
Usher Syndrome: Genetics of a Human Ciliopathy

 , and
, and
Abstract
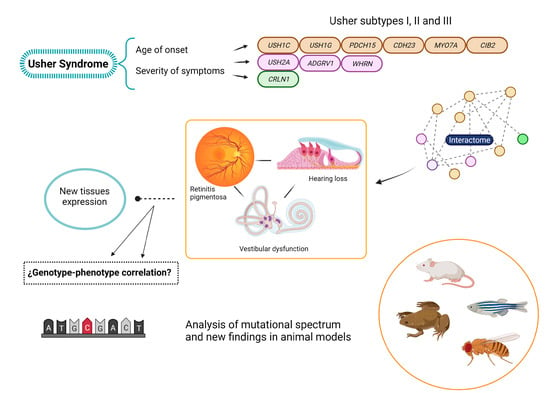
:
1. Introduction
2. The Genetic Heterogeneity of USH
3. The USH Genes
3.1. MYO7A
3.2. USH1C
3.3. CDH23
3.4. PCDH15
3.5. USH1G
3.6. CIB2
3.7. USH2A
3.8. ADGRV1
3.9. WHRN
3.10. CLRN1
3.11. Other Related Genes
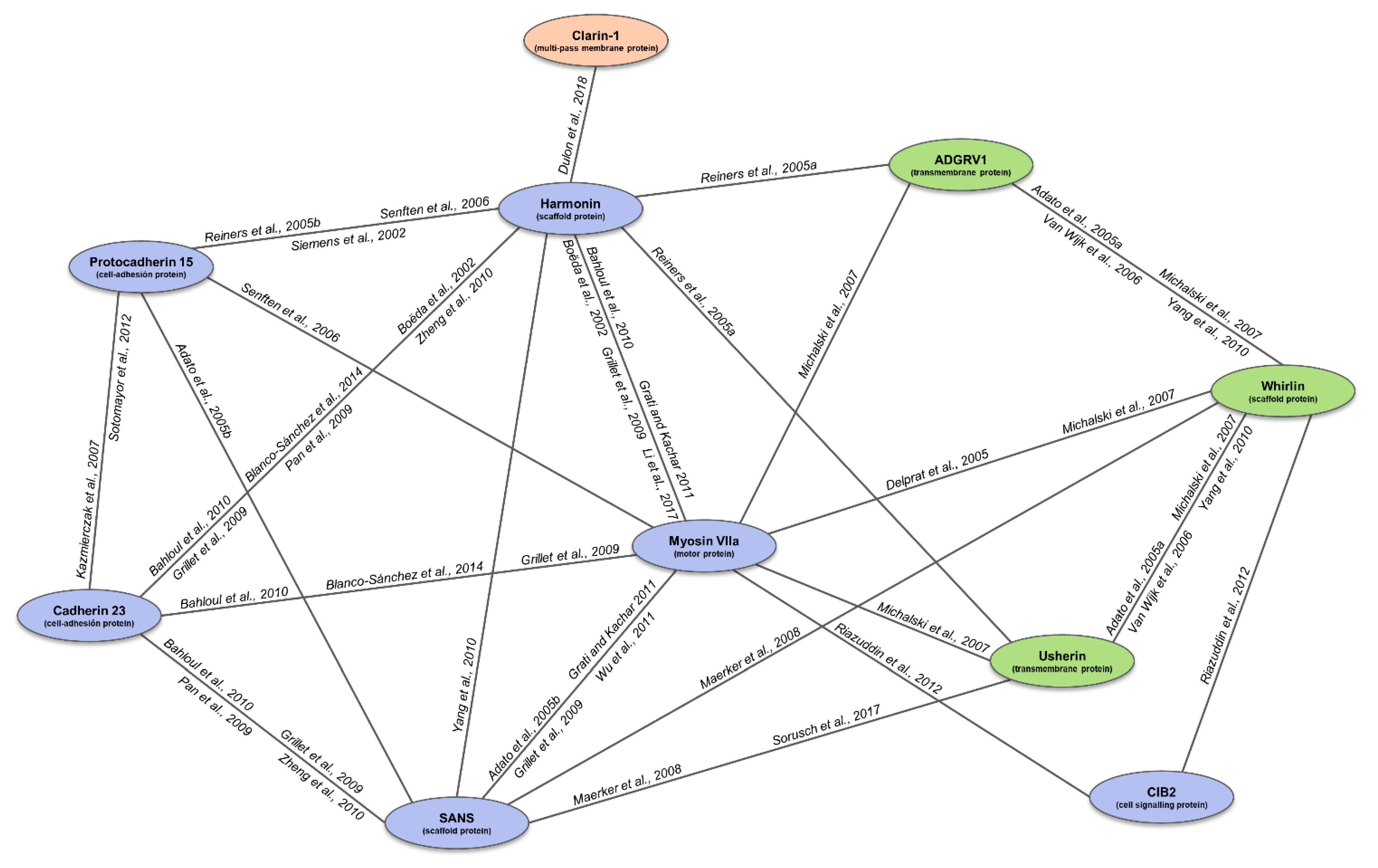
4. The USH Interactome
5. Is USH Actually a Ciliopathy?
6. Animal Models
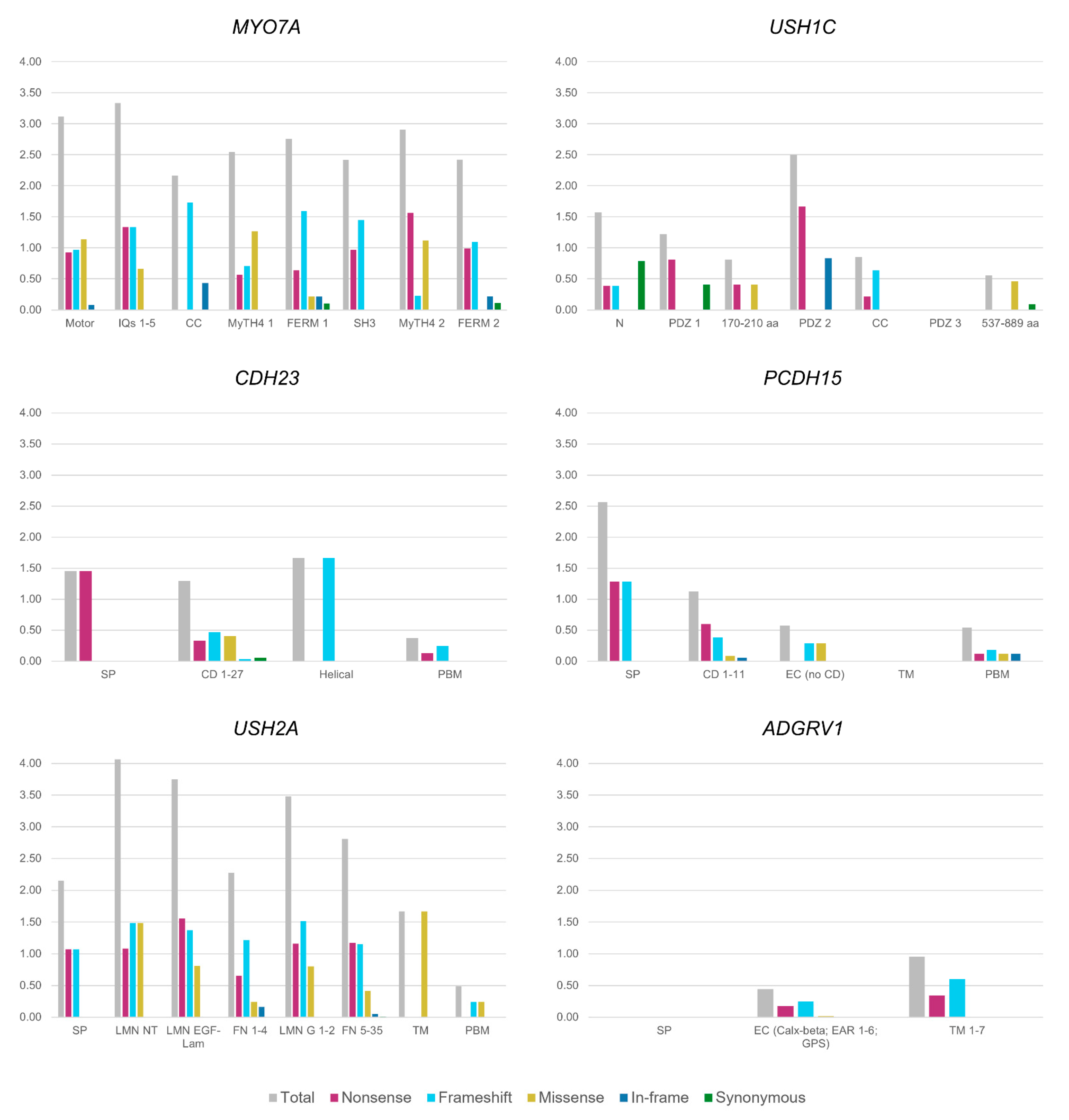
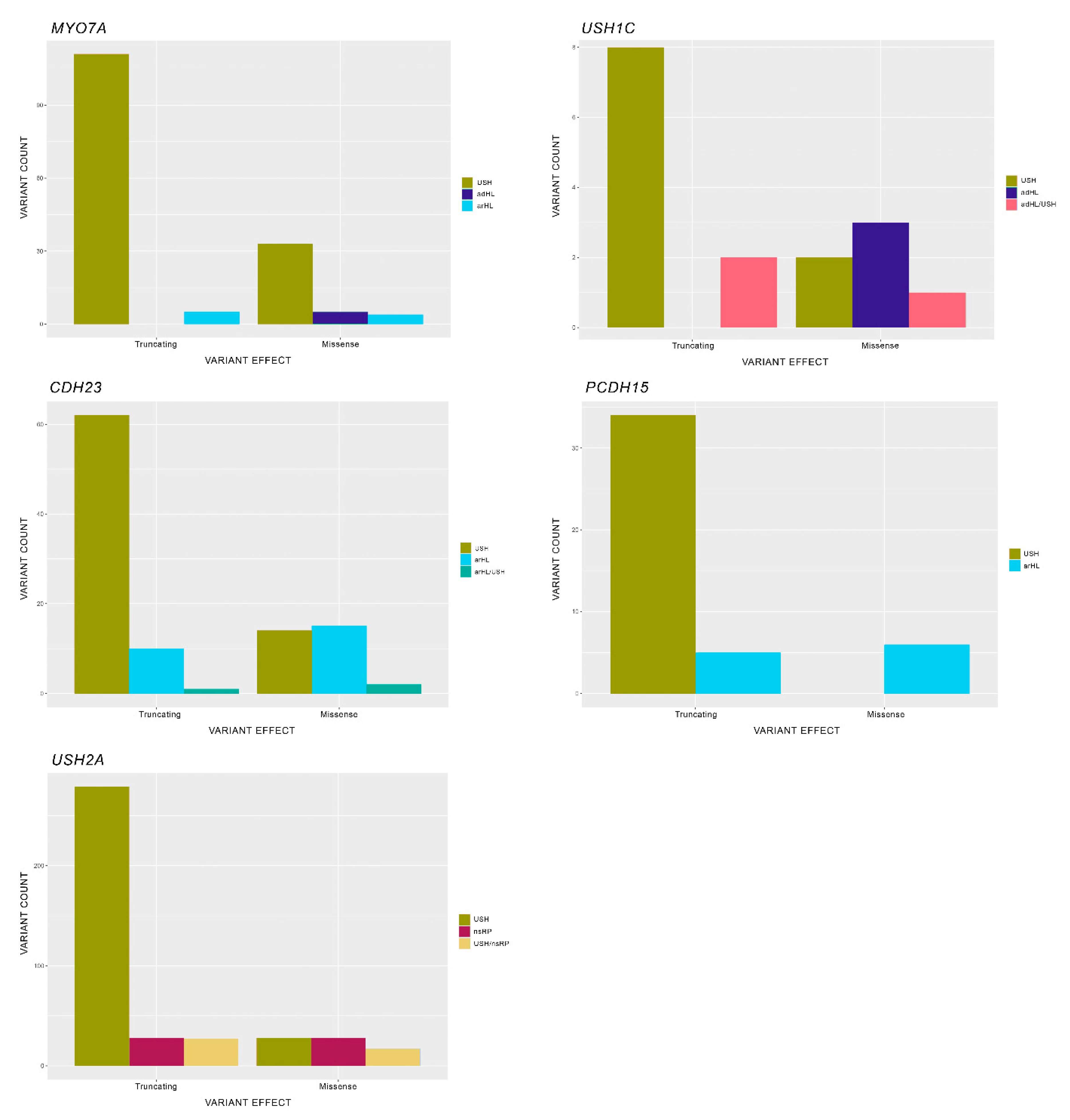
7. Mutational Spectra in the USH Genes: Types, Distribution and Genotype-Phenotype Correlations
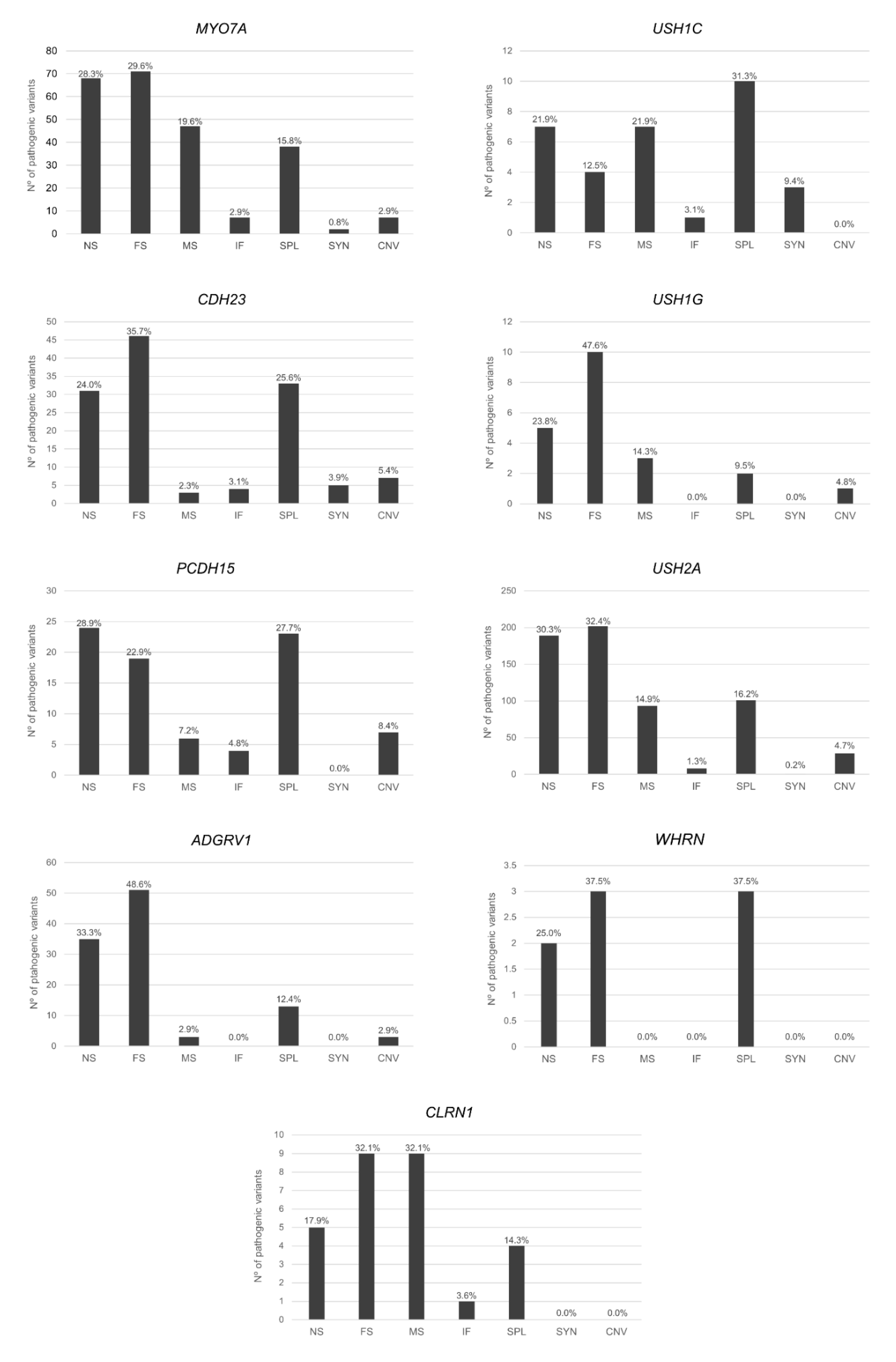
7.1. Type of Mutations
7.2. Mutation Distribution among the USH Proteins Domains
7.2.1. USH2A
7.2.2. PCDH15
7.2.3. MYO7A
7.2.4. ADGRV1
7.2.5. USH1C
7.2.6. CDH23
7.3. Types of Mutations and Their Influence in the Phenotype
7.3.1. USH2A
7.3.2. PCDH15
7.3.3. MYO7A
7.3.4. CDH23
7.3.5. USH1C
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Millán, J.M.; Aller, E.; Jaijo, T.; Blanco-Kelly, F.; Gimenez-Pardo, A.; Ayuso, C. An Update on the Genetics of Usher Syndrome. J. Ophthalmol. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wafa, T.T.; Faridi, R.; King, K.A.; Zalewski, C.; Yousaf, R.; Schultz, J.M.; Morell, R.J.; Muskett, J.; Turriff, A.; Tsilou, E.; et al. Vestibular phenotype-genotype correlation in a cohort of 90 patients with Usher syndrome. Clin. Genet. 2021, 99, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Davenport, S.L.H.; Omenn, G.S. The Heterogeneity of Usher Syndrome. In Proceedings of the 5th International Conference of Birth Defects, Montreal, QC, Canada, 21–27 August 1977. [Google Scholar]
- Weil, D.; Küssel, P.; Blanchard, S.; Levy, G.G.; Levi-Acobas, F.; Drira, M.; Ayadi, H.; Petit, C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet. 1997, 16, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Bork, J.M.; Peters, L.; Riazuddin, S.; Bernstein, S.L.; Ahmed, Z.M.; Ness, S.L.; Polomeno, R.; Ramesh, A.; Schloss, M.; Srisailpathy, C.R.S.; et al. Usher Syndrome 1D and Nonsyndromic Autosomal Recessive Deafness DFNB12 Are Caused by Allelic Mutations of the Novel Cadherin-Like Gene CDH23. Am. J. Hum. Genet. 2001, 68, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.M.; Smith, T.N.; Riazuddin, S.; Makishima, T.; Ghosh, M.; Bokhari, S.; Menon, P.S.; Deshmukh, D.; Griffith, A.J.; Riazuddin, S.; et al. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Qual. Life Res. 2002, 110, 527–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.M.; Riazuddin, S.; Ahmad, J.; Bernstein, S.L.; Guo, Y.; Sabar, M.F.; Sieving, P.; Riazuddin, S.; Griffith, A.J.; Friedman, T.B.; et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 2003, 12, 3215–3223. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, X.; Xia, X.; Verpy, E.; Du, L.; Pandya, A.; Petit, C.; Balkany, T.; Nance, W.E.; Liu, X. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Qual. Life Res. 2002, 111, 26–30. [Google Scholar] [CrossRef]
- Mburu, P.; Mustapha, M.; Varela, A.; Weil, D.; El-Amraoui, A.; Holme, R.H.; Rump, A.; Hardisty, R.E.; Blanchard, S.; Coimbra, R.S.; et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB. Nat. Genet. 2003, 34, 421–428. [Google Scholar] [CrossRef]
- Riazuddin, S.; Belyantseva, I.A.; Giese, A.P.J.; Lee, K.; Indzhykulian, A.A.; Nandamuri, S.P.; Yousaf, R.; Sinha, G.P.; Lee, S.; Terrell, D.; et al. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat. Genet. 2012, 44, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Oonk, A.M.M.; van Huet, R.A.C.; Leijendeckers, J.M.; Oostrik, J.; Venselaar, H.; van Wijk, E.; Beynon, A.; Kunst, H.P.M.; Hoyng, C.B.; Kremer, H.; et al. Nonsyndromic Hearing Loss Caused by USH1G Mutations. Ear Hear. 2015, 36, 205–211. [Google Scholar] [CrossRef]
- Vona, B.; Lechno, S.; Hofrichter, M.A.H.; Hopf, S.; Läßig, A.K.; Haaf, T.; Keilmann, A.; Zechner, U.; Bartsch, O. Confirmation of PDZD7 as a Nonsyndromic Hearing Loss Gene. Ear Hear. 2016, 37, e238–e246. [Google Scholar] [CrossRef]
- Liu, X.-Z.; Walsh, J.; Tamagawa, Y.; Kitamura, K.; Nishizawa, M.; Steel, K.P.; Brown, S.D. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 1997, 17, 268–269. [Google Scholar] [CrossRef]
- Khan, M.I.; Kersten, F.F.; Azam, M.; Collin, R.W.; Hussain, A.; Shah, S.T.-A.; Keunen, J.E.; Kremer, H.; Cremers, F.P.; Qamar, R.; et al. CLRN1 Mutations Cause Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2011, 118, 1444–1448. [Google Scholar] [CrossRef]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, C.; Grati, M.; Marlin, S.; Levilliers, J.; Hardelin, J.-P.; Parodi, M.; Niasme-Grare, M.; Zelenika, D.; Délépine, M.; Feldmann, D.; et al. Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J. Rare Dis. 2011, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparisi, M.J.; Aller, E.; Fuster-García, C.; García-García, G.; Rodrigo, R.; Vazquez-Manrique, R.P.; Blanco-Kelly, F.; Ayuso, C.; Roux, A.-F.; Jaijo, T.; et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J. Rare Dis. 2014, 9, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, L.; George, S.; Greenberg, J.; Ramesar, R.S. A Founder Mutation inMYO7AUnderlies a Significant Proportion of Usher Syndrome in Indigenous South Africans: Implications for the African Diaspora. Investig. Opthalmol. Vis. Sci. 2015, 56, 6671–6678. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Chen, K.; Wu, X.; Liu, M.; Jiang, H. Compound heterozygous MYO7A mutations segregating Usher syndrome type 2 in a Han family. Int. J. Pediatr. Otorhinolaryngol. 2016, 90, 150–155. [Google Scholar] [CrossRef]
- Fuster-García, C.; García-García, G.; Jaijo, T.; Fornés, N.; Ayuso, C.; Fernández-Burriel, M.; La Morena, A.S.-D.; Aller, E.; Millán, J.M. High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep. 2018, 8, 17113. [Google Scholar] [CrossRef] [PubMed]
- Kimberling, W.; Möller, C.; Davenport, S.; Priluck, I.; Beighton, P.; Greenberg, J.; Reardon, W.; Weston, M.; Kenyon, J.; Grunkemeyer, J.; et al. Linkage of usher syndrome type I gene (USH1B) to the long arm of chromosome 11. Genomics 1992, 14, 988–994. [Google Scholar] [CrossRef]
- Well, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type IB. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Jaijo, T.; Aller, E.; Beneyto, M.; Najera, C.; Graziano, C.; Turchetti, D.; Seri, M.; Ayuso, C.; Baiget, M.; Moreno, F.; et al. MYO7A mutation screening in Usher syndrome type I patients from diverse origins. J. Med. Genet. 2006, 44, e71. [Google Scholar] [CrossRef] [Green Version]
- Jouret, G.; Poirsier, C.; Spodenkiewicz, M.; Jaquin, C.; Gouy, E.; Arndt, C.; Labrousse, M.; Gaillard, D.; Doco-Fenzy, M.; Lebre, A.-S. Genetics of Usher Syndrome: New Insights from a Meta-analysis. Otol. Neurotol. 2019, 40, 121–129. [Google Scholar] [CrossRef]
- Liu, X.-Z.; Hope, C.; Walsh, J.; Newton, V.; Ke, X.M.; Liang, C.Y.; Xu, L.R.; Zhou, J.M.; Trump, D.; Steel, K.P.; et al. Mutations in the Myosin VIIA Gene Cause a Wide Phenotypic Spectrum, Including Atypical Usher Syndrome. Am. J. Hum. Genet. 1998, 63, 909–912. [Google Scholar] [CrossRef] [Green Version]
