
Transcriptional Stress Induces Chromatin Relocation of the Nucleotide Excision Repair Factor XPG

 , ,
, ,
Abstract
:1. Introduction
2. Results
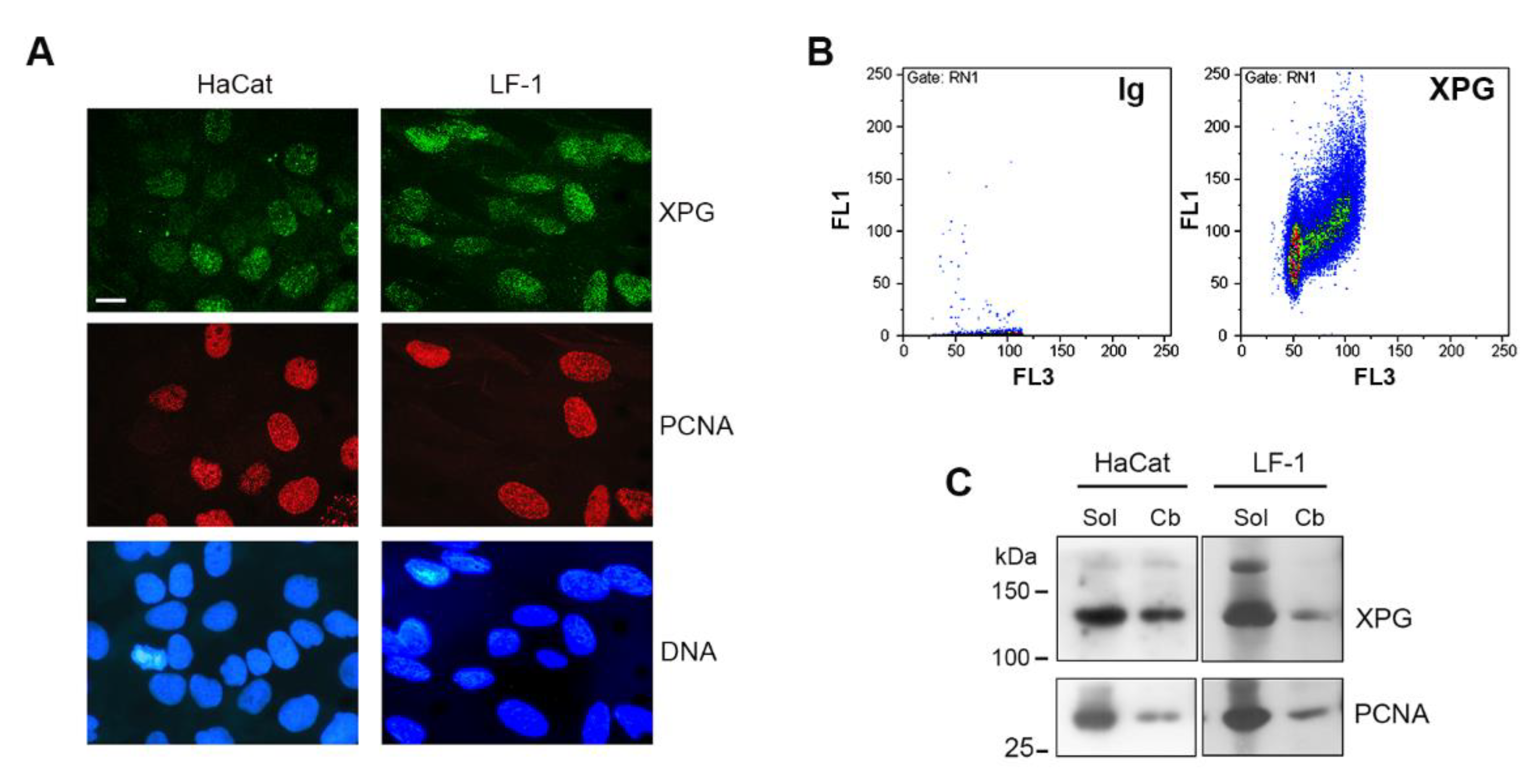
2.1. Chromatin Association of XPG
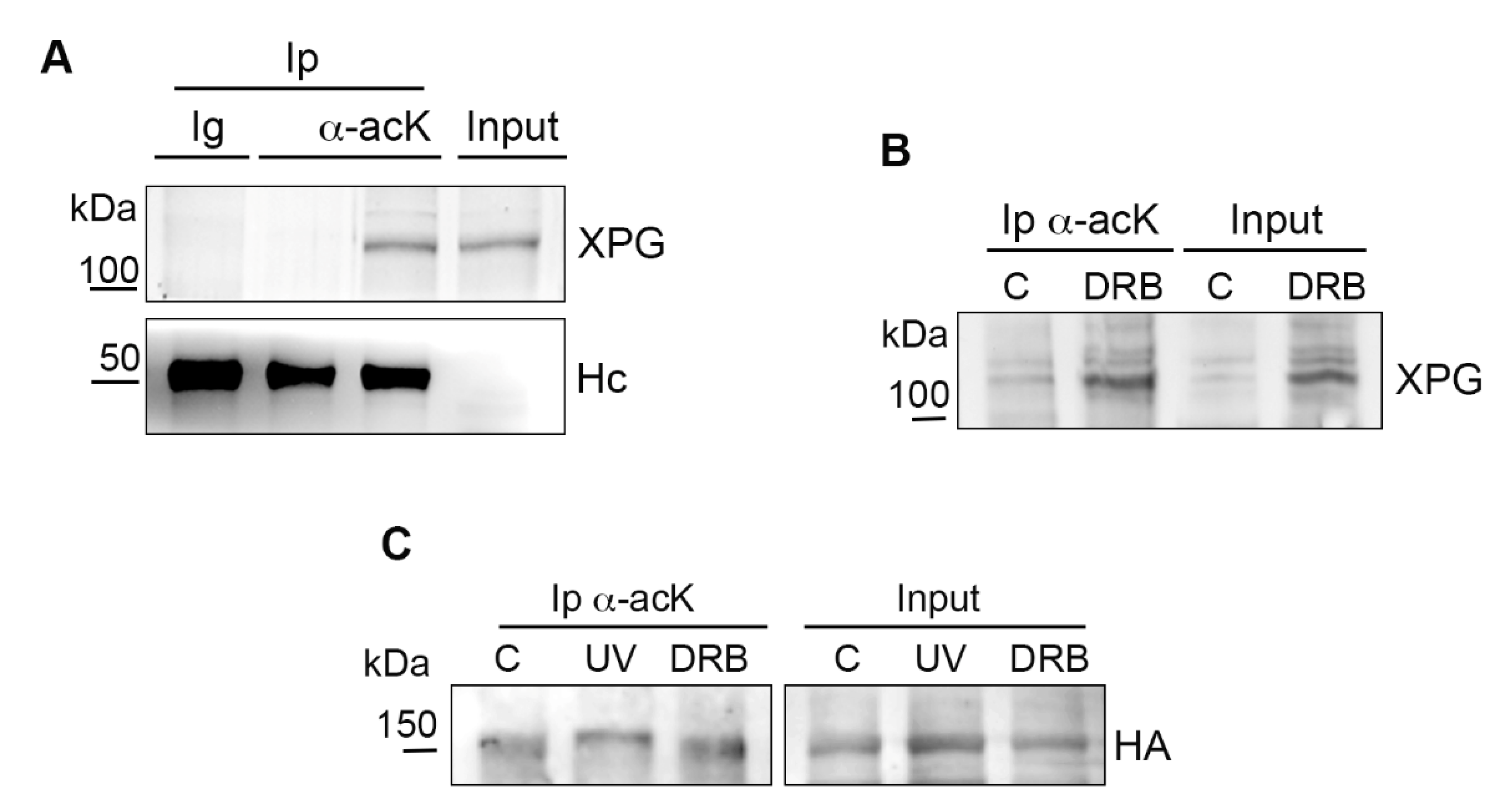
2.2. Acetylation of Chromatin-Bound XPG
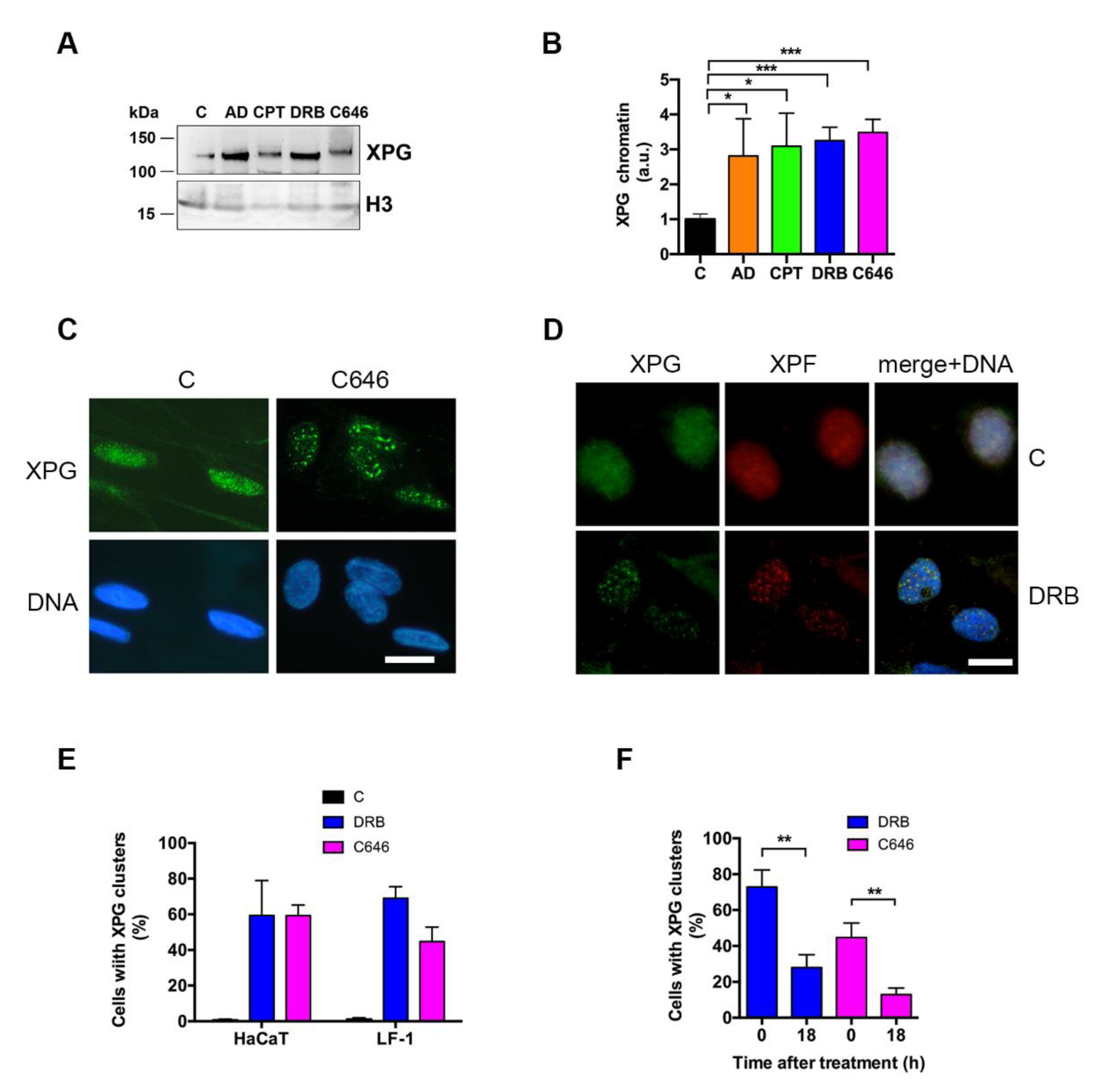
2.3. Inhibition of Transcription Induces XPG Accumulation and Relocation
2.4. XPG Relocation Occurs in Concomitance with Histone γ-H2AX and R-Loop Formation
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. Flow Cytometry
4.3. Western Blot and Immunoprecipitation
4.4. Immunofluorescence
4.5. Proximity Ligation Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarkson, S. The XPG story. Biochimie 2003, 85, 1113–1121. [Google Scholar] [CrossRef]
- Schärer, O.D. XPG: Its products and biological roles. Adv. Exp. Med. Biol. 2009, 637, 83–92. [Google Scholar] [CrossRef]
- O’Donovan, A.; Davies, A.A.; Moggs, J.G.; West, S.; Wood, R.D. XPG endonuclease makes the 3′ incision in human DNA nucleotide excision repair. Nat. Cell Biol. 1994, 371, 432–435. [Google Scholar] [CrossRef]
- Evans, E.; Fellows, J.; Coffer, A.; Wood, R.D. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J. 1997, 16, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009, 28, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Fagbemi, A.F.; Orelli, B.; Schärer, O.D. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair 2011, 10, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Marteijn, J.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Lehmann, A.R. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003, 85, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T. Nucleotide excision repair and neurological diseases. DNA Repair 2008, 7, 1155–1167. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Bohr, V.A.; Sander, M.; Kraemer, K.H. Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: Molecules to patients. Mech. Ageing Dev. 2011, 132, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-K.; Yu, S.