CrossRef] [Green Version]
- English, A.R.; Zurek, N.; Voeltz, G.K. Peripheral ER structure and function. Curr. Opin. Cell Biol. 2009, 21, 596–602. [Google Scholar] [CrossRef] [Green Version]
- Gurel, P.S.; Hatch, A.L.; Higgs, H.N. Connecting the cytoskeleton to the endoplasmic reticulum and Golgi. Curr. Biol. 2014, 24, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celli, J.; Tsolis, R.M. Bacteria, the endoplasmic reticulum and the unfolded protein response: Friends or foes? Nat. Rev. Microbiol. 2015, 13, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, M.S.; Isberg, R.R. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 1995, 63, 3609–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhavsar, A.P.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef]
- Omotade, T.O.; Roy, C.R. Manipulation of Host Cell Organelles by Intracellular Pathogens. Microbiol. Spectr. 2019, 7, 179–196. [Google Scholar] [CrossRef]
- Celli, J.; de Chastellier, C.; Franchini, D.M.; Pizarro-Cerda, J.; Moreno, E.; Gorvel, J.P. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 2003, 198, 545–556. [Google Scholar] [CrossRef]
- Elwell, C.; Mirrashidi, K.; Engel, J. Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 2016, 14, 385–400. [Google Scholar] [CrossRef]
- Dorer, M.S.; Kirton, D.; Bader, J.S.; Isberg, R.R. RNA interference analysis of Legionella in Drosophila cells: Exploitation of early secretory apparatus dynamics. PLoS Pathog. 2006, 2, e34. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Roy, C.R. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 2002, 4, 945–954. [Google Scholar] [CrossRef]
- Hubber, A.; Roy, C.R. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 2010, 26, 261–283. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Ding, C.; Sun, Y. Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections. Int. J. Mol. Sci. 2020, 21, 3689. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Strunze, S.; Engelke, M.F.; Wang, I.H.; Puntener, D.; Boucke, K.; Schleich, S.; Way, M.; Schoenenberger, P.; Burckhardt, C.J.; Greber, U.F. Kinesin-1-mediated capsid disassembly and disruption of the nuclear pore complex promote virus infection. Cell Host Microbe 2011, 10, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Tsai, B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 2011, 7, e1002037. [Google Scholar] [CrossRef] [Green Version]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio 2013, 4, e00801-13. [Google Scholar] [CrossRef] [Green Version]
- Sinai, A.P.; Webster, P.; Joiner, K.A. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: A high affinity interaction. J. Cell Sci. 1997, 110, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Nishi, M.; Hu, K.; Murray, J.M.; Roos, D.S. Organellar dynamics during the cell cycle of Toxoplasma gondii. J. Cell Sci. 2008, 121, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Augusto, L.; Martynowicz, J.; Amin, P.H.; Alakhras, N.S.; Kaplan, M.H.; Wek, R.C.; Sullivan, W.J., Jr. Toxoplasma gondii Co-opts the Unfolded Protein Response to Enhance Migration and Dissemination of Infected Host Cells. mBio 2020, 11, e00915-20. [Google Scholar] [CrossRef] [PubMed]
- Laliberte, J.; Carruthers, V.B. Host cell manipulation by the human pathogen Toxoplasma gondii. Cell. Mol. Life Sci. 2008, 65, 1900–1915. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Ahn, H.J.; Ryu, K.J.; Nam, H.W. Interaction between parasitophorous vacuolar membrane-associated GRA3 and calcium modulating ligand of host cell endoplasmic reticulum in the parasitism of Toxoplasma gondii. Korean J. Parasitol. 2008, 46, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.H.; Seemann, J. Unraveling the Golgi ribbon. Traffic 2010, 11, 1391–1400. [Google Scholar] [CrossRef]