- Mathur, P.D.; Yang, J. Usher syndrome and non-syndromic deafness: Functions of different whirlin isoforms in the cochlea, vestibular organs, and retina. Hear. Res. 2019, 375, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.; Nazli, S.; Ahmed, Z.M.; Yang, Y.; Zulfiqar, F.; Shaikh, R.S.; Zafar, A.U.; Khan, S.N.; Sabar, F.; Javid, F.T.; et al. Mutation spectrum ofMYO7Aand evaluation of a novel nonsyndromic deafnessDFNB2allele with residual function. Hum. Mutat. 2008, 29, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Kelley, P.M.; Weston, M.D.; Chen, Z.-Y.; Orten, D.J.; Hasson, T.; Overbeck, L.D.; Pinnt, J.; Talmadge, C.B.; Ing, P.; Mooseker, M.S.; et al. The Genomic Structure of the Gene Defective in Usher Syndrome Type Ib (MYO7A). Genome 1997, 40, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, A.; Ikebe, M. Characterization of the Motor Activity of Mammalian Myosin VIIA. J. Biol. Chem. 2003, 278, 5478–5487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; He, Y.; Weck, M.L.; Lu, Q.; Tyska, M.J.; Zhang, M. Structure of Myo7b/USH1C complex suggests a general PDZ domain binding mode by MyTH4-FERM myosins. Proc. Natl. Acad. Sci. USA 2017, 114, E3776–E3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Pan, L.; Wei, Z.; Zhang, M. Structure of MyTH4-FERM Domains in Myosin VIIa Tail Bound to Cargo. Science 2011, 331, 757–760. [Google Scholar] [CrossRef]
- Gibbs, D.; Diemer, T.; Khanobdee, K.; Hu, J.; Bok, D.; Williams, D.S. Function of MYO7A in the Human RPE and the Validity of Shaker1 Mice as a Model for Usher Syndrome 1B. Investig. Opthalmol. Vis. Sci. 2010, 51, 1130–1135. [Google Scholar] [CrossRef]
- Liu, X.; Udovichenko, I.P.; Brown, S.D.; Steel, K.P.; Williams, D.S. Myosin VIIa Participates in Opsin Transport through the Photoreceptor Cilium. J. Neurosci. 1999, 19, 6267–6274. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, U.; Schmitt, A. Rhodopsin transport in the membrane of the connecting cilium of mammalian photoreceptor cells. Cell Motil. Cytoskelet. 2000, 46, 95–107. [Google Scholar] [CrossRef]
- El-Amraoui, A.; Schonn, J.; Küssel-Andermann, P.; Blanchard, S.; Desnos, C.; Henry, J.; Wolfrum, U.; Darchen, F.; Petit, C. MyRIP, a novel Rab effector, enables myosin VIIa recruitment to retinal melanosomes. EMBO Rep. 2002, 3, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, D.; Kitamoto, J.; Williams, D.S. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc. Natl. Acad. Sci. USA 2003, 100, 6481–6486. [Google Scholar] [CrossRef] [Green Version]
- Boëda, B.; El-Amraoui, A.; Bahloul, A.; Goodyear, R.; Daviet, L.; Blanchard, S.; Perfettini, I.; Fath, K.R.; Shorte, S.; Reiners, J.; et al. Myosin VIIa, Harmonin and Cadherin 23, Three Usher I Gene Products That Cooperate to Shape the Sensory Hair Cell Bundle. EMBO J. 2002, 21, 6689–6699. [Google Scholar] [CrossRef] [Green Version]
- Grati, M.; Kachar, B. Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. USA 2011, 108, 11476–11481. [Google Scholar] [CrossRef] [Green Version]
- Kros, C.J.; Marcotti, W.; Van Netten, S.M.; Self, T.J.; Libby, R.T.; Brown, S.D.M.; Richardson, G.P.; Steel, K.P. Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat. Neurosci. 2001, 5, 41–47. [Google Scholar] [CrossRef]
- Bitner-Glindzicz, M.; Lindley, K.J.; Rutland, P.; Blaydon, D.; Smith, V.V.; Milla, P.J.; Hussain, K.; Furth-Lavi, J.; Cosgrove, K.E.; Shepherd, R.M.; et al. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat. Genet. 2000, 26, 56–60. [Google Scholar] [CrossRef]
- Verpy, E.; Leibovici, M.; Zwaenepoel, I.; Liu, X.-Z.; Gal, A.; Salem, N.; Mansour, A.M.; Blanchard, S.; Kobayashi, I.; Keats, B.J.; et al. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet. 2000, 26, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.K.; Lalwani, A.K.; Li, X.C.; Singleton, T.L.; Smith, T.N.; Chen, A.; Deshmukh, D.; Verma, I.C.; Smith, R.J.; Wilcox, E.R. A Gene for Recessive Nonsyndromic Sensorineural Deafness (DFNB18) Maps to the Chromosomal Region 11p14–p15.1 Containing the Usher Syndrome Type 1C Gene. Genomics 1998, 50, 290–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiners, J.; Van Wijk, E.; Märker, T.; Zimmermann, U.; Jürgens, K.; Brinke, H.T.; Overlack, N.; Roepman, R.; Knipper, M.; Kremer, H.; et al. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type. Hum. Mol. Genet. 2005, 14, 3933–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baux, D.; Vaché, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugère, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef] [Green Version]
- Khateb, S.; Zelinger, L.; Ben-Yosef, T.; Merin, S.; Crystal-Shalit, O.; Gross, M.; Banin, E.; Sharon, D. Exome Sequencing Identifies a Founder Frameshift Mutation in an Alternative Exon of USH1C as the Cause of Autosomal Recessive Retinitis Pigmentosa with Late-Onset Hearing Loss. PLoS ONE 2012, 7, e51566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, M.; Sala, C. PDZ Domains and the Organization of Supramolecular Complexes. Annu. Rev. Neurosci. 2001, 24, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Reiners, J.; Nagel-Wolfrum, K.; Jürgens, K.; Märker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Bolz, H.; Von Brederlow, B.; Ramírez, A.; Bryda, E.C.; Kutsche, K.; Nothwang, H.G.; Seeliger, M.; Cabrera, M.D.C.-S.; Vila, M.C.; Molina, O.P.; et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 2001, 27, 108–112. [Google Scholar] [CrossRef]
- Astuto, L.M.; Bork, J.M.; Weston, M.D.; Askew, J.W.; Fields, R.R.; Orten, D.J.; Ohliger, S.J.; Riazuddin, S.; Morell, R.; Khan, S.; et al. CDH23 Mutation and Phenotype Heterogeneity: A Profile of 107 Diverse Families with Usher Syndrome and Nonsyndromic Deafness. Am. J. Hum. Genet. 2002, 71, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Baux, D.; Faugère, V.; Larrieu, L.; Le Guédard-Méreuze, S.; Hamroun, D.; Béroud, C.; Malcolm, S.; Claustres, M.; Roux, A.-F. UMD-USHbases: A comprehensive set of databases to record and analyse pathogenic mutations and unclassified variants in seven Usher syndrome causing genes. Hum. Mutat. 2008, 29, E76–E87. [Google Scholar] [CrossRef]
- Becirovic, E.; Ebermann, I.; Nagy, D.; Zrenner, E.; Seeliger, M.W.; Bolz, H.J. Usher syndrome type 1 due to missense mutations on bothCDH23 alleles: Investigation of mRNA splicing. Hum. Mutat. 2008, 29, 452. [Google Scholar] [CrossRef]
- Schultz, J.M.; Bhatti, R.; Madeo, A.C.; Turriff, A.; Muskett, J.A.; Zalewski, C.K.; King, K.A.; Ahmed, Z.M.; Riazuddin, S.; Ahmad, N.; et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J. Med. Genet. 2011, 48, 767–775. [Google Scholar] [CrossRef]
- Jaiganesh, A.; Narui, Y.; Araya-Secchi, R.; Sotomayor, M. Beyond Cell–Cell Adhesion: Sensational Cadherins for Hearing and Balance. Cold Spring Harb. Perspect. Biol. 2017, 10, a029280. [Google Scholar] [CrossRef]
- Sahly, I.; Dufour, E.; Schietroma, C.; Michel, V.; Bahloul, A.; Perfettini, I.; Pepermans, E.; Estivalet, A.; Carette, D.; Aghaie, A.; et al. Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol. 2012, 199, 381–399. [Google Scholar] [CrossRef]
- Schietroma, C.; Parain, K.; Estivalet, A.; Aghaie, A.; De Monvel, J.B.; Picaud, S.; Sahel, J.-A.; Perron, M.; El-Amraoui, A.; Petit, C. Usher syndrome type 1–associated cadherins shape the photoreceptor outer segment. J. Cell Biol. 2017, 216, 1849–1864. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Mui, V.J.; Rosenberg, S.K.; Homma, K.; Cheatham, M.A.; Zheng, J. Cadherin 23-C Regulates Microtubule Networks by Modifying CAMSAP3’s Function. Sci. Rep. 2016, 6, 28706. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.M.; Riazuddin, S.; Bernstein, S.L.; Ahmed, Z.; Khan, S.; Griffith, A.J.; Morell, R.; Friedman, T.B.; Riazuddin, S.; Wilcox, E.R. Mutations of the Protocadherin Gene PCDH15 Cause Usher Syndrome Type 1F. Am. J. Hum. Genet. 2001, 69, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.M.; Goodyear, R.; Riazuddin, S.; Lagziel, A.; Legan, P.K.; Behra, M.; Burgess, S.M.; Lilley, K.S.; Wilcox, E.R.; Griffith, A.J.; et al. The Tip-Link Antigen, a Protein Associated with the Transduction Complex of Sensory Hair Cells, Is Protocadherin-15. J. Neurosci. 2006, 26, 7022–7034. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.M.; Riazuddin, S.; Aye, S.; Ali, R.A.; Venselaar, H.; Anwar, S.; Belyantseva, P.P.; Qasim, M.; Riazuddin, S.; Friedman, T.B. Gene structure and mutant alleles of PCDH15: Nonsyndromic deafness DFNB23 and type 1 Usher syndrome. Qual. Life Res. 2008, 124, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagramam, K.N.; Yuan, H.; Kuehn, M.H.; Murcia, C.L.; Wayne, S.; Srisailpathy, C.S.; Lowry, R.B.; Knaus, R.; Van Laer, L.; Bernier, F.; et al. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum. Mol. Genet. 2001, 10, 1709–1718. [Google Scholar] [CrossRef] [Green Version]