-L.; Prakash, L.; Prakash, S. Requirement of Yeast RAD2, a Homolog of Human XPG Gene, for Efficient RNA Polymerase II Transcription: Implications for Cockayne Syndrome. Cell 2002, 109, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Kuraoka, I.; Chymkowitch, P.; Compe, E.; Takedachi, A.; Ishigami, C.; Coin, F.; Egly, J.-M.; Tanaka, K. XPG Stabilizes TFIIH, Allowing Transactivation of Nuclear Receptors: Implications for Cockayne Syndrome in XP-G/CS Patients. Mol. Cell 2007, 26, 231–243. [Google Scholar] [CrossRef]
- Le May, N.; Mota-Fernandes, D.; Velez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER Factors Are Recruited to Active Promoters and Facilitate Chromatin Modification for Transcription in the Absence of Exogenous Genotoxic Attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Le May, N.; Fradin, D.; Iltis, I.; Bougnères, P.; Egly, J.-M. XPG and XPF Endonucleases Trigger Chromatin Looping and DNA Demethylation for Accurate Expression of Activated Genes. Mol. Cell 2012, 47, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Trego, K.S.; Chernikova, S.B.; Davalos, A.R.; Perry, J.J.P.; Finger, L.D.; Ng, C.; Tsai, M.-S.; Yannone, S.M.; Tainer, J.A.; Campisi, J.; et al. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle 2011, 10, 1998–2007. [Google Scholar] [CrossRef] [Green Version]
- Trego, K.S.; Groesser, T.; Davalos, A.R.; Parplys, A.C.; Zhao, W.; Nelson, M.R.; Hlaing, A.; Shih, B.; Rydberg, B.; Pluth, J.M.; et al. Non-catalytic roles for XPG with BRCA1 and BRCA2 in homologous recombination and genome stability. Mol. Cell 2016, 61, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickramasinghe, V.O.; Venkitaraman, A.R. RNA Processing and Genome Stability: Cause and Consequence. Mol. Cell 2016, 61, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Cristini, A.; Ricci, G.; Britton, S.; Salimbeni, S.; Huang, S.-Y.N.; Marinello, J.; Calsou, P.; Pommier, Y.; Favre, G.; Capranico, G.; et al. Dual Processing of R-Loops and Topoisomerase I Induces Transcription-Dependent DNA Double-Strand Breaks. Cell Rep. 2019, 28, 3167–3181.e6. [Google Scholar] [CrossRef] [Green Version]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11. [Google Scholar] [CrossRef] [Green Version]
