
SIRT3 Overexpression Ameliorates Asbestos-Induced Pulmonary Fibrosis, mt-DNA Damage, and Lung Fibrogenic Monocyte Recruitment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
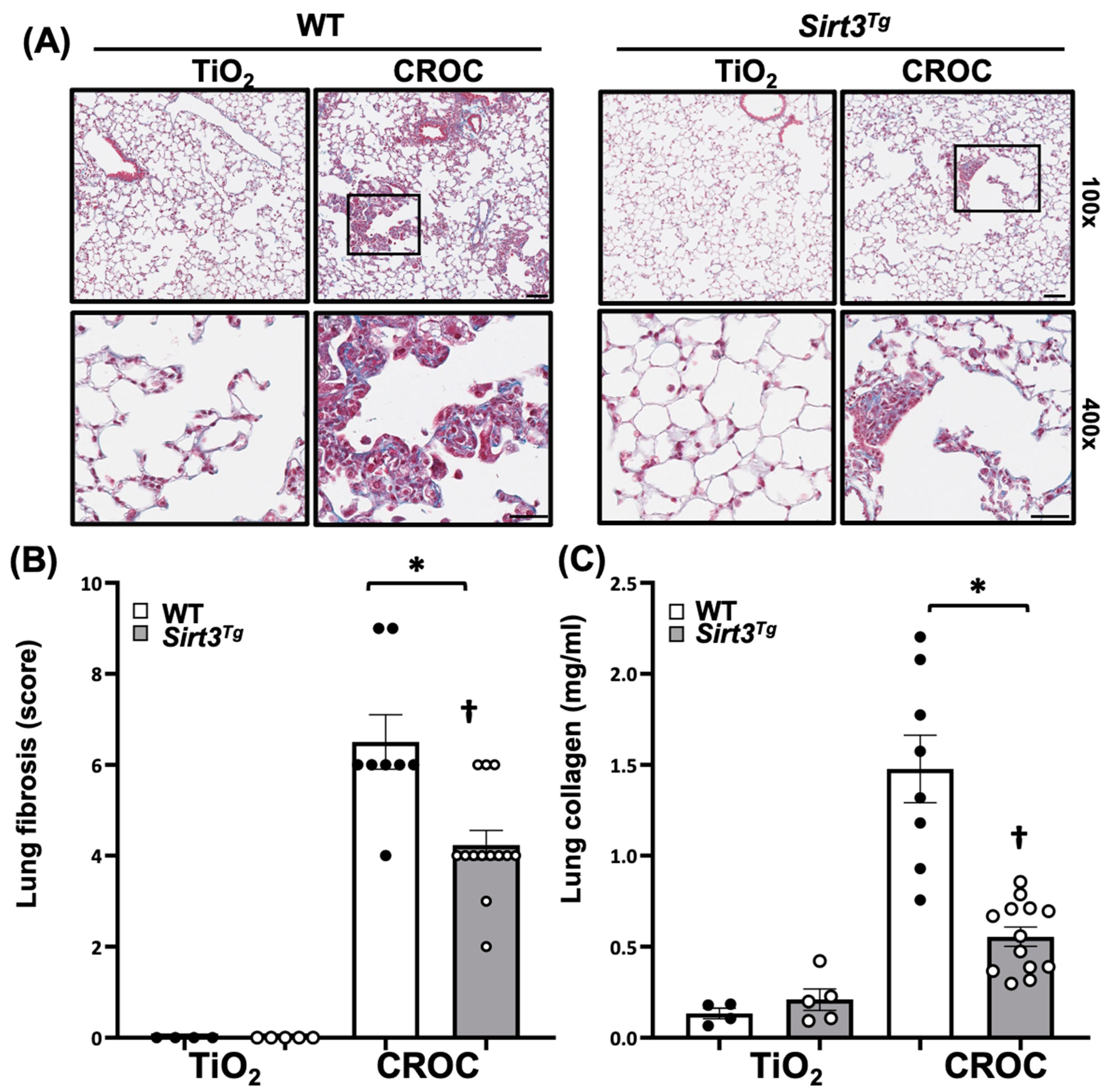
2.1. SIRT3 Overexpression Attenuates Asbestos-Induced Pulmonary Fibrosis
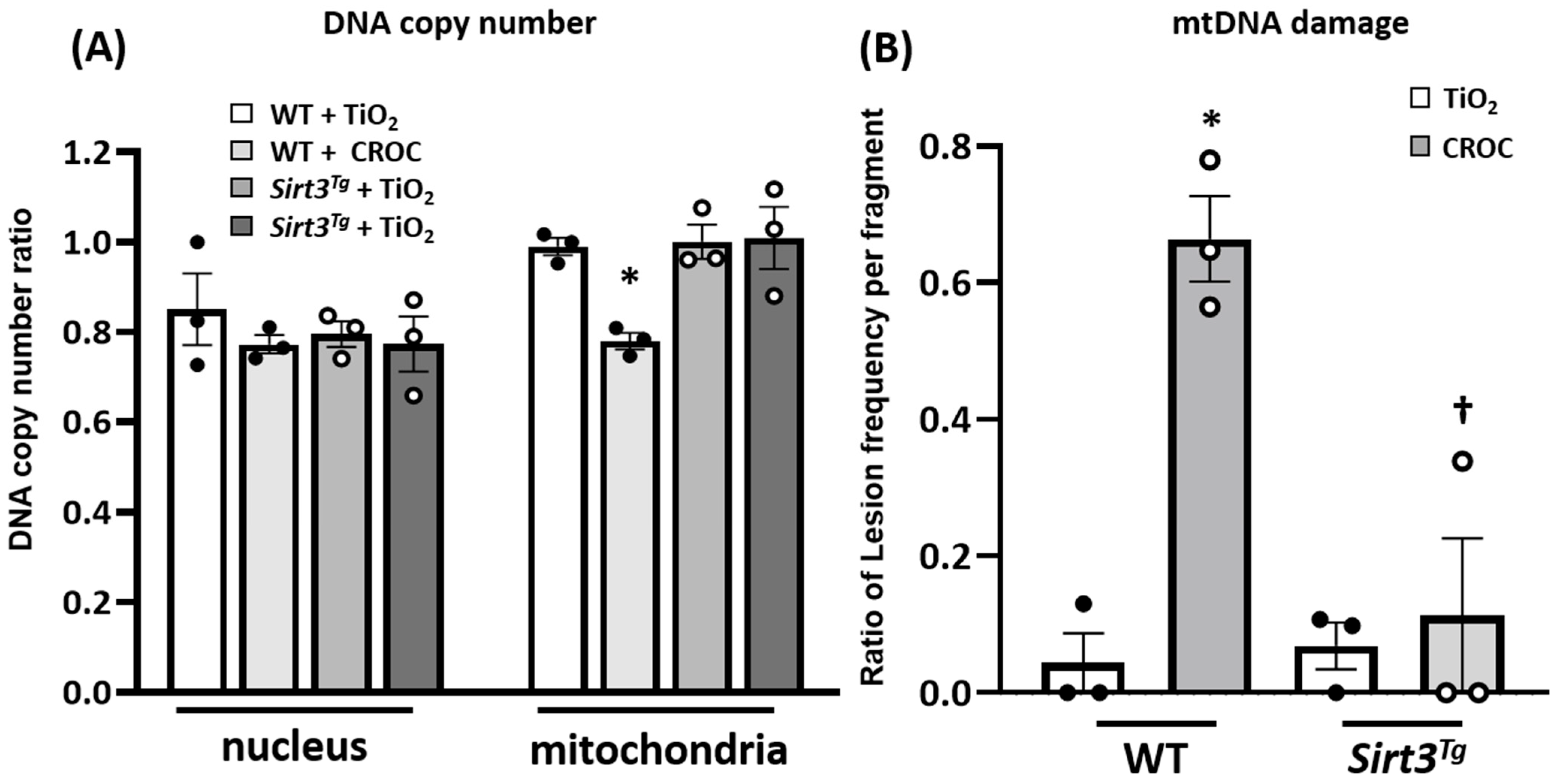
2.2. SIRT3 Overexpression Mitigates Asbestos-Induced mtDNA Damage in Mouse Lungs
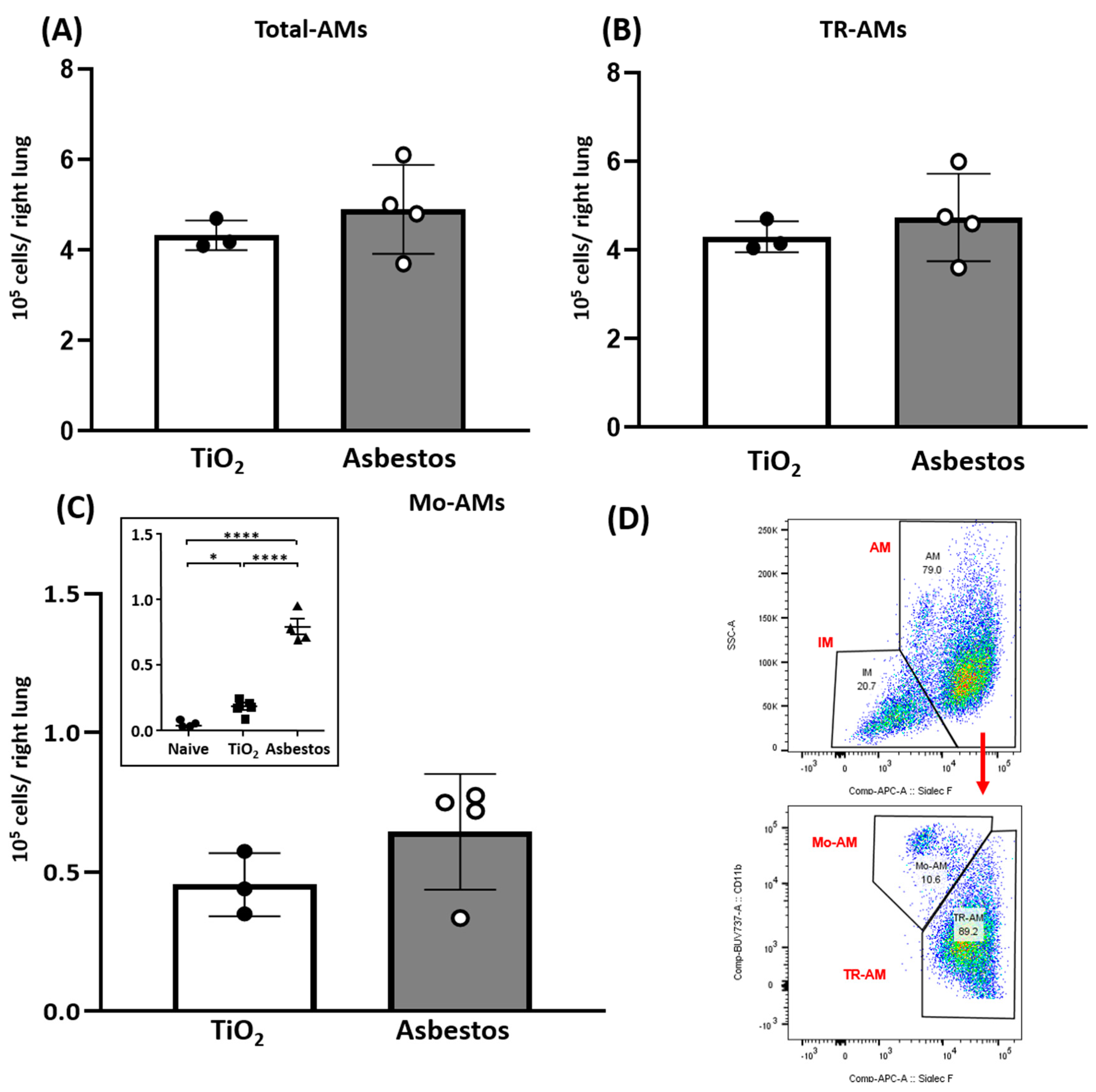
2.3. SIRT3 Overexpression Diminishes Recruitment of Pro-Fibrotic Mo-AMs
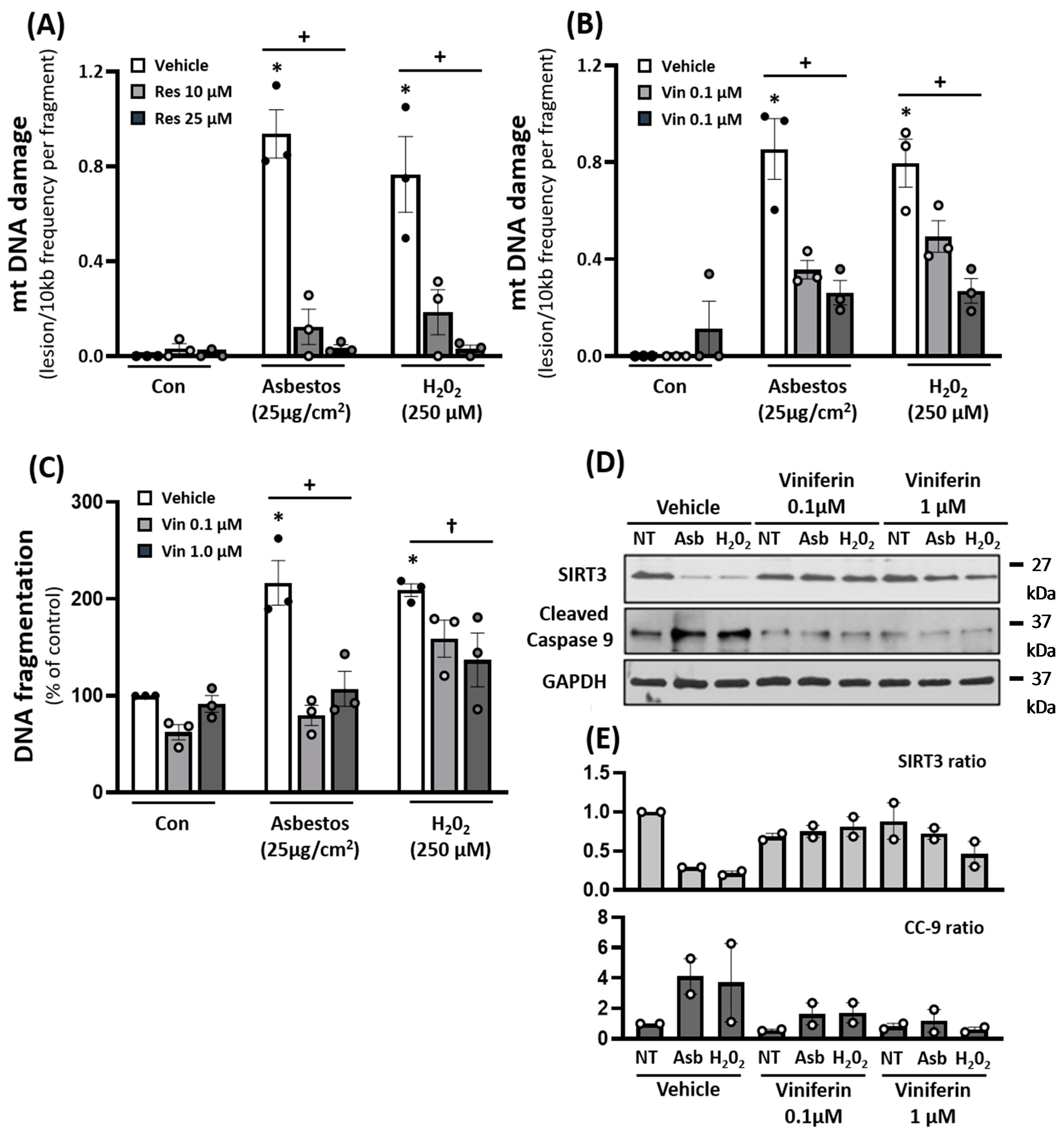
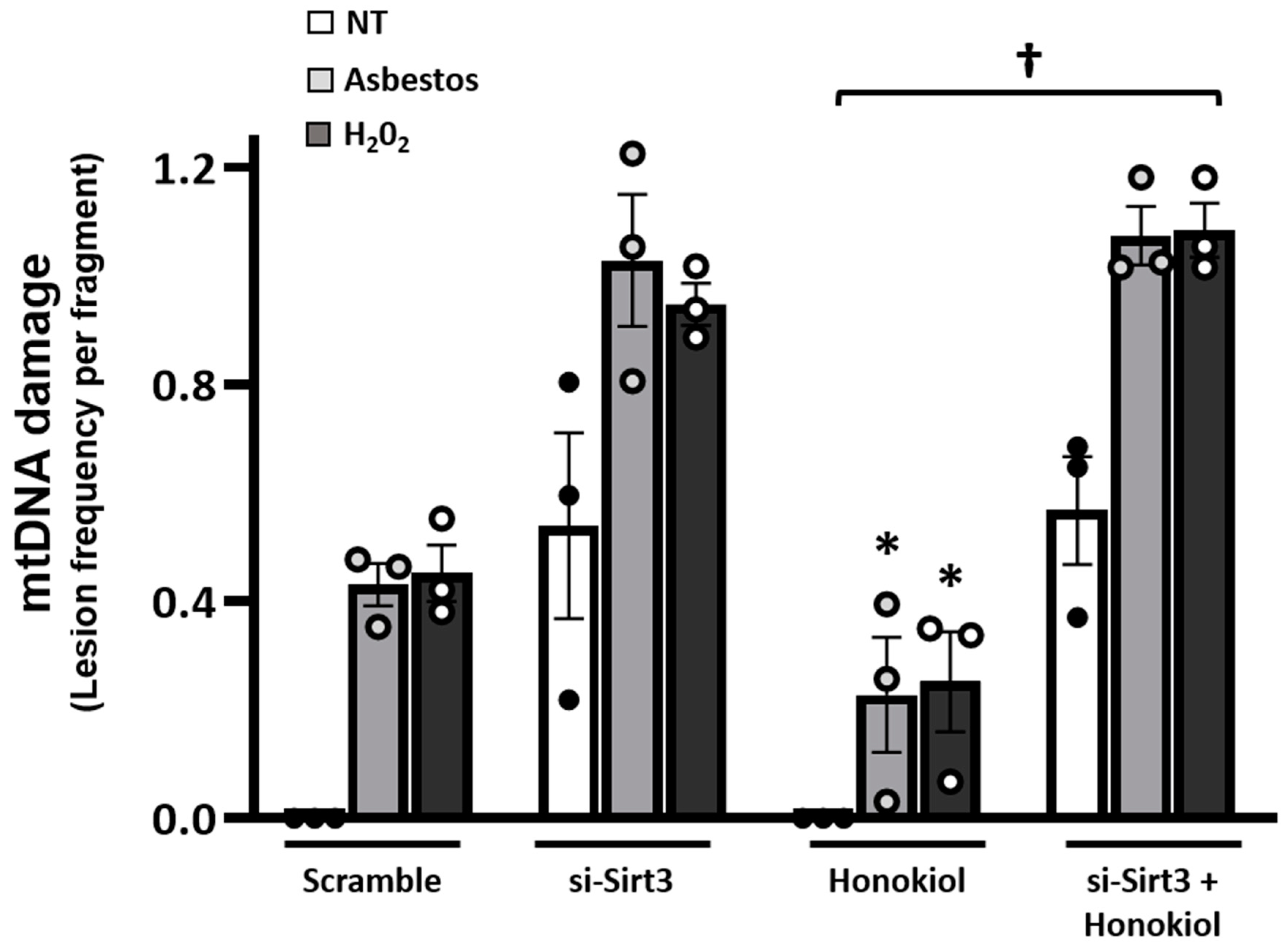
2.4. Pharmacologic Agents That Augment SIRT3 Levels Diminish Oxidant-Induced AEC mtDNA Damage
3. Discussion
4. Methods
4.1. Reagents
4.2. Animals
4.3. Asbestos Instillation into Mice
4.4. Titanium Dioxide and Asbestos Preparation for Instillation into Mice
4.5. Lung Harvest and Histology
4.6. Lung Collagen Detection
4.7. Fibrosis Scoring System
4.8. Quantitative mtDNA Damage Assay via PCR
4.9. DNA Fragmentation
4.10. Western Blotting
4.11. Cell Culture
4.12. Sirt3 Gene Silencing
4.13. Cell Isolation, Staining, Flow for Mo-AMs
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SIRT3 | sirtuin-3 |
| Sirt3Tg | transgenic whole body SIRT3 overexpression |
| WT | wild-type |
| AEC | alveolar epithelial cell |
| AT2 | alveolar type II cell |
| ROS | reactive oxygen species |
| mtDNA | mitochondrial DNA |
| mtOGG1 | mitochondrial 8-oxoguanine-DNA glycosylase 1 |
| OGG1 | 8-oxyguanine DNA glycosylase |
| BER | base excision repair |
| BALF | bronchoalveolar lavage fluid |
| MnSOD | manganese superoxide dismutase |
| EVs | extracellular vesicles |
| ER | endoplasmic reticulum |
| H&E | hematoxylin and eosin |
| HLF | human lung fibroblasts |
| mtOgg1tg | human mitochondria-targeted OGG1 subunit 1-alpha transgene |