- Kazmierczak, P.; Sakaguchi, H.; Tokita, J.; Wilson-Kubalek, E.M.; Milligan, R.A.; Müller, U.; Kachar, B. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nat. Cell Biol. 2007, 449, 87–91. [Google Scholar] [CrossRef]
- McGrath, J.; Roy, P.; Perrin, B.J. Stereocilia morphogenesis and maintenance through regulation of actin stability. Semin. Cell Dev. Biol. 2017, 65, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya-Secchi, R.; Neel, B.L.; Sotomayor, M. An elastic element in the protocadherin-15 tip link of the inner ear. Nat. Commun. 2016, 7, 13458. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, T.F.; Hengel, F.E.; Oswald, A.; Dionne, G.; Chipendo, I.V.; Mangat, S.S.; El Shatanofy, M.; Shapiro, L.; Müller, U.; Hudspeth, A.J. Elasticity of individual protocadherin 15 molecules implicates tip links as the gating springs for hearing. Proc. Natl. Acad. Sci. USA 2019, 116, 11048–11056. [Google Scholar] [CrossRef] [Green Version]
- Powers, R.E.; Gaudet, R.; Sotomayor, M. A Partial Calcium-Free Linker Confers Flexibility to Inner-Ear Protocadherin-15. Structure 2017, 25, 482–495. [Google Scholar] [CrossRef]
- Weil, D.; El-Amraoui, A.; Masmoudi, S.; Mustapha, M.; Kikkawa, Y.; Lainé, S.; Delmaghani, S.; Adato, A.; Nadifi, S.; Ben Zina, Z.; et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum. Mol. Genet. 2003, 12, 463–471. [Google Scholar] [CrossRef]
- Bashir, R.; Fatima, A.; Naz, S. A frameshift mutation in SANS results in atypical Usher syndrome. Clin. Genet. 2010, 78, 601–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalay, E.; De Brouwer, A.P.M.; Caylan, R.; Nabuurs, S.B.; Wollnik, B.; Karaguzel, A.; Heister, J.G.A.M.; Erdol, H.; Cremers, F.P.M.; Cremers, C.W.R.J.; et al. A novel D458V mutation in the SANS PDZ binding motif causes atypical Usher syndrome. J. Mol. Med. 2005, 83, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Bauß, K.; Knapp, B.; Jores, P.; Roepman, R.; Kremer, H.; Wijk, E.V.; Märker, T.; Wolfrum, U. Phosphorylation of the Usher syndrome 1G protein SANS controls Magi2-mediated endocytosis. Hum. Mol. Genet. 2014, 23, 3923–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberlotto, E.; Michel, V.; Foucher, I.; Bahloul, A.; Goodyear, R.J.; Pepermans, E.; Michalski, N.; Perfettini, I.; Alegria-Prévot, O.; Chardenoux, S.; et al. Usher type 1G protein sans is a critical component of the tip-link complex, a structure controlling actin polymerization in stereocilia. Proc. Natl. Acad. Sci. USA 2011, 108, 5825–5830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maerker, T.; Van Wijk, E.; Overlack, N.; Kersten, F.F.; McGee, J.; Goldmann, T.; Sehn, E.; Roepman, R.; Walsh, E.J.; Kremer, H.; et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet. 2007, 17, 71–86. [Google Scholar] [CrossRef]
- Overlack, N.; Kilic, D.; Bauß, K.; Märker, T.; Kremer, H.; van Wijk, E.; Wolfrum, U. Direct interaction of the Usher syndrome 1G protein SANS and myomegalin in the retina. Biochim. Biophys. Acta Bioenerg. 2011, 1813, 1883–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorusch, N.; Wunderlich, K.; Bauss, K.; Nagel-Wolfrum, K.; Wolfrum, U. Usher Syndrome Protein Network Functions in the Retina and their Relation to Other Retinal Ciliopathies. Adv. Exp. Med. Biol. 2014, 801, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Sorusch, N.; Bauß, K.; Plutniok, J.; Samanta, A.; Knapp, B.; Nagel-Wolfrum, K.; Wolfrum, U. Characterization of the ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum. Mol. Genet. 2017, 26, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Jacoszek, A.; Pollak, A.; Płoski, R.; Ołdak, M. Advances in genetic hearing loss: CIB2 gene. Eur. Arch. Oto-Rhino-Laryngol. 2016, 274, 1791–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, A.P.J.; Tang, Y.-Q.; Sinha, G.P.; Bowl, M.; Goldring, A.C.; Parker, A.; Freeman, M.; Brown, S.D.M.; Riazuddin, S.; Fettiplace, R.; et al. CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Booth, K.T.; Patni, P.; Cortese, M.; Azaiez, H.; Bahloul, A.; Kahrizi, K.; Labbé, M.; Emptoz, A.; Lelli, A.; et al. CIB2, defective in isolated deafness, is key for auditory hair cell mechanotransduction and survival. EMBO Mol. Med. 2017, 9, 1711–1731. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Yao, X.; Li, W.; Du, H.; Tang, M.; Xiong, W.; Chai, R.; Xu, Z. Loss of CIB2 Causes Profound Hearing Loss and Abolishes Mechanoelectrical Transduction in Mice. Front. Mol. Neurosci. 2017, 10, 401. [Google Scholar] [CrossRef] [Green Version]
- Booth, K.; Kahrizi, K.; Babanejad, M.; Daghagh, H.; Bademci, G.; Arzhangi, S.; Zareabdollahi, D.; Duman, D.; El-Amraoui, A.; Tekin, M.; et al. Variants in CIB2 cause DFNB48 and not USH1J. Clin. Genet. 2018, 93, 812–821. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Weston, M.D.; Möller, C.; Davenport, S.L.; Shugart, Y.Y.; Priluck, I.A.; Martini, A.; Milani, M.; Smith, R.J. Localization of Usher syndrome type II to chromosome 1q. Genomics 1990, 7, 245–249. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Weston, M.D.; Möller, C.; Van Aarem, A.; Cremers, C.W.R.J.; Sumegi, J.; Ing, P.S.; Connolly, C.; Martini, A.; Milani, M.; et al. Gene Mapping of Usher Syndrome Type IIa: Localization of the Gene to a 2.1-cM Segment on Chromosome 1q41. Am. J. Hum. Genet. 1995, 56, 216–223. [Google Scholar]
- Dreyer, B.; Tranebjærg, L.; Brox, V.; Rosenberg, T.; Möller, C.; Beneyto, M.; Weston, M.D.; Kimberling, W.J.; Nilssen, Ø. A Common Ancestral Origin of the Frequent and Widespread 2299delG USH2A Mutation. Am. J. Hum. Genet. 2001, 69, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Aller, E.; Larrieu, L.; Jaijo, T.; Baux, D.; Espinós, C.; González-Candelas, F.; Nájera, C.; Palau, F.; Claustres, M.; Roux, A.-F.; et al. The USH2A c.2299delG mutation: Dating its common origin in a Southern European population. Eur. J. Hum. Genet. 2010, 18, 788–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGee, T.L.; Seyedahmadi, B.J.; Sweeney, M.O.; Dryja, T.P.; Berson, E.L. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J. Med. Genet. 2010, 47, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense Mutation in the USH2A Gene: Association with Recessive Retinitis Pigmentosa without Hearing Loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eudy, J.D.; Weston, M.D.; Yao, S.; Hoover, D.M.; Rehm, H.L.; Ma-Edmonds, M.; Yan, D.; Ahmad, I.; Cheng, J.J.; Ayuso, C.; et al. Mutation of a Gene Encoding a Protein with Extracellular Matrix Motifs in Usher Syndrome Type IIa. Science 1998, 280, 1753–1757. [Google Scholar] [CrossRef]
- Weston, M.; Eudy, J.; Fujita, S.; Yao, S.-F.; Usami, S.; Cremers, C.; Greenburg, J.; Ramesar, R.; Martini, A.; Moller, C.; et al. Genomic Structure and Identification of Novel Mutations in Usherin, the Gene Responsible for Usher Syndrome Type IIa. Am. J. Hum. Genet. 2000, 66, 1199–1210. [Google Scholar] [CrossRef] [Green Version]
