
Targeting Mitochondria in Diabetes

 ,
,  , ,
, ,
Abstract
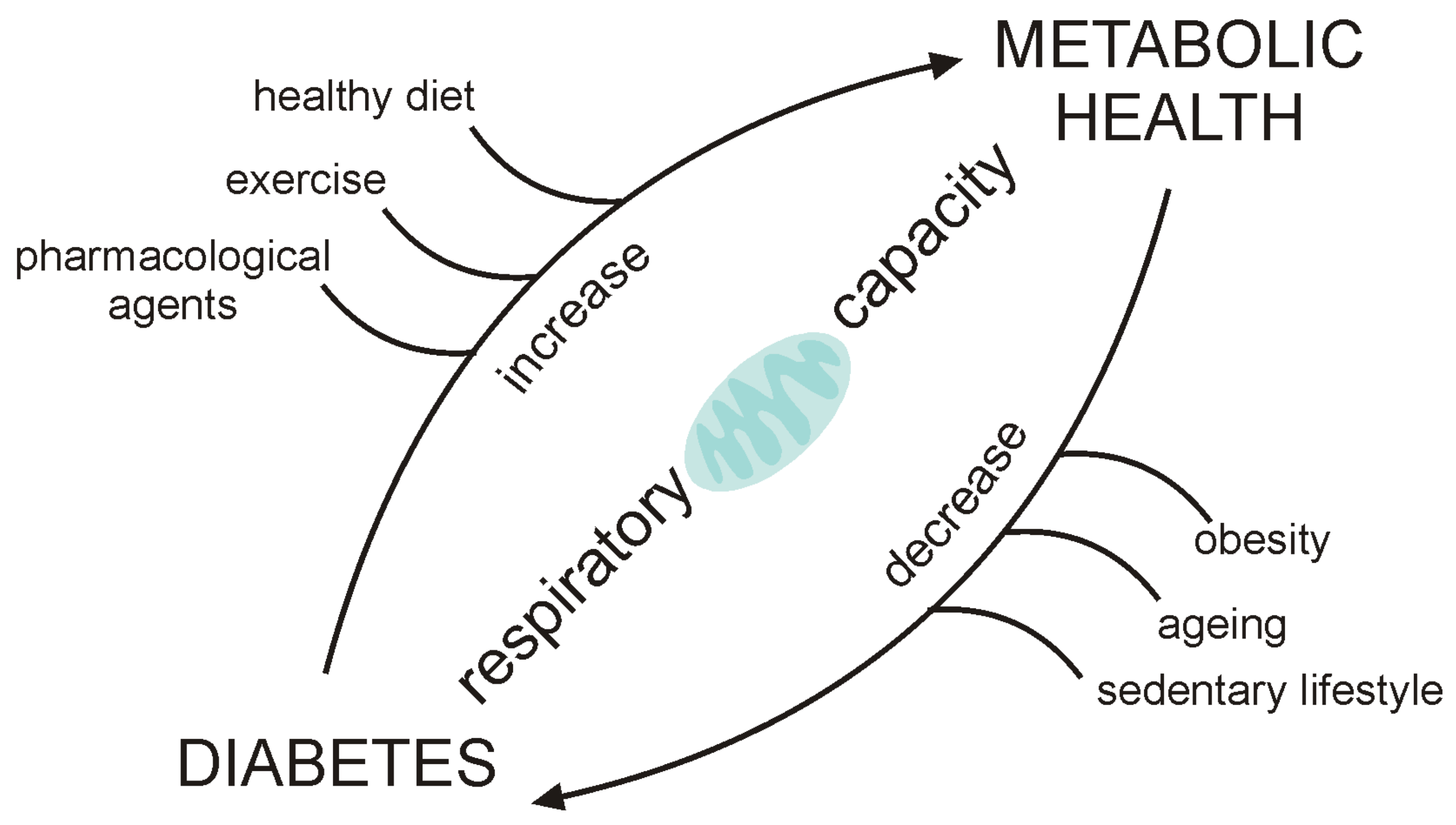
:1. Introduction
2. Role of Mitochondria in Diabetes
2.1. Skeletal Muscle
2.2. Liver
2.3. Blood Cells
3. Interventions Targeting Mitochondria in Diabetes
3.1. Exercise
3.2. Pharmacological Therapy
3.3. Novel Therapeutic Approaches That Target Mitochondria
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- A DeFronzo, R. Lilly lecture: The triumvirate: Beta cell, muscle, liver, a collusion responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef]
- DeFronzo, R.A. From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [Green Version]
- Lowell, B.B. Mitochondrial Dysfunction and Type 2 Diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [Green Version]
- Blake, R.; Trounce, I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 1404–1412. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in Mitochondrial Oxidative Stress and Mitophagy in Subjects with Prediabetes and Type 2 Diabetes Mellitus. Front. Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.-E.; Corvera, S. The Role of Mitochondria in the Pathogenesis of Type 2 Diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, M.L.; Wong, R.; Datta, M.; Fazio, T.N.; Ebrahim, M.M.; McNamara, E.C.; De Jong, G.; Gilfillan, C. Mitochondrial disease: An uncommon but important cause of diabetes mellitus. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Lanza, I.; Nair, K.S. Mitochondrial metabolic function assessed in vivo and in vitro. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef]
- Gnaiger, E.; Group, M.T. Mitochondrial physiology. Bioenerg. Commun. 2020. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Quantification of Mitochondrial Oxidative Phosphorylation in Metabolic Disease: Application to Type 2 Diabetes. Int. J. Mol. Sci. 2019, 20, 5271. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]
- Prompers, J.J.; Wessels, B.B.; Kemp, G.J.; Nicolay, K. MITOCHONDRIA: Investigation of in vivo muscle mitochondrial function by 31P magnetic resonance spectroscopy. Int. J. Biochem. Cell Biol. 2014, 50, 67–72. [Google Scholar] [CrossRef]
- Jacobs, R.A.; Lundby, C. Contextualizing the biological relevance of standardized high-resolution respirometry to assess mitochondrial function in permeabilized human skeletal muscle. Acta Physiol. 2021, 231, e13625. [Google Scholar] [CrossRef]
- Simoneau, J.-A.; Kelley, D.E. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J. Appl. Physiol. 1997, 83, 166–171. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role ofPGC1andNRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.J.; Kooi, M.M.E.; Moonen-Kornips, E.; Sels, J.-P.; Hesselink, M.K.; et al. Lower Intrinsic ADP-Stimulated Mitochondrial Respiration Underlies In Vivo Mitochondrial Dysfunction in Muscle of Male Type 2 Diabetic Patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Hey-Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial Respiration Is Decreased in Skeletal Muscle of Patients With Type 2 Diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsøe, R.; Dela, F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Stride, N.; Hey-Mogensen, M.; Hansen, C.N.; Andersen, J.L.; Madsbad, S.; Worm, D.; Helge, J.; Dela, F. Increased mitochondrial substrate sensitivity in skeletal muscle of patients with type 2 diabetes. Diabetologia 2011, 54, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Rabøl, R.; Højberg, P.M.V.; Almdal, T.; Boushel, R.; Haugaard, S.B.; Madsbad, S.; Dela, F. Improved glycaemic control decreases inner mitochondrial membrane leak in type 2 diabetes. Diabetes Obes. Metab. 2009, 11, 355–360. [Google Scholar] [CrossRef]