| IPF | idiopathic pulmonary fibrosis |
| IP | intra-peritoneal |
| IT | intra-tracheal |
| MCAT | mitochondrial catalase enforced-expression |
| α-SMA | alpha-smooth muscle actin |
| TGFβ | transforming growth factor-beta |
References
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Olson, A.L.; Swigris, J.J.; Lezotte, D.C.; Norris, J.M.; Wilson, C.G.; Brown, K.K. Mortality from Pulmonary Fibrosis Increased in the United States from 1992 to 2003. Am. J. Respir. Crit. Care Med. 2007, 176, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.X.L.; Jaurand, M.-C.; Kamp, D.W.; Whysner, J.; Hei, T.K. Role of Mutagenicity in Asbestos Fiber-Induced Carcinogenicity and Other Diseases. J. Toxicol. Environ. Health Part B 2011, 14, 179–245. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Cheresh, P.; Kamp, D.W. Molecular Basis of Asbestos-Induced Lung Disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 161–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhal, B.D.; Nguyen, H. The Witschi Hypothesis revisited after 35 years: Genetic proof from SP-C BRICHOS domain mutations. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L906–L911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selman, M.; Pardo, A. Revealing the Pathogenic and Aging-related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis. An Integral Model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Murthy, M.; Balch, W.E.; Chandel, N.S.; Meiners, S.; Eickelberg, O.; Selman, M.; Pardo, A.; White, E.S.; Levy, B.D.; et al. Blue Journal Conference. Aging and Susceptibility to Lung Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Williams, D.B.; Kamp, D.W. The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis. Int. J. Mol. Sci. 2015, 16, 21486–21519. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Cheresh, P.; Williams, D.; Cheng, Y.; Ridge, K.; Schumacker, P.T.; Weitzman, S.; Bohr, V.A.; Kamp, D.W. Mitochondria-targeted Ogg1 and Aconitase-2 Prevent Oxidant-induced Mitochondrial DNA Damage in Alveolar Epithelial Cells. J. Biol. Chem. 2014, 289, 6165–6176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Morales-Nebreda, L.; Cheng, Y.; Hogan, E.; Yeldandi, A.; Chi, M.; Piseaux, R.; Ridge, K.; et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic. Biol. Med. 2016, 101, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Malsin, E.S.; Kamp, D.W. The mitochondria in lung fibrosis: Friend or foe? Transl. Res. 2018, 202, 1–23. [Google Scholar] [CrossRef]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; De Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; Croix, C.S.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial Cell Mitochondrial Dysfunction and PINK1 Are Induced by Transforming Growth Factor-Beta1 in Pulmonary Fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, D.W.; Liu, G.; Cheresh, P.; Kim, S.-J.; Mueller, A.; Lam, A.P.; Trejo, H.; Williams, D.; Tulasiram, S.; Baker, M.; et al. Asbestos-Induced Alveolar Epithelial Cell Apoptosis. The Role of Endoplasmic Reticulum Stress Response. Am. J. Respir. Cell Mol. Biol. 2013, 49, 892–901. [Google Scholar] [CrossRef]
- Cheresh, P.; Morales-Nebreda, L.; Kim, S.-J.; Yeldandi, A.; Williams, D.B.; Cheng, Y.; Mutlu, G.M.; Budinger, G.R.S.; Ridge, K.; Schumacker, P.T.; et al. Asbestos-Induced Pulmonary Fibrosis Is Augmented in 8-Oxoguanine DNA Glycosylase Knockout Mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Panduri, V.; Liu, G.; Surapureddi, S.; Kondapalli, J.; Soberanes, S.; De Souza-Pinto, N.; Bohr, V.; Budinger, G.; Schumacker, P.T.; Weitzman, S.; et al. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic. Biol. Med. 2009, 47, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Rachek, L.; Yeldandi, A.; Piseaux-Aillon, R.; Ciesielski, M.J.; Ridge, K.M.; Gottardi, C.J.; Lam, A.P.; et al. Mitochondrial 8-oxoguanine DNA glycosylase mitigates alveolar epithelial cell PINK1 deficiency, mitochondrial DNA damage, apoptosis, and lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L1084–L1096. [Google Scholar] [CrossRef]
- Ryu, C.; Sun, H.; Gulati, M.; Herazo-Maya, J.D.; Chen, Y.; Osafo-Addo, A.; Brandsdorfer, C.; Winkler, J.; Blaul, C.; Faunce, J.; et al. Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1571–1581. [Google Scholar] [CrossRef]
- Bueno, M.; Zank, D.; Buendia-Roldán, I.; Fiedler, K.; Mays, B.G.; Alvarez, D.; Sembrat, J.; Kimball, B.; Bullock, J.K.; Martin, J.L.; et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS ONE 2019, 14, e0218003. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Alt, F.W.; Cheng, H.-L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 Is a Mitochondria-Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism during Stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; Coleman, M.; Pennington, J.D.; Ozden, O.; Park, S.-H.; Jiang, H.; Kim, H.-S.; Flynn, C.R.; Hill, S.; McDonald, W.H.; et al. Sirt3-Mediated Deacetylation of Evolutionarily Conserved Lysine 122 Regulates MnSOD Activity in Response to Stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.-Y.; Guan, K.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.-S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Vassilopoulos, A.; Parisiadou, L.; Yan, Y.; Gius, D. Regulation of MnSOD Enzymatic Activity by Sirt3 Connects the Mitochondrial Acetylome Signaling Networks to Aging and Carcinogenesis. Antioxid. Redox Signal. 2014, 20, 1646–1654. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Bindu, S.; Pillai, V.B.; Samant, S.; Pan, Y.; Huang, J.-Y.; Gupta, M.; Nagalingam, R.S.; Wolfgeher, D.; Verdin, E.; et al. SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3β. Mol. Cell. Biol. 2015, 36, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, R.P.; Kim, S.J.; Cheresh, P.; Williams, D.B.; Morales-Nebreda, L.; Cheng, Y.; Yeldandi, A.; Bhorade, S.; Pardo, A.; Selman, M.; et al. SIRT3 Deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondria DNA damage and apoptosis. FASEB J. 2017, 31, 2520–2532. [Google Scholar] [CrossRef] [Green Version]
- Bindu, S.; Pillai, V.B.; Kanwal, A.; Samant, S.; Mutlu, G.M.; Verdin, E.; Dulin, N.; Gupta, M.P. SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L68–L78. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Feghali-Bostwick, C.; Lasky, J.A.; Sanchez, C.G. Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 72, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Rehan, M.; Kurundkar, D.; Kurundkar, A.R.; Logsdon, N.J.; Smith, S.R.; Chanda, D.; Bernard, K.; Sanders, Y.Y.; Deshane, J.S.; Dsouza, K.G.; et al. Restoration of SIRT3 gene expression by airway delivery resolves age-associated persistent lung fibrosis in mice. Nat. Aging 2021, 1, 205–217. [Google Scholar] [CrossRef]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Miyamoto, A.; Suzuki, S.; Fujimori, S.; Kohno, T.; et al. Extracellular Vesicles from Fibroblasts Induce Epithelial Cell Senescence in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Akamata, K.; Wei, J.; Bhattacharyya, M.; Cheresh, P.; Bonner, M.Y.; Arbiser, J.L.; Raparia, K.; Gupta, M.P.; Kamp, D.W.; Varga, J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget 2016, 7, 69321–69336. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-X.; Li, Y.