- van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 Novel Exons of the Usher Syndrome Type 2A (USH2A) Gene That Encode Multiple Conserved Functional Domains and That Are Mutated in Patients with Usher Syndrome Type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adato, A.; Lefèvre, G.; Delprat, B.; Michel, V.; Michalski, N.; Chardenoux, S.; Weil, D.; El-Amraoui, A.; Petit, C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 2005, 14, 3921–3932. [Google Scholar] [CrossRef]
- Michalski, N.; Michel, V.; Bahloul, A.; Lefèvre, G.; Barral, J.; Yagi, H.; Chardenoux, S.; Weil, D.; Martin, P.; Hardelin, J.-P.; et al. Molecular Characterization of the Ankle-Link Complex in Cochlear Hair Cells and Its Role in the Hair Bundle Functioning. J. Neurosci. 2007, 27, 6478–6488. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, L.; Song, H.; Sokolov, M. Current Understanding of Usher Syndrome Type II. Front. Biosci. 2012, 17, 1165–1183. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, G.; Miller, C.; Kimberling, W.J.; Jablonski, M.M.; Cosgrove, D. Localization and expression of usherin: A novel basement membrane protein defective in people with Usher’s syndrome type IIa. Hear. Res. 2002, 163, 1–11. [Google Scholar] [CrossRef]
- Weston, M.D.; Luijendijk, M.W.; Humphrey, K.D.; Möller, C.; Kimberling, W.J. Mutations in the VLGR1 Gene Implicate G-Protein Signaling in the Pathogenesis of Usher Syndrome Type II. Am. J. Hum. Genet. 2004, 74, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, D.R.; Kayes-Wandover, K.M.; Richardson, J.A.; White, P.C. Very Large G Protein-coupled Receptor-1, the Largest Known Cell Surface Protein, Is Highly Expressed in the Developing Central Nervous System. J. Biol. Chem. 2002, 277, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.-P.; Li, R.; Ren, H.-Z.; Xu, A.-T.; Yu, X.; Xu, Z.-G. The Very Large G Protein Coupled Receptor (Vlgr1) in Hair Cells. J. Mol. Neurosci. 2012, 50, 204–214. [Google Scholar] [CrossRef]
- Yagi, H.; Tokano, H.; Maeda, M.; Takabayashi, T.; Nagano, T.; Kiyama, H.; Fujieda, S.; Kitamura, K.; Sato, M. Vlgr1 is required for proper stereocilia maturation of cochlear hair cells. Genes Cells 2007, 12, 235–250. [Google Scholar] [CrossRef]
- Ebermann, I.; Scholl, H.P.N.; Issa, P.C.; Becirovic, E.; Lamprecht, J.; Jurklies, B.; Millán, J.M.; Aller, E.; Mitter, D.; Bolz, H. A novel gene for Usher syndrome type 2: Mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Qual. Life Res. 2006, 121, 203–211. [Google Scholar] [CrossRef]
- Van Wijk, E.; Van Der Zwaag, B.; Peters, T.; Zimmermann, U.; Brinke, H.T.; Kersten, F.F.; Märker, T.; Aller, E.; Hoefsloot, L.H.; Cremers, C.W.; et al. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet. 2006, 15, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef] [Green Version]
- Tlili, A.; Charfedine, I.; Lahmar, I.; Benzina, Z.; Mohamed, B.A.; Weil, D.; Idriss, N.; Drira, M.; Masmoudi, S.; Ayadi, H. Identification of a novel frameshift mutation in the DFNB31/WHRN gene in a Tunisian consanguineous family with hereditary non-syndromic recessive hearing loss. Hum. Mutat. 2005, 25, 503. [Google Scholar] [CrossRef]
- Audo, I.; Bujakowska, K.; Mohand-Saïd, S.; Tronche, S.; Lancelot, M.-E.; Antonio, A.; Germain, A.; Lonjou, C.; Carpentier, W.; Sahel, J.-A.; et al. A novel DFNB31 mutation associated with Usher type 2 syndrome showing variable degrees of auditory loss in a consanguineous Portuguese family. Mol. Vis. 2011, 17, 1598–1606. [Google Scholar]
- Besnard, T.; Vaché, C.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Odent, S.; Blanchet, P.; Calvas, P.; Hamel, C.; et al. Non-USH2A mutations in USH2 patients. Hum. Mutat. 2012, 33, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liang, X.; Li, Y.; Wang, J.; Zaneveld, J.E.; Wang, H.; Xu, S.; Wang, K.; Wang, B.; Chen, R.; et al. Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: High rate of ethnicity specific mutations in Chinese USH patients. Orphanet J. Rare Dis. 2015, 10, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Wei, X.; Chai, Y.; Li, L.; Wu, H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J. Rare Dis. 2013, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Ebrahim, S.; Ingham, N.J.; Lewis, M.A.; Rogers, M.J.; Cui, R.; Kachar, B.; Pass, J.C.; Steel, K.P. Alternative Splice Forms Influence Functions of Whirlin in Mechanosensory Hair Cell Stereocilia. Cell Rep. 2016, 15, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, X.; Zhao, Y.; Adamian, M.; Pawlyk, B.S.; Sun, X.; McMillan, D.R.; Liberman, M.C.; Li, T. Ablation of Whirlin Long Isoform Disrupts the USH2 Protein Complex and Causes Vision and Hearing Loss. PLoS Genet. 2010, 6, e1000955. [Google Scholar] [CrossRef]
- Delprat, B.; Michel, V.; Goodyear, R.; Yamasaki, Y.; Michalski, N.; El-Amraoui, A.; Perfettini, I.; Legrain, P.; Richardson, G.; Hardelin, J.-P.; et al. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum. Mol. Genet. 2004, 14, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Holme, R.H.; Kiernan, B.W.; Brown, S.D.; Steel, K.P. Elongation of hair cell stereocilia is defective in the mouse mutant whirler. J. Comp. Neurol. 2002, 450, 94–102. [Google Scholar] [CrossRef]
- Kikkawa, Y.; Mburu, P.; Morse, S.; Kominami, R.; Townsend, S.; Brown, S.D. Mutant analysis reveals whirlin as a dynamic organizer in the growing hair cell stereocilium. Hum. Mol. Genet. 2004, 14, 391–400. [Google Scholar] [CrossRef]
- Manor, U.; Disanza, A.; Grati, M.; Andrade, L.; Lin, H.; Di Fiore, P.P.; Scita, G.; Kachar, B. Regulation of Stereocilia Length by Myosin XVa and Whirlin Depends on the Actin-Regulatory Protein Eps8. Curr. Biol. 2011, 21, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Kersten, F.F.J.; Van Wijk, E.; Van Reeuwijk, J.; Van Der Zwaag, B.; Märker, T.; Peters, T.A.; Katsanis, N.; Wolfrum, U.; Keunen, J.E.E.; Roepman, R.; et al. Association of Whirlin with Cav1.3 (α1D) Channels in Photoreceptors, Defining a Novel Member of the Usher Protein Network. Investig. Opthalmol. Vis. Sci. 2010, 51, 2338–2346. [Google Scholar] [CrossRef] [Green Version]
- Sankila, E.-M.; Pakarinen, L.; Kääriäinen, H.; Aittomäki, K.; Karjalainen, S.; Sistonen, P.; De La Chapelle, A. Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. Hum. Mol. Genet. 1995, 4, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, T.; Hämäläinen, R.; Yuan, B.; Johnson, C.; Tegelberg, S.; Gasparini, P.; Zelante, L.; Pirvola, U.; Pakarinen, L.; Lehesjoki, A.-E.; et al. Mutations in a Novel Gene with Transmembrane Domains Underlie Usher Syndrome Type 3. Am. J. Hum. Genet. 2001, 69, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Adato, A.; Vreugde, S.; Joensuu, T.; Avidan, N.; Hamalainen, R.H.; Belenkiy, O.; Olender, T.; Bonne-Tamir, B.; Ben-Asher, E.; Espinos, C.; et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 2002, 10, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Västinsalo, H.; Jalkanen, R.; Dinculescu, A.; Isosomppi, J.; Geller, S.; Flannery, J.G.; Hauswirth, W.W.; Sankila, E.-M. Alternative splice variants of the USH3A gene Clarin 1 (CLRN1). Eur. J. Hum. Genet. 2010, 19, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogun, O.; Zallocchi, M. Clarin-1 acts as a modulator of mechanotransduction activity and presynaptic ribbon assembly. J. Cell Biol. 2014, 207, 375–391. [Google Scholar] [CrossRef] [Green Version]