- Thyberg, J.; Moskalewski, S. Role of microtubules in the organization of the Golgi complex. Exp. Cell Res. 1999, 246, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.H.; Seemann, J. Golgi ribbon disassembly during mitosis, differentiation and disease progression. Curr. Opin. Cell Biol. 2017, 47, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Mochizuki, A. Golgi apparatus self-organizes into the characteristic shape via postmitotic reassembly dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, 5177–5182. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wang, Y. Golgi structure formation, function, and post-translational modifications in mammalian cells. F1000Research 2017, 6, 2050. [Google Scholar] [CrossRef]
- Guizzunti, G.; Seemann, J. Mitotic Golgi disassembly is required for bipolar spindle formation and mitotic progression. Proc. Natl. Acad. Sci. USA 2016, 113, 6590–6599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colanzi, A.; Corda, D. Mitosis controls the Golgi and the Golgi controls mitosis. Curr. Opin. Cell Biol. 2007, 19, 386–393. [Google Scholar] [CrossRef]
- Glick, B.S.; Nakano, A. Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 2009, 25, 113–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bexiga, M.G.; Simpson, J.C. Human diseases associated with form and function of the Golgi complex. Int. J. Mol. Sci. 2013, 14, 18670–18681. [Google Scholar] [CrossRef] [PubMed]
- Brandizzi, F.; Barlowe, C. Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 2013, 14, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Crump, C.M.; Thomas, G. Trans-Golgi network sorting. Cell. Mol. Life Sci. 2001, 58, 1067–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Yang, Y.; Zhou, R.; Gong, T. Golgi apparatus: An Emerging Platform for Innate Immunity. Trends Cell Biol. 2020, 30, 467–477. [Google Scholar] [CrossRef]
- Traeger, M.S.; Regan, J.J.; Humpherys, D.; Mahoney, D.L.; Martinez, M.; Emerson, G.L.; Tack, D.M.; Geissler, A.; Yasmin, S.; Lawson, R.; et al. Rocky mountain spotted fever characterization and comparison to similar illnesses in a highly endemic area-Arizona, 2002-2011. Clin. Infect. Dis. 2015, 60, 1650–1658. [Google Scholar] [CrossRef]
- Clark, T.R.; Noriea, N.F.; Bublitz, D.C.; Ellison, D.W.; Martens, C.; Lutter, E.I.; Hackstadt, T. Comparative genome sequencing of Rickettsia rickettsii strains that differ in virulence. Infect. Immun. 2015, 83, 1568–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, S.S.; Noriea, N.F.; Aistleitner, K.; Clark, T.R.; Dooley, C.A.; Nair, V.; Kaur, S.J.; Rahman, M.S.; Gillespie, J.J.; Azad, A.F.; et al. The Rickettsial Ankyrin Repeat Protein 2 Is a Type IV Secreted Effector That Associates with the Endoplasmic Reticulum. mBio 2018, 9, e00975-18. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Alvaro-Benito, M.; Stolzenberg, S.; Noe, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.H.; Olano, J.P.; Feng, H.M. Critical role of cytotoxic T lymphocytes in immune clearance of rickettsial infection. Infect. Immun. 2001, 69, 1841–1846. [Google Scholar] [CrossRef] [Green Version]
- Sahni, S.K.; Narra, H.P.; Sahni, A.; Walker, D.H. Recent molecular insights into rickettsial pathogenesis and immunity. Future Microbiol. 2013, 8, 1265–1288. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, M.L.; Malarde, V.; Grassart, A.; Salavessa, L.; Nigro, G.; Descorps-Declere, S.; Rohde, J.R.; Schnupf, P.; Masson, V.; Arras, G.; et al. Shigella promotes major alteration of gut epithelial physiology and tissue invasion by shutting off host intracellular transport. Proc. Natl. Acad. Sci. USA 2019, 116, 13582–13591. [Google Scholar] [CrossRef] [Green Version]
- Mounier, J.; Boncompain, G.; Senerovic, L.; Lagache, T.; Chretien, F.; Perez, F.; Kolbe, M.; Olivo-Marin, J.C.; Sansonetti, P.J.; Sauvonnet, N. Shigella effector IpaB-induced cholesterol relocation disrupts the Golgi complex and recycling network to inhibit host cell secretion. Cell Host Microbe 2012, 12, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Oh, C.; Kim, Y.; Chang, K.O. Proteases facilitate the endosomal escape of porcine epidemic diarrhea virus during entry into host cells. Virus Res. 2019, 272, 197730. [Google Scholar] [CrossRef] [PubMed]