-N.; Wang, X.-L.; Ye, C.-L.; Zhu, X.-Y.; Li, H.-P.; Yang, T.; Liu, Y.-J. Probucol ameliorates EMT and lung fibrosis through restoration of SIRT3 expression. Pulm. Pharmacol. Ther. 2019, 57, 101803. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, F.; Wang, D.; Li, L.; Si, H.; Wang, C.; Liu, J.; Chen, Y.; Cheng, J.; Lu, Y. Mesenchymal Stem Cells Attenuate Diabetic Lung Fibrosis via Adjusting Sirt3-Mediated Stress Responses in Rats. Oxid. Med. Cell. Longev. 2020, 2020, 1–15. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Watanabe, S.; Verma, R.; Jablonski, R.P.; Chen, C.-I.; Cheresh, P.; Markov, N.; Reyfman, P.A.; McQuattie-Pimentel, A.C.; Sichizya, L.; et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J. 2019, 55, 1900646. [Google Scholar] [CrossRef] [Green Version]
- Cheresh, P.; Kim, S.-J.; Huang, L.S.; Watanabe, S.; Joshi, N.; Williams, K.J.; Chi, M.; Lu, Z.; Harijith, A.; Yeldandi, A.; et al. The Sphingosine Kinase 1 Inhibitor, PF543, Mitigates Pulmonary Fibrosis by Reducing Lung Epithelial Cell mtDNA Damage and Recruitment of Fibrogenic Monocytes. Int. J. Mol. Sci. 2020, 21, 5595. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal Models of Fibrotic Lung Disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Şener, G.; Topaloğlu, N.; Şehirli, A. Özer; Ercan, F.; Gedik, N. Resveratrol alleviates bleomycin-induced lung injury in rats. Pulm. Pharmacol. Ther. 2007, 20, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Ricardo, S.D.; Bertram, J.; Nikolic-Paterson, D.J. Resveratrol Inhibits Renal Fibrosis in the Obstructed Kidney: Potential Role in Deacetylation of Smad3. Am. J. Pathol. 2010, 177, 1065–1071. [Google Scholar] [CrossRef]