- Geng, R.; Geller, S.F.; Hayashi, T.; Ray, C.A.; Reh, T.A.; Bermingham-McDonogh, O.; Jones, S.M.; Wright, C.G.; Melki, S.; Imanishi, Y.; et al. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum. Mol. Genet. 2009, 18, 2748–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zallocchi, M.; Meehan, D.T.; Delimont, D.; Askew, C.; Garige, S.; Gratton, M.A.; Rothermund-Franklin, C.A.; Cosgrove, D. Localization and expression of clarin-1, the Clrn1 gene product, in auditory hair cells and photoreceptors. Hear. Res. 2009, 255, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Eisenberger, T.; Slim, R.; Mansour, A.; Nauck, M.; Nürnberg, G.; Nürnberg, P.; Decker, C.; Dafinger, C.; Ebermann, I.; Bergmann, C.; et al. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J. Rare Dis. 2012, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puffenberger, E.G.; Jinks, R.N.; Sougnez, C.; Cibulskis, K.; Willert, R.A.; Achilly, N.P.; Cassidy, R.P.; Fiorentini, C.J.; Heiken, K.F.; Lawrence, J.J.; et al. Genetic Mapping and Exome Sequencing Identify Variants Associated with Five Novel Diseases. PLoS ONE 2012, 7, e28936. [Google Scholar] [CrossRef] [Green Version]
- Vester, A.; Velez-Ruiz, G.; McLaughlin, H.M.; Program, T.N.C.S.; Lupski, J.R.; Talbot, K.; Vance, J.; Züchner, S.; Roda, R.H.; Fischbeck, K.H.; et al. A Loss-of-Function Variant in the Human Histidyl-tRNA Synthetase (HARS) Gene is Neurotoxic In Vivo. Hum. Mutat. 2013, 34, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Brozkova, D.S.; Deconinck, T.; Griffin, L.B.; Ferbert, A.; Haberlova, J.; Mazanec, R.; Lassuthova, P.; Roth, C.; Pilunthanakul, T.; Rautenstrauss, B.; et al. Loss of function mutations inHARScause a spectrum of inherited peripheral neuropathies. Brain 2015, 138, 2161–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galatolo, D.; Kuo, M.E.; Mullen, P.; Meyer-Schuman, R.; Doccini, S.; Battini, R.; Lieto, M.; Tessa, A.; Filla, A.; Francklyn, C.; et al. Bi-allelic mutations in HARS1 severely impair histidyl-tRNA synthetase expression and enzymatic activity causing a novel multisystem ataxic syndrome. Hum. Mutat. 2020, 41, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schuman, R.; Antonellis, A. Evidence for a dominant-negative mechanism in HARS1-mediated peripheral neuropathy. FEBS J. 2021, 288, 91–94. [Google Scholar] [CrossRef]
- Khateb, S.; Zelinger, L.; Mizrahi-Meissonnier, L.; Ayuso, C.; Koenekoop, R.K.; Laxer, U.; Gross, M.; Banin, E.; Sharon, D. A homozygous nonsense CEP250 mutation combined with a heterozygous nonsense C2orf71 mutation is associated with atypical Usher syndrome. J. Med. Genet. 2014, 51, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Kubota, D.; Gocho, K.; Kikuchi, S.; Akeo, K.; Miura, M.; Yamaki, K.; Takahashi, H.; Kameya, S. CEP250 mutations associated with mild cone-rod dystrophy and sensorineural hearing loss in a Japanese family. Ophthalmic Genet. 2018, 39, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Khateb, S.; Kowalewski, B.; Bedoni, N.; Damme, M.; Pollack, N.; Saada, A.; Obolensky, A.; Ben-Yosef, T.; Gross, M.; Dierks, T.; et al. A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet. Med. 2018, 20, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad-Morales, V.; Navarro, R.; Burés-Jelstrup, A.; Pomares, E. Identification of a novel homozygous ARSG mutation as the second cause of Usher syndrome type 4. Am. J. Ophthalmol. Case Rep. 2020, 19, 100736. [Google Scholar] [CrossRef]
- Peter, V.G.; Quinodoz, M.; Sadio, S.; Held, S.; Rodrigues, M.; Soares, M.; Sousa, A.B.; Santos, L.C.; Damme, M.; Rivolta, C. New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV. Hum. Mutat. 2021, 42, 261–271. [Google Scholar] [CrossRef]
- Namburi, P.; Ratnapriya, R.; Khateb, S.; Lazar, C.H.; Kinarty, Y.; Obolensky, A.; Erdinest, I.; Marks-Ohana, D.; Pras, E.; Ben-Yosef, T.; et al. Bi-allelic Truncating Mutations in CEP78, Encoding Centrosomal Protein 78, Cause Cone-Rod Degeneration with Sensorineural Hearing Loss. Am. J. Hum. Genet. 2016, 99, 777–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikopoulos, K.; Farinelli, P.; Giangreco, B.; Tsika, C.; Royer-Bertrand, B.; Mbefo, M.K.; Bedoni, N.; Kjellström, U.; El Zaoui, I.; Di Gioia, S.A.; et al. Mutations in CEP78 Cause Cone-Rod Dystrophy and Hearing Loss Associated with Primary-Cilia Defects. Am. J. Hum. Genet. 2016, 99, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Xu, M.; Chen, X.; Sheng, X.; Yuan, Z.; Liu, Y.; Li, H.; Sun, Z.; Li, H.; Yang, L.; et al. CEP78is mutated in a distinct type of Usher syndrome. J. Med. Genet. 2017, 54, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.M.; Jaworek, T.J.; Sarangdhar, G.N.; Zheng, L.; Gul, K.; Khan, S.N.; Friedman, T.B.; Sisk, R.A.; Bartles, J.R.; Riazuddin, S.; et al. Inframe deletion of human ESPN is associated with deafness, vestibulopathy and vision impairment. J. Med. Genet. 2018, 55, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wei, B.; Fu, X.; Wang, Y.; Sui, Y.; Ma, J.; Gong, X.; Hao, J.; Xing, S. Identification of whirlin domains interacting with espin: A study of the mechanism of Usher syndrome type II. Mol. Med. Rep. 2019, 20, 5111–5117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebermann, I.; Phillips, J.B.; Liebau, M.C.; Koenekoop, R.K.; Schermer, B.; Lopez, I.; Schäfer, E.; Roux, A.-F.; Dafinger, C.; Bernd, A.; et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Investig. 2010, 120, 1812–1823. [Google Scholar] [CrossRef] [Green Version]
- García, C.F. Therapeutic Approaches and Development of Genomic Diagnostic Tools for Usher Syndrome; Universitat Politecnica de Valencia: Valencia, Spain, 2020. [Google Scholar]
- Siemens, J.; Kazmierczak, P.; Reynolds, A.; Sticker, M.; Littlewood-Evans, A.; Müller, U. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc. Natl. Acad. Sci. USA 2002, 99, 14946–14951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adato, A.; Michel, V.; Kikkawa, Y.; Reiners, J.; Alagramam, K.N.; Weil, D.; Yonekawa, H.; Wolfrum, U.; El-Amraoui, A.; Petit, C. Interactions in the network of Usher syndrome type 1 proteins. Hum. Mol. Genet. 2004, 14, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Reiners, J.; Märker, T.; Jürgens, K.; Reidel, B.; Wolfrum, U. Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C). Mol. Vis. 2005, 11, 347–355. [Google Scholar]
- Senften, M.; Schwander, M.; Kazmierczak, P.; Lillo, C.; Shin, J.-B.; Hasson, T.; Géléoc, G.S.G.; Gillespie, P.G.; Williams, D.; Holt, J.R.; et al. Physical and Functional Interaction between Protocadherin 15 and Myosin VIIa in Mechanosensory Hair Cells. J. Neurosci. 2006, 26, 2060–2071. [Google Scholar] [CrossRef]
- Dulon, D.; Papal, S.; Patni, P.; Cortese, M.; Vincent, M.; Tertrais, M.; Emptoz, A.; Tlili, A.; Bouleau, Y.; Michel, V.; et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J. Clin. Investig. 2018, 128, 3382–3401. [Google Scholar] [CrossRef]
- Grillet, N.; Xiong, W.; Reynolds, A.; Kazmierczak, P.; Sato, T.; Lillo, C.; Dumont, R.A.; Hintermann, E.; Sczaniecka, A.; Schwander, M.; et al. Harmonin Mutations Cause Mechanotransduction Defects in Cochlear Hair Cells. Neuron 2009, 62, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahloul, A.; Michel, V.; Hardelin, J.-P.; Nouaille, S.; Hoos, S.; Houdusse, A.; England, P.; Petit, C. Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Hum. Mol. Genet. 2010, 19, 3557–3565. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Sánchez, B.; Clément, A.; Junior, J.F.; Washbourne, P.; Westerfield, M. Usher protein complexes preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis. Model. Mech. 2014, 7, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Yan, J.; Wu, L.; Zhang, M. Assembling stable hair cell tip link complex via multidentate interactions between harmonin and cadherin. Proc. Natl. Acad. Sci. USA 2009, 106, 5575–5580. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Zheng, J.; Whitlon, D.S.; García-Añoveros, J.; Bartles, J.R. Targeting of the Hair Cell Proteins Cadherin 23, Harmonin, Myosin XVa, Espin, and Prestin in an Epithelial Cell Model. J. Neurosci. 2010, 30, 7187–7201. [Google Scholar] [CrossRef]