- Ritter, O.; Jelenik, T.; Roden, M. Lipid-mediated muscle insulin resistance: Different fat, different pathways? J. Mol. Med. 2015, 93, 831–843. [Google Scholar] [CrossRef]
- Cree-Green, M.; Scalzo, R.L.; Harrall, K.; Newcomer, B.R.; Schauer, I.E.; Huebschmann, A.G.; McMillin, S.; Brown, M.S.; Orlicky, D.; Knaub, L.; et al. Supplemental Oxygen Improves In Vivo Mitochondrial Oxidative Phosphorylation Flux in Sedentary Obese Adults With Type 2 Diabetes. Diabetes 2018, 67, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef] [Green Version]
- Kotronen, A.; Westerbacka, J.; Bergholm, R.; Pietiläinen, K.H.; Yki-Järvinen, H. Liver Fat in the Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3490–3497. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Cusi, K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019, 1, 312–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, H.; Fukura, M.; Tsuchishima, M.; Takase, S. Mutation of Mitochondrial DNA in Livers from Patients with Alcoholic Hepatitis and Nonalcoholic Steatohepatitis. Alcohol. Clin. Exp. Res. 2007, 31, S54–S60. [Google Scholar] [CrossRef]
- Sevastianova, K.; Hakkarainen, A.; Kotronen, A.; Cornér, A.; Arkkila, P.; Arola, J.; Westerbacka, J.; Bergholm, R.; Lundbom, J.; Lundbom, N.; et al. Nonalcoholic Fatty Liver Disease: Detection of Elevated Nicotinamide Adenine Dinucleotide Phosphate with in Vivo 3.0-T31P MR Spectroscopy with Proton Decoupling. Radiology 2010, 256, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Grieco, A.; Armuzzi, A.; Candelli, M.; Forgione, A.; Gasbarrini, A.; Gasbarrini, G. Hepatic Mitochondrial Beta-Oxidation in Patients With Nonalcoholic Steatohepatitis Assessed by 13C-Octanoate Breath Test. Am. J. Gastroenterol. 2003, 98, 2335–2336. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; E Hespenheide, E.; Parks, J.K.; Parker, W. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Szendroedi, J.; Chmelik, M.; Schmid, A.I.; Nowotny, P.; Brehm, A.; Krssak, M.; Moser, E.; Roden, M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology 2009, 50, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in Liver ATP Homeostasis in Human Nonalcoholic Steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [Green Version]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krššák, M.; Moser, E.; Roden, M. Liver ATP Synthesis Is Lower and Relates to Insulin Sensitivity in Patients With Type 2 Diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Brøns, C.; Jacobsen, S.; Hiscock, N.; White, A.; Nilsson, E.; Dunger, D.; Astrup, A.; Quistorff, B.; Vaag, A.A.; Dunger, P.D. Effects of high-fat overfeeding on mitochondrial function, glucose and fat metabolism, and adipokine levels in low-birth-weight subjects. Am. J. Physiol. Metab. 2012, 302, E43–E51. [Google Scholar] [CrossRef] [Green Version]
- Brøns, C.; Grunnet, L.G. Mechanisms in Endocrinology: Skeletal muscle lipotoxicity in insulin resistance and type 2 diabetes: A causal mechanism or an innocent bystander? Eur. J. Endocrinol. 2017, 176, R67–R78. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Kupriyanova, Y.; Zaharia, O.P.; Bobrov, P.; Karusheva, Y.; Burkart, V.; Szendroedi, J.; Hwang, J.-H.; Roden, M.; GDS Group. Early changes in hepatic energy metabolism and lipid content in recent-onset type 1 and 2 diabetes mellitus. J. Hepatol. 2021, 74, 1028–1037. [Google Scholar] [CrossRef]