- Arauújo, S.J.; Nigg, E.A.; Wood, R.D. Strong Functional Interactions of TFIIH with XPC and XPG in Human DNA Nucleotide Excision Repair, without a Preassembled Repairosome. Mol. Cell. Biol. 2001, 21, 2281–2291. [Google Scholar] [CrossRef] [Green Version]
- Thorel, F.; Constantinou, A.; Dunand-Sauthier, I.; Nouspikel, T.; Lalle, P.; Raams, A.; Jaspers, N.G.J.; Vermeulen, W.; Shivji, M.K.K.; Wood, R.D.; et al. Definition of a Short Region of XPG Necessary for TFIIH Interaction and Stable Recruitment to Sites of UV Damage. Mol. Cell. Biol. 2004, 24, 10670–10680. [Google Scholar] [CrossRef] [Green Version]
- Mocquet, V.; Lainé, J.P.; Riedl, T.; Yajin, Z.; Lee, M.Y.; Egly, J.M. Sequential recruitment of the repair factors during NER: The role of XPG in initiating the resynthesis step. EMBO J. 2007, 27, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Eyboulet, F.; Cibot, C.; Eychenne, T.; Neil, H.; Alibert, O.; Werner, M.; Soutourina, J. Mediator links transcription and DNA repair by facilitating Rad2/XPG recruitment. Genes Dev. 2013, 27, 2549–2562. [Google Scholar] [CrossRef] [Green Version]
- Puumalainen, M.-R.; Lessel, D.; Rüthemann, P.; Kaczmarek, N.; Bachmann, K.; Ramadan, K.; Naegeli, H. Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Van Cuijk, L.; Van Belle, G.J.; Turkyilmaz, Y.; Poulsen, S.L.; Janssens, R.C.; Theil, A.F.; Sabatella, M.; Lans, H.; Mailand, N.; Houtsmuller, A.B.; et al. SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat. Commun. 2015, 6, 7499. [Google Scholar] [CrossRef] [Green Version]
- Tillhon, M.; Cazzalini, O.; Nardo, T.; Necchi, D.; Sommatis, S.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair 2012, 11, 844–852. [Google Scholar] [CrossRef]