- Mehndiratta, M.M.; Mehndiratta, P.; Pande, R. Poliomyelitis: Historical facts, epidemiology, and current challenges in eradication. Neurohospitalist 2014, 4, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, I.V.; Carrasco, L. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro-090179. J. Virol. 1997, 71, 4679–4693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Gil, L.; Bano-Polo, M.; Redondo, N.; Sanchez-Martinez, S.; Nieva, J.L.; Carrasco, L.; Mingarro, I. Membrane integration of poliovirus 2B viroporin. J. Virol. 2011, 85, 11315–11324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teterina, N.L.; Pinto, Y.; Weaver, J.D.; Jensen, K.S.; Ehrenfeld, E. Analysis of poliovirus protein 3A interactions with viral and cellular proteins in infected cells. J. Virol. 2011, 85, 4284–4296. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, 3462–3471. [Google Scholar] [CrossRef] [Green Version]
- Ferlin, J.; Farhat, R.; Belouzard, S.; Cocquerel, L.; Bertin, A.; Hober, D.; Dubuisson, J.; Rouille, Y. Investigation of the role of GBF1 in the replication of positive-sense single-stranded RNA viruses. J. Gen. Virol. 2018, 99, 1086–1096. [Google Scholar] [CrossRef]
- Talapko, J.; Skrlec, I.; Alebic, T.; Jukic, M.; Vcev, A. Malaria: The past and the present. Microorganisms 2019, 7, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudencio, M.; Rodriguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006, 4, 849–856. [Google Scholar] [CrossRef]
- De Niz, M.; Kaiser, G.; Zuber, B.; Do Heo, W.; Heussler, V.T.; Agop-Nersesian, C. Hijacking of the host cell Golgi by Plasmodium berghei liver stage parasites. J. Cell Sci. 2020, 15. [Google Scholar] [CrossRef]
- Welz, T.; Wellbourne-Wood, J.; Kerkhoff, E. Orchestration of cell surface proteins by Rab11. Trends Cell Biol. 2014, 24, 407–415. [Google Scholar] [CrossRef]
- Romano, J.D.; Nolan, S.J.; Porter, C.; Ehrenman, K.; Hartman, E.J.; Hsia, R.C.; Coppens, I. The parasite Toxoplasma sequesters diverse Rab host vesicles within an intravacuolar network. J. Cell Biol. 2017, 216, 4235–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, K.; Chehade, S.; Werkmeister, E.; Barois, N.; Periz, J.; Lafont, F.; Tardieux, I.; Khalife, J.; Langsley, G.; Meissner, M.; et al. Rab11A regulates dense granule transport and secretion during Toxoplasma gondii invasion of host cells and parasite replication. PLoS Pathog. 2020, 16, e1008106. [Google Scholar] [CrossRef]
- Margulis, L. Recombination of non-chromosomal genes in Chlamydomonas: Assortment of mitochondria and chloroplasts? J. Theor. Biol. 1970, 26, 337–342. [Google Scholar] [CrossRef]
- Fielden, L.F.; Kang, Y.; Newton, H.J.; Stojanovski, D. Targeting mitochondria: How intravacuolar bacterial pathogens manipulate mitochondria. Cell Tissue Res. 2017, 367, 141–154. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Kuhlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharczyk, R.; Zick, M.; Bietenhader, M.; Rak, M.; Couplan, E.; Blondel, M.; Caubet, S.D.; di Rago, J.P. Mitochondrial ATP synthase disorders: Molecular mechanisms and the quest for curative therapeutic approaches. Biochim. Biophys. Acta 2009, 1793, 186–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell 2003, 112, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Siegl, C.; Prusty, B.K.; Karunakaran, K.; Wischhusen, J.; Rudel, T. Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Rep. 2014, 9, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Rosas-Lemus, M.; Patel, D.; Fang, X.; Tuz, K.; Juarez, O. Dynamic energy dependency of Chlamydia trachomatis on host cell metabolism during intracellular growth: Role of sodium-based energetics in chlamydial ATP generation. J. Biol. Chem. 2018, 293, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocchiaro, J.L.; Kumar, Y.; Fischer, E.R.; Hackstadt, T.; Valdivia, R.H. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. USA 2008, 105, 9379–9384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rother, M.; Gonzalez, E.; da Costa, A.R.T.; Wask, L.; Gravenstein, I.; Pardo, M.; Pietzke, M.; Gurumurthy, R.K.; Angermann, J.; Laudeley, R.; et al. Combined Human Genome-wide RNAi and Metabolite Analyses Identify IMPDH as a Host-Directed Target against Chlamydia Infection. Cell Host Microbe 2018, 23, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef] [Green Version]
- Pirbhai, M.; Dong, F.; Zhong, Y.; Pan, K.Z.; Zhong, G. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 2006, 281, 31495–31501. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.F.; Vier, J.; Kirschnek, S.; Klos, A.; Hess, S.; Ying, S.; Hacker, G. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J. Exp. Med. 2004, 200, 905–916. [Google Scholar] [CrossRef]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, M.L.; Parker, M.L.; Ramaswamy, R.; Powell, C.J.; English, E.D.; Adomako-Ankomah, Y.; Pernas, L.F.; Workman, S.D.; Boothroyd, J.C.; Boulanger, M.J.; et al. A Toxoplasma gondii locus required for the direct manipulation of host mitochondria has maintained multiple ancestral functions. Mol. Microbiol. 2018, 108, 519–535. [Google Scholar] [CrossRef] [Green Version]
- Pernas, L.; Bean, C.; Boothroyd, J.C.; Scorrano, L. Mitochondria Restrict Growth of the Intracellular Parasite Toxoplasma gondii by Limiting Its Uptake of Fatty Acids. Cell Metab. 2018, 27, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid droplet biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Choudhary, V.; Ojha, N.; Golden, A.; Prinz, W.A. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J. Cell Biol. 2015, 211, 261–271. [Google Scholar] [CrossRef]
- Kadereit, B.; Kumar, P.; Wang, W.J.; Miranda, D.; Snapp, E.L.; Severina, N.; Torregroza, I.; Evans, T.; Silver, D.L. Evolutionarily conserved gene family important for fat storage. Proc. Natl. Acad. Sci. USA 2008, 105, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, T.; Parton, R.G. Not just fat: The structure and function of the lipid droplet. Cold Spring Harb. Perspect. Biol. 2011, 3, a004838. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, A.D.; Siniossoglou, S. Function of lipid droplet-organelle interactions in lipid homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1459–1468. [Google Scholar] [CrossRef]
- Henne, W.M.; Reese, M.L.; Goodman, J.M. The assembly of lipid droplets and their roles in challenged cells. EMBO J. 2018, 37, e98947. [Google Scholar] [CrossRef]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003. [Google Scholar] [CrossRef] [Green Version]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Emre, S.D. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Pennisi, E.M.; Arca, M.; Bertini, E.; Bruno, C.; Cassandrini, D.; D’Amico, A.; Garibaldi, M.; Gragnani, F.; Maggi, L.; Massa, R.; et al. Neutral Lipid Storage Diseases: Clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J. R. Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herker, E.; Ott, M. Emerging role of lipid droplets in host/pathogen interactions. J. Biol. Chem. 2012, 287, 2280–2287. [Google Scholar] [CrossRef] [Green Version]
- Delogu, G.; Sali, M.; Fadda, G. The biology of Mycobacterium tuberculosis infection. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013070. [Google Scholar] [CrossRef] [Green Version]
- Libbing, C.L.; McDevitt, A.R.; Azcueta, R.P.; Ahila, A.; Mulye, M. Lipid Droplets: A Significant but Understudied Contributor of Host(-)Bacterial Interactions. Cells 2019, 8, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirado, E.; Schlesinger, L.S. Modeling the Mycobacterium tuberculosis Granuloma - the Critical Battlefield in Host Immunity and Disease. Front. Immunol. 2013, 4, 98. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.W.; Jacobs, W.R., Jr. Postprimary Tuberculosis and Macrophage Necrosis: Is There a Big ConNECtion? mBio 2016, 7, e01589-15. [Google Scholar] [CrossRef] [Green Version]