- Akgedik, R.; Akgedik, Ş.; Karamanlı, H.; Uysal, S.; Bozkurt, B.; Özol, D.; Armutcu, F.; Yıldırım, Z.; Karamanli, H. Effect of Resveratrol on Treatment of Bleomycin-Induced Pulmonary Fibrosis in Rats. Inflammation 2012, 35, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Jin, J.; Cichewicz, R.H.; Hageman, S.A.; Ellis, T.K.; Xiang, L.; Peng, Q.; Jiang, M.; Arbez, N.; Hotaling, K.; et al. Trans-(-)-e-viniferin increases mitochondrial SIRT3, activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington disease. J. Biol. Chem. 2012, 287, 24460–24472. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Li, J.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Zeng, M.; Zhang, Y.; Bu, P. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am. J. Physiol. Circ. Physiol. 2015, 308, H424–H434. [Google Scholar] [CrossRef]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.S.; Sudhadevi, P.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, M.; Miller, J.J.; Thapa, D.; Stiewe, S.; Timm, K.N.; Aparicio, C.N.M.; Scott, I.; Tyler, D.J.; Heather, L.C. Rescue of myocardial energetic dysfunction in diabetes through the correction of mitochondrial hyperacetylation by honokiol. JCI Insight 2020, 5, e140326. [Google Scholar] [CrossRef]
- Liao, G.; Zhao, Z.; Yang, H.; Li, X. Honokiol ameliorates radiation-induced brain injury via the activation of SIRT3. J. Int. Med. Res. 2020, 48, 10. [Google Scholar] [CrossRef]
- Bause, A.S.; Matsui, M.S.; Haigis, M.C. The protein deacetylase SIRT3 prevents oxidative stress-induced keratinocyte differentiation. J. Biol. Chem. 2013, 288, 36484–36491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. SIRT3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Larson-Casey, J.L.; Davis, D.; Hanumanthu, V.S.; Longhini, A.L.F.; Thannickal, V.J.; Gu, L.; Carter, A.B. NOX4 modulates macrophage phenotype and mitochondrial biogenesis in asbestosis. JCI Insight 2019, 4, e126551. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Jiang, D.; Banerjee, S.; Xie, N.; Kulkarni, T.; Liu, R.M.; Duncan, S.R.; Liu, G. Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. JCI Insight 2020, 5, e134539. [Google Scholar] [CrossRef] [PubMed]