- Han, C.; Wani, G.; Zhao, R.; Qian, J.; Sharma, N.; He, J.; Zhu, Q.; Wang, Q.-E.; A Wani, A. Cdt2-mediated XPG degradation promotes gap-filling DNA synthesis in nucleotide excision repair. Cell Cycle 2015, 14, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual Ligand Screening of the p300/CBP Histone Acetyltransferase: Identification of a Selective Small Molecule Inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Scovassi, A.I.; Prosperi, E. Analysis of Proliferating Cell Nuclear Antigen (PCNA) Associated with DNA Excision Repair Sites in Mammalian Cells. In DNA Repair Protocols; Methods in Molecular Biology; Henderson, D.S., Ed.; Humana Press: Totowa, NJ, USA, 2006; Volume 314, pp. 457–475. [Google Scholar]
- Cazzalini, O.; Sommatis, S.; Tillhon, M.; Dutto, I.; Bachi, A.; Rapp, A.; Nardo, T.; Scovassi, A.I.; Necchi, D.; Cardoso, M.C.; et al. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 2014, 42, 8433–8448. [Google Scholar] [CrossRef] [Green Version]
- Dutto, I.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. An improved method for the detection of nucleotide excision repair factors at local UV DNA damage sites. DNA Repair 2017, 51, 79–84. [Google Scholar] [CrossRef]
- Paunovic, J.; Vucevic, D.; Radosavljevic, T.; Djurdjevic, B.V.; Stankovic, S.; Pantic, I. Effects of Iron Oxide Nanoparticles on Structural Organization of Hepatocyte Chromatin: Gray Level Co-occurrence Matrix Analysis. Microsc. Microanal. 2021, 1–8. [Google Scholar] [CrossRef]
- Dancy, B.M.; Cole, P.A. Protein Lysine Acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Shav-Tal, Y.; Blechman, J.; Darzacq, X.; Montagna, C.; Dye, B.T.; Patton, J.G.; Singer, R.H.; Zipori, D. Dynamic Sorting of Nuclear Components into Distinct Nucleolar Caps during Transcriptional Inhibition. Mol. Biol. Cell 2005, 16, 2395–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mischo, H.E.; Hemmerich, P.; Grosse, F.; Zhang, S. Actinomycin D induces histone gamma-H2AX foci and complex formation of gamma-H2AX with Ku70 and nuclear DNA helicase II. J. Biol. Chem. 2005, 280, 9586–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, T.; Takemura, H.; Liao, Z.