- Sotomayor, M.; Weihofen, W.A.; Gaudet, R.; Corey, D.P. Structure of a force-conveying cadherin bond essential for inner-ear mechanotransduction. Nat. Cell Biol. 2012, 492, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Frolenkov, G.I.; Belyantseva, I.A.; Friedman, T.B.; Griffith, A.J. Genetic insights into the morphogenesis of inner ear hair cells. Nat. Rev. Genet. 2004, 5, 489–498. [Google Scholar] [CrossRef]
- Petit, C.; Richardson, G.P. Linking genes underlying deafness to hair-bundle development and function. Nat. Neurosci. 2009, 12, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C. Educational paper. Eur. J. Nucl. Med. Mol. Imaging 2011, 171, 1285–1300. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Shivanna, M.; Anand, M.; Chakrabarti, S.; Khanna, H. Ocular Ciliopathies: Genetic and Mechanistic Insights into Developing Therapies. Curr. Med. Chem. 2019, 26, 3120–3131. [Google Scholar] [CrossRef]
- Bujakowska, K.M.; Liu, Q.; Pierce, E.A. Photoreceptor Cilia and Retinal Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028274. [Google Scholar] [CrossRef]
- Gerth-Kahlert, C.; Koller, S. Retinale Ziliopathien. Klinische Monatsblätter Augenheilkunde 2018, 235, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Usher Syndrome. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 1085, pp. 167–170. [Google Scholar]
- Bonneau, D.; Raymond, F.; Kremer, C.; Klossek, J.M.; Kaplan, J.; Patte, F. Usher syndrome type I associated with bronchiectasis and immotile nasal cilia in two brothers. J. Med. Genet. 1993, 30, 253–254. [Google Scholar] [CrossRef]
- Ribeiro, J.C.; Oliveiros, B.; Pereira, P.; António, N.; Hummel, T.; Paiva, A.; Silva, E.D. Accelerated age-related olfactory decline among type 1 Usher patients. Sci. Rep. 2016, 6, 28309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zrada, S.E.; Braat, K.; Doty, R.L.; Laties, A.M. Olfactory loss in Usher syndrome: Another sensory deficit? Am. J. Med. Genet. 1996, 64, 602–603. [Google Scholar] [CrossRef]
- Aparisi, M.J.; García-García, G.; Aller, E.; Sequedo, M.D.; de la Camara, C.M.-F.; Rodrigo, R.; Armengot, M.; Cortijo, J.; Milara, J.; Díaz-Llopis, M.; et al. Study of USH1 Splicing Variants through Minigenes and Transcript Analysis from Nasal Epithelial Cells. PLoS ONE 2013, 8, e57506. [Google Scholar] [CrossRef]
- Ciardo, M.G.; Borderia, D.A.; Cuesta, N.; Valente, P.; Camprubí-Robles, M.; Yang, J.; Planells-Cases, R.; Ferrer-Montiel, A. Whirlin increases TRPV1 channel expression and cellular stability. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.G.; Fishman, G.A.; Mehta, R.S.; Kretzer, F.L. Abnormal Sperm and Photoreceptor Axonemes in Usher’s Syndrome. Arch. Ophthalmol. 1986, 104, 385–389. [Google Scholar] [CrossRef]
- Frenzel, H.; Bohlender, J.; Pinsker, K.; Wohlleben, B.; Tank, J.; Lechner, S.G.; Schiska, D.; Jaijo, T.; Rüschendorf, F.; Saar, K.; et al. A Genetic Basis for Mechanosensory Traits in Humans. PLoS Biol. 2012, 10, e1001318. [Google Scholar] [CrossRef] [Green Version]
- Moshourab, R.; Bégay, V.; Wetzel, C.; Walcher, J.; Middleton, S.; Gross, M.; Lewin, G.R. Congenital deafness is associated with specific somatosensory deficits in adolescents. Sci. Rep. 2017, 7, 4251. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, F.; Bégay, V.; García-García, G.; Taberner, F.J.; Moshourab, R.; McDonald, B.; Docter, T.; Kühnemund, J.; Ojeda-Alonso, J.; Paricio-Montesinos, R.; et al. USH2A is a Meissner’s corpuscle protein necessary for normal vibration sensing in mice and humans. Nat. Neurosci. 2021, 24, 74–81. [Google Scholar] [CrossRef]
- Géléoc, G.G.; El-Amraoui, A. Disease mechanisms and gene therapy for Usher syndrome. Hear. Res. 2020, 394, 107932. [Google Scholar] [CrossRef] [PubMed]
- Lentz, J.J.; Gordon, W.C.; Farris, H.E.; Macdonald, G.H.; Cunningham, D.E.; Robbins, C.A.; Tempel, B.L.; Bazan, N.G.; Rubel, E.W.; Oesterle, E.C.; et al. Deafness and retinal degeneration in a novel USH1C knock-in mouse model. Dev. Neurobiol. 2010, 70, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Zhang, L.; Qi, L.-S.; Liu, W.; An, J.; Wang, B.; Xue, J.-H.; Zhang, Z.-M. The Time Course of Deafness and Retinal Degeneration in a Kunming Mouse Model for Usher Syndrome. PLoS ONE 2016, 11, e0155619. [Google Scholar] [CrossRef] [PubMed]
- Dona, M.; Slijkerman, R.; Lerner, K.; Broekman, S.; Wegner, J.; Howat, T.; Peters, T.; Hetterschijt, L.; Boon, N.; de Vrieze, E.; et al. Usherin defects lead to early-onset retinal dysfunction in zebrafish. Exp. Eye Res. 2018, 173, 148–159. [Google Scholar] [CrossRef]
- McGee, J.; Goodyear, R.J.; McMillan, D.R.; Stauffer, E.A.; Holt, J.R.; Locke, K.G.; Birch, D.G.; Legan, P.K.; White, P.C.; Walsh, E.J.; et al. The Very Large G-Protein-Coupled Receptor VLGR1: A Component of the Ankle Link Complex Required for the Normal Development of Auditory Hair Bundles. J. Neurosci. 2006, 26, 6543–6553. [Google Scholar] [CrossRef]
- Blanco-Sánchez, B.; Clément, A.; Phillips, J.; Westerfield, M. Zebrafish models of human eye and inner ear diseases. Meth. Cell Biol. 2017, 138, 415–467. [Google Scholar] [CrossRef]
- Han, S.; Liu, X.; Xie, S.; Gao, M.; Liu, F.; Yu, S.; Sun, P.; Wang, C.; Archacki, S.; Lu, Z.; et al. Knockout of ush2a gene in zebrafish causes hearing impairment and late onset rod-cone dystrophy. Qual. Life Res. 2018, 137, 779–794. [Google Scholar] [CrossRef]
- Ernest, S.; Rauch, G.-J.; Haffter, P.; Geisler, R.; Petit, C.; Nicolson, T. Mariner is defective in myosin VIIA: A zebrafish model for human hereditary deafness. Hum. Mol. Genet. 2000, 9, 2189–2196. [Google Scholar] [CrossRef] [Green Version]
- Wasfy, M.M.; Matsui, J.I.; Miller, J.; Dowling, J.E.; Perkins, B.D. myosin 7aa−/− mutant zebrafish show mild photoreceptor degeneration and reduced electroretinographic responses. Exp. Eye Res. 2014, 122, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Gibson, F.; Walsh, J.P.; Mburu, P.; Varela, A.; Brown, K.A.; Antonio, M.R.D.S.; Beisel, K.W.; Steel, K.P.; Brown, S.D.M. A type VII myosin encoded by the mouse deafness gene shaker-1. Nat. Cell Biol. 1995, 374, 62–64. [Google Scholar] [CrossRef]
- Mburu, P.; Liu, X.Z.; Walsh, J.; Saw, D.; Cope, J.; Gibson, F.; Kendrick-Jones, J.; Steel, K.; Brown, S. Mutation analysis of the mouse myosin VIIA deafness gene. Genes Funct. 1997, 1, 191–203. [Google Scholar] [CrossRef]
- Rhodes, C.R.; Hertzano, R.; Fuchs, H.; Bell, R.E.; de Angelis, M.H.; Steel, K.P.; Avraham, K.B. A Myo7a mutation cosegregates with stereocilia defects and low-frequency hearing impairment. Mamm. Genome 2004, 15, 686–697. [Google Scholar] [CrossRef]