- Pagan, A.J.; Ramakrishnan, L. The Formation and Function of Granulomas. Annu. Rev. Immunol. 2018, 36, 639–665. [Google Scholar] [CrossRef]
- Ehlers, S.; Schaible, U.E. The granuloma in tuberculosis: Dynamics of a host-pathogen collusion. Front. Immunol. 2012, 3, 411. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G.; Cardona, P.J.; Kim, M.J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Salamon, H.; Bruiners, N.; Lakehal, K.; Shi, L.; Ravi, J.; Yamaguchi, K.D.; Pine, R.; Gennaro, M.L. Cutting edge: Vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J. Immunol. 2014, 193, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef] [PubMed]
- Saka, H.A.; Thompson, J.W.; Chen, Y.S.; Dubois, L.G.; Haas, J.T.; Moseley, A.; Valdivia, R.H. Chlamydia trachomatis infection leads to defined alterations to the lipid droplet proteome in epithelial cells. PLoS ONE 2015, 10, e0124630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.M.; Noriea, N.F.; Bauler, L.D.; Lam, J.L.; Sager, J.; Wesolowski, J.; Paumet, F.; Hackstadt, T. A functional core of IncA is required for Chlamydia trachomatis inclusion fusion. J. Bacteriol. 2016, 198, 1347–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Ogawa, K.; Hishiki, T.; Shimizu, Y.; Funami, K.; Sugiyama, K.; Miyanari, Y.; Shimotohno, K. Hepatitis C virus utilizes lipid droplet for production of infectious virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloherty, A.P.M.; Olmstead, A.D.; Ribeiro, C.M.S.; Jean, F. Hijacking of Lipid Droplets by Hepatitis C, Dengue and Zika Viruses-From Viral Protein Moonlighting to Extracellular Release. Int. J. Mol. Sci. 2020, 21, 7901. [Google Scholar] [CrossRef]
- Meng, Z.; Liu, Q.; Sun, F.; Qiao, L. Hepatitis C virus nonstructural protein 5A perturbs lipid metabolism by modulating AMPK/SREBP-1c signaling. Lipids Health Dis. 2019, 18, 191. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Jun, H.J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar] [CrossRef] [Green Version]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assuncao-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe 2018, 23, 819–831. [Google Scholar] [CrossRef] [Green Version]
- Hyrina, A.; Meng, F.; McArthur, S.J.; Eivemark, S.; Nabi, I.R.; Jean, F. Human Subtilisin Kexin Isozyme-1 (SKI-1)/Site-1 Protease (S1P) regulates cytoplasmic lipid droplet abundance: A potential target for indirect-acting anti-dengue virus agents. PLoS ONE 2017, 12, e0174483. [Google Scholar] [CrossRef]
- Nolan, S.J.; Romano, J.D.; Coppens, I. Host lipid droplets: An important source of lipids salvaged by the intracellular parasite Toxoplasma gondii. PLoS Pathog. 2017, 13, e1006362. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Membrane-bound organelles as targets of manipulation by intracellular pathogens. To initiate infection, intracellular pathogens manipulate the plasma membrane (PM) to enter host cells, or are phagocytosed. Subsequently, intracellular pathogens manipulate endosomes and phagosomes to either escape into cytoplasm, or for transport to further intracellular targets such as the endoplasmic reticulum (ER), the Golgi, mitochondria, or lipid droplets (LDs). The ER is mainly targeted by viral pathogens as they can exploit host cell ribosomes for protein synthesis. Manipulations targeting the Golgi often include disruptions of the secretory pathway. Mitochondria, as energy-generating organelles, are exploited to either decrease or increase cellular energy levels, with both processes promoting proliferation of distinct pathogens. LDs are mainly targeted by pathogens to utilize their lipid content as nutritional supply. However, viruses are able to utilize the vesicles for cellular spread.