- Krako Jakovljevic, N.; Pavlovic, K.; Zujovic, T.; Kravic-Stevovic, T.; Jotic, A.; Markovic, I.; Lalic, N.M. In vitro models of insulin resistance: Mitochondrial coupling is differently affected in liver and muscle cells. Mitochondrion 2021. in review. [Google Scholar]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate Induces Insulin Resistance in H4IIEC3 Hepatocytes through Reactive Oxygen Species Produced by Mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nat. Cell Biol. 2001, 413, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.-H.; Satoh, H.; Herzig, S.; Lee, C.-H.; Hedrick, S.; Kulkarni, R.N.; Evans, R.M.; Olefsky, J.M.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-α-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Guo, S.; Copps, K.D.; Dong, X.; Kollipara, R.; Rodgers, J.T.; A DePinho, R.; Puigserver, P.; White, M.F. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, M.; Cai, W.; Wang, C.-H.; Cederquist, C.T.; Damasio, M.; Homan, E.P.; Batista, T.; Ramirez, A.K.; Gupta, M.K.; Steger, M.; et al. FoxK1 and FoxK2 in insulin regulation of cellular and mitochondrial metabolism. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Is Required for Insulin Signaling and Is Implicated in Hepatic Insulin Resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Gang, X.; He, G.; Liu, Y.; Wang, Y.; Zhao, X.; Wang, G. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Front. Endocrinol. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Wang, J.; Shenouda, S.M.; Hagen, T.M.; Smith, A.R.; Kizhakekuttu, T.J.; Kluge, M.A.; Weihrauch, D.; Gutterman, D.D.; Vita, J.A. Altered mitochondrial membrane potential, mass, and morphology in the mononuclear cells of humans with type 2 diabetes. Transl. Res. 2010, 156, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Avila, C.; Huang, R.J.; Stevens, M.V.; Aponte, A.M.; Tripodi, D.; Kim, K.Y.; Sack, M.N. Platelet Mitochondrial Dysfunction is Evident in Type 2 Diabetes in Association with Modifications of Mitochondrial Anti-Oxidant Stress Proteins. Exp. Clin. Endocrinol. Diabetes 2011, 120, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.-L.; Shirihai, O.S.; Holbrook, M.; Xu, G.; Kocherla, M.; Shah, A.; Fetterman, J.L.; Kluge, M.A.; Frame, A.A.; Hamburg, N.M.; et al. Relation of mitochondrial oxygen consumption in peripheral blood mononuclear cells to vascular function in type 2 diabetes mellitus. Vasc. Med. 2014, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Tyrrell, D.J.; Bharadwaj, M.S.; Van Horn, C.G.; Marsh, A.P.; Nicklas, B.J.; Molina, A.J. Blood-cell bioenergetics are associated with physical function and inflammation in overweight/obese older adults. Exp. Gerontol. 2015, 70, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, G.; Smith, S.C.; Hughes, T.M.; Wagner, B.; Maldjian, J.A.; Freedman, B.I.; Molina, A.J.A. Blood-based bioenergetic profiling is related to differences in brain morphology in African Americans with Type 2 diabetes. Clin. Sci. 2018, 132, 2509–2518. [Google Scholar] [CrossRef]
- Rose, S.; Carvalho, E.; Diaz, E.C.; Cotter, M.; Bennuri, S.C.; Azhar, G.; Frye, R.E.; Adams, S.H.; Børsheim, E. A comparative study of mitochondrial respiration in circulating blood cells and skeletal muscle fibers in women. Am. J. Physiol. Metab. 2019, 317, E503–E512. [Google Scholar] [CrossRef]