- McCubbrey, A.L.; Barthel, L.; Mohning, M.P.; Redente, E.F.; Mould, K.J.; Thomas, S.M.; Leach, S.M.; Danhorn, T.; Gibbings, S.L.; Jakubzick, C.V.; et al. Deletion of c-FLIP from CD11b(hi) Macrophages Prevents Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 66–78. [Google Scholar] [CrossRef]
- Zhou, Y.; Lagares, D. Anti-aging therapy for pulmonary fibrosis. Nat. Aging 2021, 1, 155–156. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheresh, P.; Kim, S.-J.; Jablonski, R.; Watanabe, S.; Lu, Z.; Chi, M.; Helmin, K.A.; Gius, D.; Budinger, G.R.S.; Kamp, D.W. SIRT3 Overexpression Ameliorates Asbestos-Induced Pulmonary Fibrosis, mt-DNA Damage, and Lung Fibrogenic Monocyte Recruitment. Int. J. Mol. Sci. 2021, 22, 6856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136856
Cheresh P, Kim S-J, Jablonski R, Watanabe S, Lu Z, Chi M, Helmin KA, Gius D, Budinger GRS, Kamp DW. SIRT3 Overexpression Ameliorates Asbestos-Induced Pulmonary Fibrosis, mt-DNA Damage, and Lung Fibrogenic Monocyte Recruitment. International Journal of Molecular Sciences. 2021; 22(13):6856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136856
Chicago/Turabian StyleCheresh, Paul, Seok-Jo Kim, Renea Jablonski, Satoshi Watanabe, Ziyan Lu, Monica Chi, Kathryn A. Helmin, David Gius, G. R. Scott Budinger, and David W. Kamp. 2021. "SIRT3 Overexpression Ameliorates Asbestos-Induced Pulmonary Fibrosis, mt-DNA Damage, and Lung Fibrogenic Monocyte Recruitment" International Journal of Molecular Sciences 22, no. 13: 6856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136856