-Y.; Aune, G.; Redon, C.; Sedelnikova, O.A.; Pilch, D.R.; Rogakou, E.P.; Celeste, A.; Chen, H.T.; et al. Phosphorylation of Histone H2AX and Activation of Mre11, Rad50, and Nbs1 in Response to Replication-dependent DNA Double-strand Breaks Induced by Mammalian DNA Topoisomerase I Cleavage Complexes. J. Biol. Chem. 2003, 278, 20303–20312. [Google Scholar] [CrossRef] [Green Version]
- Bensaude, O. Inhibiting eukaryotic transcription. Which compound to choose? How to evaluate its activity? Transcription 2011, 2, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.; Laiho, M. A Targeting Modality for Destruction of RNA Polymerase I that Possesses Anticancer Activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Wienholz, F.; Vermeulen, W.; Marteijn, J.A. Amplification of unscheduled DNA synthesis signal enables fluorescence-based single cell quantification of transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2017, 45, e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance | ||||
|---|---|---|---|---|
| GLCM Feature | Sample | d = 3 | d = 6 | d = 9 |
| Angular second moment | siControl | 0.02100 ± 0.001474 | 0.01375 ± 0.001066 | 0.01073 ± 0.0008634 |
| sip300/CBP | 0.003390 ± 0.0009269 **** | 0.002188 ± 0.0006927 **** | 0.001645 ± 0.0005484 **** | |
| Correlation | siControl | 0.03145 ± 0.002600 | 0.02413 ± 0.001858 | 0.01782 ± 0.001343 |
| sip300/CBP | 0.0004503 ± 1.790 × 10−5 **** | 0.0004200 ± 1.565 × 10−5 **** | 0.0003847 ± 1.319 × 10−5 **** | |
| Inverse difference moment | siControl | 0.5568 ± 0.01724 | 0.4205 ± 0.01650 | 0.3492 ± 0.01563 |
| sip300/CBP | 0.2367 ± 0.01459 **** | 0.1446 ± 0.01191 **** | 0.1036 ± 0.009709 **** | |
| Entropy | siControl | 4.839 ± 0.1121 | 5.331 ± 0.1188 | 5.566 ± 0.1207 |
| sip300/CBP | 7.364 ± 0.1223 **** | 7.909 ± 0.1215 **** | 8.171 ± 0.1185 **** | |
| Distance | ||||
|---|---|---|---|---|
| GLCM Feature | Sample | d = 3 | d = 6 | d = 9 |