- Lambers, S.; Van Laethem, C.; Van Acker, K.; Calders, P. Influence of combined exercise training on indices of obesity, diabetes and cardiovascular risk in type 2 diabetes patients. Clin. Rehabil. 2008, 22, 483–492. [Google Scholar] [CrossRef]
- Menshikova, E.V.; Ritov, V.B.; Toledo, F.G.S.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am. J. Physiol. Metab. 2005, 288, E818–E825. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Drake, J.C.; Yan, Z. Exercise-Induced Mitophagy in Skeletal Muscle and Heart. Exerc. Sport Sci. Rev. 2019, 47, 151–156. [Google Scholar] [CrossRef]
- Hood, D.A.; Tryon, L.D.; Carter, H.N.; Kim, Y.; Chen, C.C. Unravelling the mechanisms regulating muscle mitochondrial biogenesis. Biochem. J. 2016, 473, 2295–2314. [Google Scholar] [CrossRef] [Green Version]
- Ji, L.L.; Yeo, D.; Kang, C.; Zhang, T. The role of mitochondria in redox signaling of muscle homeostasis. J. Sport Health Sci. 2020, 9, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.N.; Pauly, M.; Tryon, L.D.; Hood, D.A. Effect of contractile activity on PGC-1α transcription in young and aged skeletal muscle. J. Appl. Physiol. 2018, 124, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Wadley, G.D.; Lee-Young, R.S.; Canny, B.J.; Wasuntarawat, C.; Chen, Z.P.; Hargreaves, M.; Kemp, B.E.; McConell, G.K. Effect of exercise intensity and hypoxia on skeletal muscle AMPK signaling and substrate metabolism in humans. Am. J. Physiol. Metab. 2006, 290, E694–E702. [Google Scholar] [CrossRef]
- Taylor, C.W.; Ingham, S.A.; Hunt, J.E.A.; Martin, N.; Pringle, J.; Ferguson, R.A. Exercise duration-matched interval and continuous sprint cycling induce similar increases in AMPK phosphorylation, PGC-1α and VEGF mRNA expression in trained individuals. Graefe’s Arch. Clin. Exp. Ophthalmol. 2016, 116, 1445–1454. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Meex, R.C.; Schrauwen-Hinderling, V.B.; Moonen-Kornips, E.; Schaart, G.; Mensink, M.; Phielix, E.; Van De Weijer, T.; Sels, J.-P.; Schrauwen, P.; Hesselink, M.K. Restoration of Muscle Mitochondrial Function and Metabolic Flexibility in Type 2 Diabetes by Exercise Training Is Paralleled by Increased Myocellular Fat Storage and Improved Insulin Sensitivity. Diabetes 2009, 59, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.-M.; Adhihetty, P.J.; Leeuwenburgh, C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J. Physiol. 2016, 594, 5105–5123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.-H.; Chang, S.-C.; Chou, C.-H.; Weng, T.-P.; Hsu, C.-C.; Wang, J.-S. Exercise Training Alleviates Hypoxia-induced Mitochondrial Dysfunction in the Lymphocytes of Sedentary Males. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Holloszy, J.O. Biochemical Adaptations in Muscle: Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Coyle, E.F. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 831–838. [Google Scholar] [CrossRef]
- Pesta, D.H.; Jelenik, T.; Zaharia, O.; Bobrov, P.; Görgens, S.; Bódis, K.; Karusheva, Y.; Jakovljevic, N.K.; Lalic, N.; Daniel, F.; et al. NDUFB6 polymorphism is associated with physical activity-mediated metabolic changes in type 2 diabetes. Front. Endocrinol. 2021. in review. [Google Scholar]