- Schwander, M.; Sczaniecka, A.; Grillet, N.; Bailey, J.S.; Avenarius, M.; Najmabadi, H.; Steffy, B.M.; Federe, G.C.; Lagler, E.A.; Banan, R.; et al. A Forward Genetics Screen in Mice Identifies Recessive Deafness Traits and Reveals That Pejvakin Is Essential for Outer Hair Cell Function. J. Neurosci. 2007, 27, 2163–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwander, M.; Lopes, V.; Sczaniecka, A.; Gibbs, D.; Lillo, C.; Delano, D.; Tarantino, L.M.; Wiltshire, T.; Williams, D.S.; Müller, U. A Novel Allele of Myosin VIIa Reveals a Critical Function for the C-Terminal FERM Domain for Melanosome Transport in Retinal Pigment Epithelial Cells. J. Neurosci. 2009, 29, 15810–15818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.A.; Williams, L.H.; Rose, E.; Kuiper, M.; Dahl, H.-H.M.; Manji, S.S.M. Inner Ear Morphology Is Perturbed in Two Novel Mouse Models of Recessive Deafness. PLoS ONE 2012, 7, e51284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabro, K.R.; Boye, S.L.; Choudhury, S.; Fajardo, D.; Peterson, J.J.; Li, W.; Crosson, S.M.; Kim, M.-J.; Ding, D.; Salvi, R.; et al. A Novel Mouse Model of MYO7A USH1B Reveals Auditory and Visual System Haploinsufficiencies. Front. Neurosci. 2019, 13, 1255. [Google Scholar] [CrossRef]
- Phillips, J.B.; Blanco-Sanchez, B.; Lentz, J.J.; Tallafuss, A.; Khanobdee, K.; Sampath, S.; Jacobs, Z.G.; Han, P.F.; Mishra, M.; Titus, T.A.; et al. Harmonin (Ush1c) is required in zebrafish Müller glial cells for photoreceptor synaptic development and function. Dis. Model. Mech. 2011, 4, 786–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.R. Mouse models of USH1C and DFNB18: Phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum. Mol. Genet. 2003, 12, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, G.; Michel, V.; Weil, D.; Lepelletier, L.; Bizard, E.; Wolfrum, U.; Hardelin, J.-P.; Petit, C. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development 2008, 135, 1427–1437. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Liu, X.Z.; Han, F.; Yu, H.; Longo-Guess, C.; Yang, B.; Lu, C.; Yan, D.; Zheng, Q.Y. Ush1c gene expression levels in the ear and eye suggest different roles for Ush1c in neurosensory organs in a new Ush1c knockout mouse. Brain Res. 2010, 1328, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Lentz, J.; Pan, F.; Ng, S.S.; Deininger, P.; Keats, B. Ush1c216A knock-in mouse survives Katrina. Mutat. Res. Mol. Mech. Mutagen. 2007, 616, 139–144. [Google Scholar] [CrossRef]
- Trouillet, A.; Dubus, E.; Dégardin, J.; Estivalet, A.; Ivkovic, I.; Godefroy, D.; García-Ayuso, D.; Simonutti, M.; Sahly, I.; Sahel, J.A.; et al. Cone degeneration is triggered by the absence of USH1 proteins but prevented by antioxidant treatments. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Söllner, C.; Rauch, G.-J.; Siemens, J.; Geisler, R.; Schuster, S.C.; the Tübingen 2000 Screen Consortium; Müller, U.; Nicolson, T. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nat. Cell Biol. 2004, 428, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Glover, G.; Mueller, K.P.; Söllner, C.; Neuhauss, S.C.F.; Nicolson, T. The Usher gene cadherin 23 is expressed in the zebrafish brain and a subset of retinal amacrine cells. Molec. Vision 2012, 18, 2309–2322. [Google Scholar] [CrossRef] [PubMed]
- Di-Palma, F.; Holme, R.H.; Bryda, E.C.; Belyantseva, I.A.; Pellegrino, R.; Kachar, B.; Steel, K.P.; Noben-Trauth, K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 2001, 27, 103–107. [Google Scholar] [CrossRef]
- Seiler, C.; Finger-Baier, K.C.; Rinner, O.; Makhankov, Y.V.; Schwarz, H.; Neuhauss, S.C.F.; Nicolson, T. Duplicated genes with split functions: Independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development 2005, 132, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, R.; Sotomayor, M.; Kinder, K.J.; Gopal, S.; Gerka-Stuyt, J.; Chen, D.H.-C.; Hardisty-Hughes, R.E.; Ball, G.; Parker, A.; Gaudet, R.; et al. Noddy, a mouse harboring a missense mutation in protocadherin-15, reveals the impact of disrupting a critical interaction site between tip-link cadherins in inner ear hair cells. J. Neurosci. 2013, 33, 4395–4404. [Google Scholar] [CrossRef] [Green Version]
- Kikkawa, Y.; Shitara, H.; Wakana, S.; Kohara, Y.; Takada, T.; Okamoto, M.; Taya, C.; Kamiya, K.; Yoshikawa, Y.; Tokano, H.; et al. Mutations in a New Scaffold Protein Sans Cause Deafness in Jackson Shaker Mice. Hum. Mol. Genet. 2003, 12, 453–461. [Google Scholar] [CrossRef]
- Lane, P.W. WHIRLER MICE: A Recessive Behavior Mutation in Linkage Group VIII. J. Hered. 1963, 54, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.; Rogers, M.; Brown, S.; Steel, K. Linkage Analysis of the whirler Deafness Gene on Mouse Chromosome 4. Genome 1994, 21, 42–48. [Google Scholar] [CrossRef]
- Rogers, M.J.; Fleming, J.; Kiernan, B.W.; Mburu, P.; Varela, A.; Brown, S.D.; Steel, K.P. Genetic mapping of the whirler mutation. Mamm. Genome 1999, 10, 513–519. [Google Scholar] [CrossRef]
- Paige, A.J.; Kiernan, B.W.; Varela, A.; Rogers, M.J.; Hughes, D.; Steel, K.P.; Brown, S.D. A deletion on chromosome 4 cosegregates with the whirler deafness mutation: Exclusion of Orm1 as a candidate. Mamm. Genome 2000, 11, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.R.; Chen, D.H.-C.; Chou, S.-W.; Zang, J.; Neuhauss, S.C.; Stepanyan, R.; McDermott, B.M.; Alagramam, K.N. Zebrafish Models for the Mechanosensory Hair Cell Dysfunction in Usher Syndrome 3 Reveal That Clarin-1 Is an Essential Hair Bundle Protein. J. Neurosci. 2015, 35, 10188–10201. [Google Scholar] [CrossRef] [Green Version]
- Geller, S.F.; Guerin, K.I.; Visel, M.; Pham, A.; Lee, E.S.; Dror, A.A.; Avraham, K.B.; Hayashi, T.; Ray, C.A.; Reh, T.A.; et al. CLRN1 Is Nonessential in the Mouse Retina but Is Required for Cochlear Hair Cell Development. PLoS Genet. 2009, 5, e1000607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, R.; Melki, S.; Chen, D.H.-C.; Tian, G.; Furness, D.N.; Oshima-Takago, T.; Neef, J.; Moser, T.; Askew, C.; Horwitz, G.; et al. The Mechanosensory Structure of the Hair Cell Requires Clarin-1, a Protein Encoded by Usher Syndrome III Causative Gene. J. Neurosci. 2012, 32, 9485–9498. [Google Scholar] [CrossRef]
- Geng, R.; Omar, A.; Gopal, S.R.; Chen, D.H.-C.; Stepanyan, R.; Basch, M.L.; Dinculescu, A.; Furness, D.N.; Saperstein, D.; Hauswirth, W.; et al. Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Xu, L.; Bolch, S.N.; Santiago, C.P.; Dyka, F.M.; Akil, O.; Lobanova, E.; Wang, Y.; Martemyanov, K.A.; Hauswirth, W.W.; Smith, W.C.; et al. Clarin-1 expression in adult mouse and human retina highlights a role of Müller glia in Usher syndrome. J. Pathol. 2020, 250, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Vaché, C.; Besnard, T.; le Berre, P.; García-García, G.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Bolz, H.J.; Millan, J.; et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: Implications for diagnosis and therapy. Hum. Mutat. 2012, 33, 104–108. [Google Scholar] [CrossRef]
- Liquori, A.; Vaché, C.; Baux, D.; Blanchet, C.; Hamel, C.P.; Malcolm, S.; Koenig, M.; Claustres, M.; Roux, A.-F. WholeUSH2AGene Sequencing Identifies Several New Deep Intronic Mutations. Hum. Mutat. 2016, 37, 184–193. [Google Scholar] [CrossRef]
- Khan, A.O.; Becirovic, E.; Betz, C.; Neuhaus, C.; Altmüller, J.; Riedmayr, L.M.; Motameny, S.; Nürnberg, G.; Nürnberg, P.; Bolz, H.J. A deep intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe Usher syndrome on the Arabian Peninsula. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Le Guédard, S.; Faugère, V.; Malcolm, S.; Claustres, M.; Roux, A.-F. Large genomic rearrangements within the PCDH15 gene are a significant cause of USH1F syndrome. Mol. Vis. 2007, 13, 102–107. [Google Scholar] [PubMed]
- Aller, E.; Jaijo, T.; García-García, G.; Aparisi, M.J.; Blesa, D.; Díaz-Llopis, M.; Ayuso, C.; Millán, J.M. Identification of Large Rearrangements of thePCDH15Gene by Combined MLPA and a CGH: Large Duplications Are Responsible for Usher Syndrome. Investig. Opthalmol. Vis. Sci. 2010, 51, 5480–5485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-García, G.; Aller, E.; Jaijo, T.; Aparisi, M.J.; Larrieu, L.; Faugère, V.; Blanco-Kelly, F.; Ayuso, C.; Roux, A.-F.; Millán, J.M. Novel deletions involving the USH2A gene in patients with Usher syndrome and retinitis pigmentosa. Mol. Vis. 2014, 20, 1398–1410. [Google Scholar]