Figure 1.
Membrane-bound organelles as targets of manipulation by intracellular pathogens. To initiate infection, intracellular pathogens manipulate the plasma membrane (PM) to enter host cells, or are phagocytosed. Subsequently, intracellular pathogens manipulate endosomes and phagosomes to either escape into cytoplasm, or for transport to further intracellular targets such as the endoplasmic reticulum (ER), the Golgi, mitochondria, or lipid droplets (LDs). The ER is mainly targeted by viral pathogens as they can exploit host cell ribosomes for protein synthesis. Manipulations targeting the Golgi often include disruptions of the secretory pathway. Mitochondria, as energy-generating organelles, are exploited to either decrease or increase cellular energy levels, with both processes promoting proliferation of distinct pathogens. LDs are mainly targeted by pathogens to utilize their lipid content as nutritional supply. However, viruses are able to utilize the vesicles for cellular spread.

Figure 2.
Manipulations of host cell endosomal system by intracellular Salmonella enterica. Once internalized, the bacteria rearrange their vesicular compartments to bypass lysosomal degradation. Type III secretion system (T3SS)-secreted effector protein SopB mediates dephosphorylation of membrane lipid phosphatidylinositol-4,5-bisphospate (PI(4,5)P2) generating phosphatidylinositol 4-phosphate (PI4P). Further infection progress shows secretion of effector protein SifA, which associates with the Salmonella-containing vacuole (SCV) membrane and leads to Salmonella-induced filaments (SIF) formation along microtubules through interactions with SifA-Kinesin-Interacting-Protein (SKIP). SIFs provide nutritional supply through interception of vesicle containing endocytosed cargo such as proteins or lipids resulting in S. enterica replication.
Figure 2.
Manipulations of host cell endosomal system by intracellular Salmonella enterica. Once internalized, the bacteria rearrange their vesicular compartments to bypass lysosomal degradation. Type III secretion system (T3SS)-secreted effector protein SopB mediates dephosphorylation of membrane lipid phosphatidylinositol-4,5-bisphospate (PI(4,5)P2) generating phosphatidylinositol 4-phosphate (PI4P). Further infection progress shows secretion of effector protein SifA, which associates with the Salmonella-containing vacuole (SCV) membrane and leads to Salmonella-induced filaments (SIF) formation along microtubules through interactions with SifA-Kinesin-Interacting-Protein (SKIP). SIFs provide nutritional supply through interception of vesicle containing endocytosed cargo such as proteins or lipids resulting in S. enterica replication.

Figure 3.
Manipulation of endoplasmic reticulum (ER) by intracellular pathogens. Legionella pneumophila recruits small GTPase RAB1 to the Legionella-containing vacuole (LCV) membrane through T3SS-secreted effector protein DrrA resulting in redirection of vesicular transport between the ER and the Golgi. L. pneumophila thereby ensures nutritional supply for bacterial replication. Brome mosaic virus (BMV) and severe acute respiratory syndrome coronavirus (SARS) exploit the host cell protein machinery to produce viral proteins. BMVs viral protein 1a induces invaginations of ER membranes ultimately creating vesicular spherules suitable for viral replication. SARS non-structural proteins interact with ER membranes and lead to the formation of double membrane ER vesicles (DMV) that promote viral replication. T. gondii associates with the ER membrane through interaction protein GRA3 and the ER receptor calcium-modulating ligand (CAMLG) which might support manipulations by T. gondii such as inhibition of the unfolded protein response (UPR).
Figure 3.