- Liepinsh, E.; Makarova, E.; Plakane, L.; Konrade, I.; Liepins, K.; Videja, M.; Sevostjanovs, E.; Grinberga, S.; Makrecka-Kuka, M.; Dambrova, M. Low-intensity exercise stimulates bioenergetics and increases fat oxidation in mitochondria of blood mononuclear cells from sedentary adults. Physiol. Rep. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Apostolopoulou, M.; Pesta, D.; Karusheva, Y.; Gancheva, S.; Jelenik, T.; Bierwagen, A.; Müssig, K.; Szendroedi, J.; Roden, M. Effects on Insulin Sensitivity, but Not on Mitochondrial Function Are Dependent on Insulin Resistance Status after High Intensity Interval Training. Diabetes 2018, 67, 69. [Google Scholar] [CrossRef]
- Bilet, L.; Phielix, E.; Van De Weijer, T.; Gemmink, A.; Bosma, M.; Moonen-Kornips, E.; Jorgensen, J.A.; Schaart, G.; Zhang, D.; Meijer, K.; et al. One-leg inactivity induces a reduction in mitochondrial oxidative capacity, intramyocellular lipid accumulation and reduced insulin signalling upon lipid infusion: A human study with unilateral limb suspension. Diabetologia 2020, 63, 1211–1222. [Google Scholar] [CrossRef] [Green Version]
- Scalzo, R.L.; Schauer, I.E.; Rafferty, D.; Knaub, L.A.; Kvaratskhelia, N.; Johnson, T.K.; Pott, G.B.; Abushamat, L.A.; Whipple, M.O.; Huebschmann, A.G.; et al. Single-leg exercise training augments in vivo skeletal muscle oxidative flux and vascular content and function in adults with type 2 diabetes. J. Physiol. 2021, JP280603. [Google Scholar] [CrossRef]
- Bird, S.R.; Hawley, J.A. Update on the effects of physical activity on insulin sensitivity in humans. BMJ Open Sport Exerc. Med. 2017, 2, 1–26. [Google Scholar] [CrossRef] [Green Version]
- DiMenna, F.J.; Arad, A.D. Exercise as ‘precision medicine’ for insulin resistance and its progression to type 2 diabetes: A research review. BMC Sports Sci. Med. Rehabil. 2018, 10, 1–23. [Google Scholar] [CrossRef]
- Thyfault, J.P.; Bergouignan, A. Exercise and metabolic health: Beyond skeletal muscle. Diabetologia 2020, 63, 1464–1474. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, A.H.; Wiggers, H.; Dollerup, O.L.; Jespersen, N.R.; Hansson, N.H.; Frøkiær, J.; Brøsen, K.; Nørrelund, H.; Bøtker, H.E.; Moller, N.; et al. Metformin Lowers Body Weight But Fails to Increase Insulin Sensitivity in Chronic Heart Failure Patients without Diabetes: A Randomized, Double-Blind, Placebo-Controlled Study. Cardiovasc. Drugs Ther. 2021, 35, 491–503. [Google Scholar] [CrossRef]
- Larsen, S.; Rabøl, R.; Hansen, C.N.; Madsbad, S.; Helge, J.; Dela, F. Metformin-treated patients with type 2 diabetes have normal mitochondrial complex I respiration. Diabetologia 2012, 55, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; Macdonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nat. Cell Biol. 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Alshawi, A.; Agius, L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J. Biol. Chem. 2019, 294, 2839–5691. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e5. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Wiley, S.; Rogers, G.W.; Andreyev, A.Y.; Petrosyan, S.; Loviscach, M.; Wall, E.A.; Yadava, N.; Heuck, A.; Ferrick, D.A.; et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl. Acad. Sci. USA 2013, 110, 5422–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, T.; Martinou, J.-C. The mitochondrial pyruvate carrier in health and disease: To carry or not to carry? Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1863, 2436–2442. [Google Scholar] [CrossRef] [PubMed]