- Vaché, C.; Puechberty, J.; Faugère, V.; Darmaisin, F.; Liquori, A.; Baux, D.; Blanchet, C.; Garcia-Garcia, G.; Meunier, I.; Pellestor, F.; et al. A 4.6 Mb Inversion Leading to PCDH15-LINC00844 and BICC1-PCDH15 Fusion Transcripts as a New Pathogenic Mechanism Implicated in Usher Syndrome Type 1. Front. Genet. 2020, 11, 623. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zou, J.; Chen, Q.; Zhu, T.; Sui, R.; Yang, J. Structural modeling, mutation analysis, and in vitro expression of usherin, a major protein in inherited retinal degeneration and hearing loss. Comput. Struct. Biotechnol. J. 2020, 18, 1363–1382. [Google Scholar] [CrossRef]
- McMillan, D.; White, P.C. Loss of the transmembrane and cytoplasmic domains of the very large G-protein-coupled receptor-1 (VLGR1 or Mass1) causes audiogenic seizures in mice. Mol. Cell. Neurosci. 2004, 26, 322–329. [Google Scholar] [CrossRef]
- Pérez-Carro, R.; Blanco-Kelly, F.; Galbis-Martínez, L.; García-García, G.; Aller, E.; García-Sandoval, B.; Mínguez, P.; Corton, M.; Mahíllo-Fernández, I.; Martín-Mérida, I.; et al. Unravelling the pathogenic role and genotype-phenotype correlation of the USH2A p.(Cys759Phe) variant among Spanish families. PLoS ONE 2018, 13, e0199048. [Google Scholar] [CrossRef]
- Doucette, L.; Merner, N.; Cooke, S.; Ives, E.; Galutira, D.; Walsh, V.; Walsh, T.; MacLaren, L.; Cater, T.; Fernandez, B.; et al. Profound, prelingual nonsyndromic deafness maps to chromosome 10q21 and is caused by a novel missense mutation in the Usher syndrome type IF gene PCDH15. Eur. J. Hum. Genet. 2008, 17, 554–564. [Google Scholar] [CrossRef]
- Mchugh, R.K.; Friedman, R.A. Genetics of hearing loss: Allelism and modifier genes produce a phenotypic continuum. Anat. Rec. Part. A Discov. Mol. Cell. Evol. Biol. 2006, 288A, 370–381. [Google Scholar] [CrossRef]
- Khateb, S.; Mohand-Saïd, S.; Nassisi, M.; Bonnet, C.; Roux, A.-F.; Andrieu, C.; Antonio, A.; Condroyer, C.; Zeitz, C.; Devisme, C.; et al. Phenotypic characteristics of rod–cone dystrophy associated with myo7a mutations in a large french cohort. Retina 2020, 40, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation–Associated Inherited Retinal Dystrophy. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; McCaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 2013, 19, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; Peters, T.A.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| USH Type | Gene | MIM Number | Protein | GRCh38 Coordinates | Function |
|---|---|---|---|---|---|
| USH1 | MYO7A | 276900 1 276903 2 | myosin VIIA | chr11:77128246–77215241 | actin-based motor protein |
| USH1C | 276904 1 605242 2 | harmonin | chr11:17493895–17544416 | scaffold protein | |
| CDH23 | 601067 1 605516 2 | cadherin 23 | chr10:71396934–71815947 | cell adhesion | |
| PCDH15 | 602083 1 605514 2 | protocadherin 15 | chr10:53802771–55627942 | cell adhesion | |
| USH1G | 606943 1 607696 2 | SANS | chr17:74916083–74923256 | scaffold protein | |
| CIB2 | 614869 1 605564 2 | CIB2 | chr15:78104606–78131544 | calcium and integrin binding protein | |
| USH2 | USH2A | 276901 1 608400 2 | usherin | chr1:215622891–216423448 | cell adhesion |
| ADGRV1 | 605472 1 602851 2 | adhesion G protein-coupled receptor V1 | chr5:90529344–91164437 | adhesion G protein-coupled receptor | |
| WHRN | 611383 1 607928 2 | whirlin | chr9:114402080–114505473 | scaffold protein | |
| USH3 | CLRN1 | 276902 1 606397 2 | clarin 1 | chr3:150926163–150972999 | transmembrane protein |
| Gene | Animal | Model | HL | VD | RD | References |
|---|---|---|---|---|---|---|
| USH type 1 | ||||||
| MYO7A | Danio rerio | myo7am/m (mariner) | yes | yes | yes | [173,174] |
| Mus musculus | Myo7ash1/sh1 (shaker1) | yes | yes | no | [175,176] | |
| Myo7ahdb/hdb (headbanger) | yes | yes | NA | [177] | ||
| Myo7apk/pk (polka) | yes | yes | no | [178,179] | ||
| Myo7aI487N/I487N (ewaso) | yes | yes | NA | [180] | ||
| Myo7aF947I/F947I (dumbo) | yes | no | NA | [180] | ||
| Myo7a−/− | yes | yes | no | [181] | ||
| USH1C | Danio rerio | ush1cfh293/fh293 | yes | yes | yes | [182] |
| ush1c knock-down | yes | yes | yes | [182] | ||
| Mus musculus | Ush1cdfcr/dfcr (deaf circler) | yes | yes | no | [183] | |
| Ush1cdfcr/dfcr (deaf circler) | yes | yes | no | [183] | ||
| Ush1cdfcr-2J/dfcr-2J (deaf circler 2 Jackson) | yes | yes | no | [183] | ||
| Ush1c−/− | yes | yes | no | [184,185] | ||
| Ush1c knock-in [c.216G>A] | yes | yes | yes | [167,186] | ||
| Ush1c−/−C57Bl/6 J | NA | NA | no | [187] | ||
| Ush1c−/−BALB/cJ | NA | NA | yes | [187] | ||
| CDH23 | Danio rerio | cdh23s/s (sputnik) | yes | yes | no | [188,189] |
| Xenopus tropicalis | cdh23 knock-down | NA | NA | yes | [55] | |
| Mus musculus | Cdh23v/v (waltzer) | yes | yes | no | [190] | |
| PCDH15 | Danio rerio | pcdh15p/p | yes | yes | yes | [191] |
| Xenopus tropicalis | pcdh15 knock-down | yes | yes | yes | [55] | |
| Mus musculus | Pcdh15av/av (ames waltzer) | yes | yes | no | [58] | |
| Pcdh15I108N/I108N (noddy) | yes | yes | NA | [192] | ||
| USH1G | Mus musculus | Sansjs/js (jackson shaker) | yes | yes | no | [193] |
| Ush1g−/−C57Bl/6 J | NA | NA | no | [187] | ||
| Ush1g−/−BALB/cJ | NA | NA | yes | [187] | ||
| CIB2 | Drosophila melanogaster | CG9236 knock-down | NA | NA | yes | [10] |
| Danio rerio | cib2 knock-down | yes | yes | NA | [10] | |
| Mus musculus | Cib2−/− | yes | no | no | [77,78] | |
| Cib2tm1a/tm1a | yes | no | NA | [76] | ||
| Cib2 knock-in [p.F91S] | yes | no | NA | [76] | ||
| USH type 2 | ||||||
| USH2A | Danio rerio | ush2a knock-down | NA | NA | yes | [136] |
| ush2a−/− | yes | yes | yes | [172] | ||
| ush2armc1/rmc1 | NA | NA | yes | [169] | ||
| ush2ab1245/b1245 | NA | NA | yes | [169] | ||
| Mus musculus | Ush2a−/− | yes | no | yes | [93] | |
| KMush/ush (kunming) | yes | no | yes | [168] | ||
| ADGRV1 | Danio rerio | adgrv1 knock-down | NA | NA | yes | [136] |
| Mus musculus | Vlgr1del7TM/del7TM | yes | no | no | [170] | |
| Vlgr1−/− | yes | no | no | [92,97] | ||
| WHRN | Mus musculus | Whrnwi/wi (whirler) | yes | yes | no | [194,195,196,197] |
| WhrnL−/L− | yes | no | yes | [106] | ||
| USH type 3 | ||||||
| CLRN1 | Danio rerio | clrn1 knock-down | yes | yes | NA | [117] |
| clrn1−/− | yes | yes | NA | [198] | ||
| Mus musculus | Clrn1−/− | yes | yes | no | [118,199] | |
| Clrn1N48K/N48K | yes | no | NA | [200] | ||
| Clrn1ex4−/− | yes | NA | NA | [142] | ||
| Clrn1ex4fl/flMyo15-Cre+/− | yes | NA | NA | [142] | ||
| Clrn1−/− [KO-TgAC1] | yes | NA | NA | [201] | ||
| Clrn1 knock-in [N-HA] | no | NA | no | [202] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Aller, E.; Jaijo, T.; Millán, J.M.; García-García, G. Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136723
Fuster-García C, García-Bohórquez B, Rodríguez-Muñoz A, Aller E, Jaijo T, Millán JM, García-García G. Usher Syndrome: Genetics of a Human Ciliopathy. International Journal of Molecular Sciences. 2021; 22(13):6723. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136723
Chicago/Turabian StyleFuster-García, Carla, Belén García-Bohórquez, Ana Rodríguez-Muñoz, Elena Aller, Teresa Jaijo, José M. Millán, and Gema García-García. 2021. "Usher Syndrome: Genetics of a Human Ciliopathy" International Journal of Molecular Sciences 22, no. 13: 6723. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136723