Manipulation of endoplasmic reticulum (ER) by intracellular pathogens. Legionella pneumophila recruits small GTPase RAB1 to the Legionella-containing vacuole (LCV) membrane through T3SS-secreted effector protein DrrA resulting in redirection of vesicular transport between the ER and the Golgi. L. pneumophila thereby ensures nutritional supply for bacterial replication. Brome mosaic virus (BMV) and severe acute respiratory syndrome coronavirus (SARS) exploit the host cell protein machinery to produce viral proteins. BMVs viral protein 1a induces invaginations of ER membranes ultimately creating vesicular spherules suitable for viral replication. SARS non-structural proteins interact with ER membranes and lead to the formation of double membrane ER vesicles (DMV) that promote viral replication. T. gondii associates with the ER membrane through interaction protein GRA3 and the ER receptor calcium-modulating ligand (CAMLG) which might support manipulations by T. gondii such as inhibition of the unfolded protein response (UPR).

Figure 5.
Manipulation of the Golgi by intracellular pathogens. Rickettsia rickettsii inhibits transport of major histocompatibility complex 1 (MHC-I) proteins to the plasma membrane through T4SS-translocated effector protein RARP2. RARP2 binds to endoplasmic reticulum (ER)-derived vesicles and relocates to the Golgi after vesicular fusion. It promotes attenuation of trans-Golgi transport by hydrolysing cysteines belonging to a yet unknown protein. Poliovirus (PVS) exploits host cell protein machineries to synthesize Viroporin 2B and Protein 3A. Viroporin 2B forms a pore in Golgi membranes, proposed to result in Ca2+ signalling manipulation that promotes double-membrane vesicle (DMV) formation on the basis of Golgi vesicles. Protein 3A relocates the nucleotide exchange factor (NEF) GBF1 which is inhibiting ADP ribosylation factor (ARF) activation on Golgi membranes. Consequently, ER to Golgi transport is inhibited.
Figure 5.
Manipulation of the Golgi by intracellular pathogens. Rickettsia rickettsii inhibits transport of major histocompatibility complex 1 (MHC-I) proteins to the plasma membrane through T4SS-translocated effector protein RARP2. RARP2 binds to endoplasmic reticulum (ER)-derived vesicles and relocates to the Golgi after vesicular fusion. It promotes attenuation of trans-Golgi transport by hydrolysing cysteines belonging to a yet unknown protein. Poliovirus (PVS) exploits host cell protein machineries to synthesize Viroporin 2B and Protein 3A. Viroporin 2B forms a pore in Golgi membranes, proposed to result in Ca2+ signalling manipulation that promotes double-membrane vesicle (DMV) formation on the basis of Golgi vesicles. Protein 3A relocates the nucleotide exchange factor (NEF) GBF1 which is inhibiting ADP ribosylation factor (ARF) activation on Golgi membranes. Consequently, ER to Golgi transport is inhibited.

Figure 6.
Manipulation of mitochondria by intracellular pathogens. Listeria monocytogenes secretes Listeriolysin O (LLO) that forms pores in the host cell plasma membrane leading to Ca2+ influx. Subsequent decrease in mitochondrial membrane potential and mitochondrial contact site and cristae organizing system (MICOS) functionality are elemental for mitochondrial fission. C. trachomatis exploits the host cell translation machinery to synthesize cyclic adenosine monophosphate (cAMP) enriching proteins whose consequences activate cyclic AMP-dependent protein kinase (PKA). PKA specifically phosphorylates dynamin-related protein 1 (DRP1) to inhibit its function resulting in elongated mitochondria. Dengue virus (DENV) as well inhibits DRP1 function. Hepatitis C virus (HCV) activates the DRP1 phosphorylating cyclin-dependent kinase 1 (CDK1) through core proteins ultimately promoting mitochondrial fission and mitophagy.
Figure 6.
Manipulation of mitochondria by intracellular pathogens. Listeria monocytogenes secretes Listeriolysin O (LLO) that forms pores in the host cell plasma membrane leading to Ca2+ influx. Subsequent decrease in mitochondrial membrane potential and mitochondrial contact site and cristae organizing system (MICOS) functionality are elemental for mitochondrial fission. C. trachomatis exploits the host cell translation machinery to synthesize cyclic adenosine monophosphate (cAMP) enriching proteins whose consequences activate cyclic AMP-dependent protein kinase (PKA). PKA specifically phosphorylates dynamin-related protein 1 (DRP1) to inhibit its function resulting in elongated mitochondria. Dengue virus (DENV) as well inhibits DRP1 function. Hepatitis C virus (HCV) activates the DRP1 phosphorylating cyclin-dependent kinase 1 (CDK1) through core proteins ultimately promoting mitochondrial fission and mitophagy.