- Rabøl, R.; Boushel, R.; Almdal, T.; Hansen, C.N.; Ploug, T.; Haugaard, S.B.; Prats, C.; Madsbad, S.; Dela, F. Opposite effects of pioglitazone and rosiglitazone on mitochondrial respiration in skeletal muscle of patients with type 2 diabetes. Diabetes Obes. Metab. 2010, 12, 806–814. [Google Scholar] [CrossRef]
- Bajpeyi, S.; Pasarica, M.; Conley, K.E.; Newcomer, B.R.; Jubrias, S.A.; Gamboa, C.; Murray, K.; Sereda, O.; Sparks, L.M.; Smith, S.R. Pioglitazone-induced improvements in insulin sensitivity occur without concomitant changes in muscle mitochondrial function. Metabolism 2017, 69, 24–32. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Monroy, A.; Kamath, S.; Sotero, R.; Cas, M.D.; Daniele, G.; Chavez, A.O.; Abdul-Ghani, M.; Hribal, M.L.; Sesti, G.; et al. Pioglitazone corrects dysregulation of skeletal muscle mitochondrial proteins involved in ATP synthesis in type 2 diabetes. Metabolism 2021, 114, 154416. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Liao, L.; Wang, H.; Zhang, W.; Wang, T.; Xu, Z. Canagliflozin ameliorates obesity by improving mitochondrial function and fatty acid oxidation via PPARα in vivo and in vitro. Life Sci. 2020, 247, 117414. [Google Scholar] [CrossRef]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Nisr, R.B.; Affourtit, C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1837, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Shulman, G.I. Decreased Insulin-Stimulated ATP Synthesis and Phosphate Transport in Muscle of Insulin-Resistant Offspring of Type 2 Diabetic Parents. PLoS Med. 2005, 2, 0879–0884. [Google Scholar] [CrossRef]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Stride, N.; Hey-Mogensen, M.; Hansen, C.N.; Bang, L.E.; Bundgaard, H.; Nielsen, L.B.; Helge, J.; Dela, F. Simvastatin Effects on Skeletal Muscle: Relation to decreased mitochondrial function and glucose intolerance. J. Am. Coll. Cardiol. 2013, 61, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Weijer, T.; Phielix, E.; Bilet, L.; Williams, E.G.; Ropelle, E.R.; Bierwagen, A.; Livingstone, R.; Nowotny, P.; Sparks, L.M.; Paglialunga, S.; et al. Evidence for a Direct Effect of the NAD+Precursor Acipimox on Muscle Mitochondrial Function in Humans. Diabetes 2014, 64, 1193–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phielix, E.; Jelenik, T.; Nowotny, P.; Szendroedi, J.; Roden, M. Reduction of non-esterified fatty acids improves insulin sensitivity and lowers oxidative stress, but fails to restore oxidative capacity in type 2 diabetes: A randomised clinical trial. Diabetologia 2014, 57, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Timmers, S.; De Ligt, M.; Phielix, E.; Van De Weijer, T.; Hansen, J.; Moonen-Kornips, E.; Schaart, G.; Kunz, I.; Hesselink, M.K.C.; Schrauwen-Hinderling, V.B.; et al. Resveratrol as Add-on Therapy in Subjects With Well-Controlled Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care 2016, 39, 2211–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Most, J.; Timmers, S.; Warnke, I.; Jocken, J.W.E.; Van Boekschoten, M.; De Groot, P.; Bendik, I.; Schrauwen, P.; Goossens, G.H.; Blaak, E.E. Combined epigallocatechin-3-gallate and resveratrol supplementation for 12 wk increases mitochondrial capacity and fat oxidation, but not insulin sensitivity, in obese humans: A randomized controlled trial. Am. J. Clin. Nutr. 2016, 104, 215–227. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. EBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef]