| Angular second moment | Control | 0.01605 ± 0.0009294 | 0.009992 ± 0.0006226 | 0.007657 ± 0.0004850 |
| DRB | 0.01261 ± 0.0007157 ** | 0.007838 ± 0.0005743 * | 0.006063 ± 0.0004919 * | |
| C646 | 0.01288 ± 0.0006540 ** | 0.007909 ± 0.0004422 ** | 0.006070 ± 0.0003523 ** | |
| Correlation | Control | 0.03624 ± 0.002843 | 0.02972 ± 0.002082 | 0.02411 ± 0.001495 |
| DRB | 0.02652 ± 0.001212 *** | 0.02175 ± 0.0009079 *** | 0.01731 ± 0.0006764 **** | |
| C646 | 0.01612 ± 0.0009309 **** | 0.01340 ± 0.0007202 **** | 0.01073 ± 0.0005686 **** | |
| Inverse difference moment | Control | 0.6188 ± 0.007871 | 0.4613 ± 0.008658 | 0.3737 ± 0.008313 |
| DRB | 0.5411 ± 0.009005 **** | 0.3875 ± 0.008952 **** | 0.3106 ± 0.008126 **** | |
| C646 | 0.5361 ± 0.009026 **** | 0.3875 ± 0.009044 **** | 0.3115 ± 0.008228 **** | |
| Entropy | Control | 4.561 ± 0.04921 | 5.049 ± 0.05104 | 5.309 ± 0.05207 |
| DRB | 4.953 ± 0.03865 **** | 5.463 ± 0.03992 **** | 5.719 ± 0.03912 **** | |
| C646 | 5.041 ± 0.04941 **** | 5.541 ± 0.05167 **** | 5.794 ± 0.05214 **** | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalera, C.; Ticli, G.; Dutto, I.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Transcriptional Stress Induces Chromatin Relocation of the Nucleotide Excision Repair Factor XPG. Int. J. Mol. Sci. 2021, 22, 6589. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126589
Scalera C, Ticli G, Dutto I, Cazzalini O, Stivala LA, Prosperi E. Transcriptional Stress Induces Chromatin Relocation of the Nucleotide Excision Repair Factor XPG. International Journal of Molecular Sciences. 2021; 22(12):6589. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126589
Chicago/Turabian StyleScalera, Claudia, Giulio Ticli, Ilaria Dutto, Ornella Cazzalini, Lucia A. Stivala, and Ennio Prosperi. 2021. "Transcriptional Stress Induces Chromatin Relocation of the Nucleotide Excision Repair Factor XPG" International Journal of Molecular Sciences 22, no. 12: 6589. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126589