Figure 7.
Granuloma formation induced by Mycobacterium tuberculosis (MTB). After phagocytosis, MTB mediates the recruitment of diverse macrophages that serve as protection against lymphocytes such as B cells and T cells. This accumulations of immune cells as consequence of MTB infection are termed granuloma. Foamy macrophages have a specialized morphology due to high amounts of lipid droplets (LDs). MTB exploits these LDs for nutritional supply with the help of lipid import systems such as Mce4 complex and phospholipase C (PLC). Severe tissue damage can be observed as result of MTB-induced necrosis of macrophages.
Figure 7.
Granuloma formation induced by Mycobacterium tuberculosis (MTB). After phagocytosis, MTB mediates the recruitment of diverse macrophages that serve as protection against lymphocytes such as B cells and T cells. This accumulations of immune cells as consequence of MTB infection are termed granuloma. Foamy macrophages have a specialized morphology due to high amounts of lipid droplets (LDs). MTB exploits these LDs for nutritional supply with the help of lipid import systems such as Mce4 complex and phospholipase C (PLC). Severe tissue damage can be observed as result of MTB-induced necrosis of macrophages.

Figure 8.
Manipulation of lipid droplets (LDs) by Chlamydia trachomatis and Hepatitis C virus (HCV). (A) C. trachomatis redirects LDs originating from ER to the inclusion membrane through expression of lipid droplets-associated proteins (LDA) and inclusion membrane protein A (IncA). IncA mediates homotypic fusions at the inclusion membrane (IM) and is suggested to mediate the same effect on LDs. Specific LDA functions are yet to be unravel. (B) HCV exploits host cell translation machineries to synthesize non-structural protein (NSP) 5a which inhibits phosphorylation and therefore activation of regulatory enzyme adenosine monophosphate-activated protein kinase (AMPK). Subsequently, expression of genes encoding lipid metabolism regulator sterol regulator element-binding protein (SREBP) are increased, leading to higher biogenesis of LDs. HCV uses LDs to distribute its RNA throughout the organism as lipoviroparticles (LVPs) that can be secreted by cells to promote viral spread.
Figure 8.
Manipulation of lipid droplets (LDs) by Chlamydia trachomatis and Hepatitis C virus (HCV). (A) C. trachomatis redirects LDs originating from ER to the inclusion membrane through expression of lipid droplets-associated proteins (LDA) and inclusion membrane protein A (IncA). IncA mediates homotypic fusions at the inclusion membrane (IM) and is suggested to mediate the same effect on LDs. Specific LDA functions are yet to be unravel. (B) HCV exploits host cell translation machineries to synthesize non-structural protein (NSP) 5a which inhibits phosphorylation and therefore activation of regulatory enzyme adenosine monophosphate-activated protein kinase (AMPK). Subsequently, expression of genes encoding lipid metabolism regulator sterol regulator element-binding protein (SREBP) are increased, leading to higher biogenesis of LDs. HCV uses LDs to distribute its RNA throughout the organism as lipoviroparticles (LVPs) that can be secreted by cells to promote viral spread.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kellermann, M.; Scharte, F.; Hensel, M. Manipulation of Host Cell Organelles by Intracellular Pathogens. Int. J. Mol. Sci. 2021, 22, 6484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126484
AMA Style
Kellermann M, Scharte F, Hensel M. Manipulation of Host Cell Organelles by Intracellular Pathogens. International Journal of Molecular Sciences. 2021; 22(12):6484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126484
Chicago/Turabian StyleKellermann, Malte, Felix Scharte, and Michael Hensel. 2021. "Manipulation of Host Cell Organelles by Intracellular Pathogens" International Journal of Molecular Sciences 22, no. 12: 6484. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126484
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.