- Golubitzky, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for Active Small Molecules in Mitochondrial Complex I Deficient Patient’s Fibroblasts, Reveals AICAR as the Most Beneficial Compound. PLoS ONE 2011, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gao, R.; Xie, X.; Zheng, Z.; Li, H.; Li, S.; Dong, F.; Wang, L. A metabolomic study of the PPARδ agonist GW501516 for enhancing running endurance in Kunming mice. Sci. Rep. 2015, 5, 9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, K.G.; Romero-Perez, D.; Barraza-Hidalgo, M.; Cruz, M.; Rivas, M.; Cortez-Gomez, B.; Ceballos, G.; Villarreal, F. Short- and long-term effects of (−)-epicatechin on myocardial ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2008, 295, H761–H767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Vuylsteke, V.; Chastain, L.M.; Maggu, G.A.; Brown, C. Imeglimin: A Potential New Multi-Target Drug for Type 2 Diabetes. Drugs R&D 2015, 15, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Johansson, K.S.; Brønden, A.; Knop, F.K.; Christensen, M.B. Clinical pharmacology of imeglimin for the treatment of type 2 diabetes. Expert Opin. Pharmacother. 2020, 21, 871–882. [Google Scholar] [CrossRef]
- Fouqueray, P.; Leverve, X.; Fontaine, E.; Baquié, M.; Wollheim, C. Imeglimin—A New Oral Anti-Diabetic that Targets the Three Key Defects of type 2 Diabetes. J. Diabetes Metab. 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, T.S.; DeFronzo, R.A.; Ryder, R.E.J.; Bailey, C.J. Imeglimin, a novel, first in-class, blood glucose-lowering agent: A systematic review and meta-analysis of clinical evidence. Br. J. Diabetes 2020, 20, 28–31. [Google Scholar] [CrossRef]
- Fouqueray, P.; Pirags, V.; Diamant, M.; Schernthaner, G.; Lebovitz, H.E.; Inzucchi, S.E.; Bailey, C.J. The Efficacy and Safety of Imeglimin as Add-on Therapy in Patients with Type 2 Diabetes Inadequately Controlled with Sitagliptin Monotherapy. Diabetes Care 2014, 37, 1924–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouqueray, P.; Pirags, V.; Inzucchi, S.E.; Bailey, C.J.; Schernthaner, G.; Diamant, M.; Lebovitz, H.E. The Efficacy and Safety of Imeglimin as Add-on Therapy in Patients With Type 2 Diabetes Inadequately Controlled with Metformin Monotherapy. Diabetes Care 2012, 36, 565–568. [Google Scholar] [CrossRef] [Green Version]
- Vial, G.; Chauvin, M.-A.; Bendridi, N.; Durand, A.; Meugnier, E.; Madec, A.-M.; Bernoud-Hubac, N.; De Barros, J.-P.P.; Fontaine, E.; Acquaviva, C.; et al. Imeglimin Normalizes Glucose Tolerance and Insulin Sensitivity and Improves Mitochondrial Function in Liver of a High-Fat, High-Sucrose Diet Mice Model. Diabetes 2014, 64, 2254–2264. [Google Scholar] [CrossRef] [Green Version]
- Detaille, D.; Vial, G.; Borel, A.-L.; Cottet-Rouselle, C.; Hallakou-Bozec, S.; Bolze, S.; Fouqueray, P.; Fontaine, E. Imeglimin prevents human endothelial cell death by inhibiting mitochondrial permeability transition without inhibiting mitochondrial respiration. Cell Death Discov. 2016, 2, 15072. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Lamarche, F.; Cottet-Rousselle, C.; Hallakou-Bozec, S.; Borel, A.; Fontaine, E. The mechanism by which imeglimin inhibits gluconeogenesis in rat liver cells. Endocrinol. Diabetes Metab. 2021, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.G. Hepatic glucose and lipid metabolism. Diabetologia 2016, 59, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, Z.; Zhao, M.; Nie, Y.; Liu, P.; Zhu, Y.; Zhang, X. Skeletal Muscle Lipid Droplets and the Athlete’s Paradox. Cells 2019, 8, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Substance | n | Treatment | Effects on Insulin Sensitivity | Effects on Mitochondrial Function | Reference |
|---|---|---|---|---|---|
| simvastatin | 10 (hypercholesterolemia), 10 (healthy controls) | simvastatin (10 to 40 mg/day) for at least 12 months (average: 5 years) | ↓ in patients compared with controls, not measured before and after treatment | ↓ OXPHOS capacity (complex I and II-linked substrates) ↓ coenzyme Q10 content | Larsen 2013 [104] |
| acipimox | 21 (T2D) | acipimox (250 mg three times daily) or placebo for 2 weeks | ↓ | ↑ OXPHOS capacity (CI+II substrates) ↑ ET capacity (CI+II and fatty acid substrates) | Van der Weijer 2015 [105] |
| acipimox | 15 (T2D obese) | acipimox (250 mg three times daily) for 1 day, and day after one dose (250 mg) 1 h before muscle biopsy or placebo | ↑ | = basal or insulin-stimulated mitochondrial respiration in permeabilized fibers ↓OXPHOS, ET capacity (octanoyl-carnitine), LEAK on acipimox in isolated mitochondria | Phielix 2014 [106] |
| resveratrol | 17 (T2D) | resVida (150 mg/day trans-resveratrol) or placebo for 30 days | = | ↑ OXPHOS (MOct, MGSOct) and ET capacity (M+O) | Timmers 2016 [107] |
| epigallocatechin-3-gallate and resveratrol | 38 (overweight or obese, non-diabetic) | EGCG+RES (282 and 80 mg/d, respectively) or placebo for 12 weeks | = | ↑ OXPHOS capacity (CI+II substrates and CI+II+fatty acids) ↑ ET capacity | Most 2016 [108] |
| metformin, sulfonylurea | 22 (T2D), 18 (healthy controls) | T2D patients: metformin (2000 ± 200 mg/day, n = 14) or sulfonylurea (glimepiride, 2.4 ± 0.2 mg/day, n = 8) | not measured | = | Larsen 2012 [87] |
| metformin | 18 (chronic heart failure, non-diabetic) | extended-release metformin (Glucophage XR®) (starting dose 500 mg q.d, uptitration to a target dose of 1000 mg b.i.d, n = 10) or placebo (n = 8) | = | = | Larsen 2021 [86] |
| pioglitazane | 24 (T2D, obese) | pioglitazone (30 mg/day, increased to 45 mg/day if needed, n = 16) or placebo (n = 8) for 12 weeks | ↑ | = no change in muscle maximal ATP synthetic capacity (ATPmax) by 31P-MRS | Bajpeyi 2017 [95] |
| pioglitazone (PIO), rosiglitazone (ROSI) | 21 (T2D), 8 (healthy controls) | T2D patients: ROSI (4 mg/d, n = 12), PIO (30 mg/d, n = 9), for 12 weeks | ↑ | ↓ OXPHOS capacity (CI and CI+II) by ROSI treatment ↑ OXPHOS capacity (CI and CI+II)by PIO treatment ↑ protein content, complexe II and III by PIO treatment | Rabøl 2010 [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krako Jakovljevic, N.; Pavlovic, K.; Jotic, A.; Lalic, K.; Stoiljkovic, M.; Lukic, L.; Milicic, T.; Macesic, M.; Stanarcic Gajovic, J.; Lalic, N.M. Targeting Mitochondria in Diabetes. Int. J. Mol. Sci. 2021, 22, 6642. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126642
Krako Jakovljevic N, Pavlovic K, Jotic A, Lalic K, Stoiljkovic M, Lukic L, Milicic T, Macesic M, Stanarcic Gajovic J, Lalic NM. Targeting Mitochondria in Diabetes. International Journal of Molecular Sciences. 2021; 22(12):6642. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126642
Chicago/Turabian StyleKrako Jakovljevic, Nina, Kasja Pavlovic, Aleksandra Jotic, Katarina Lalic, Milica Stoiljkovic, Ljiljana Lukic, Tanja Milicic, Marija Macesic, Jelena Stanarcic Gajovic, and Nebojsa M. Lalic. 2021. "Targeting Mitochondria in Diabetes" International Journal of Molecular Sciences 22, no. 12: 6642. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126642