
Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders


Abstract
:1. Introduction
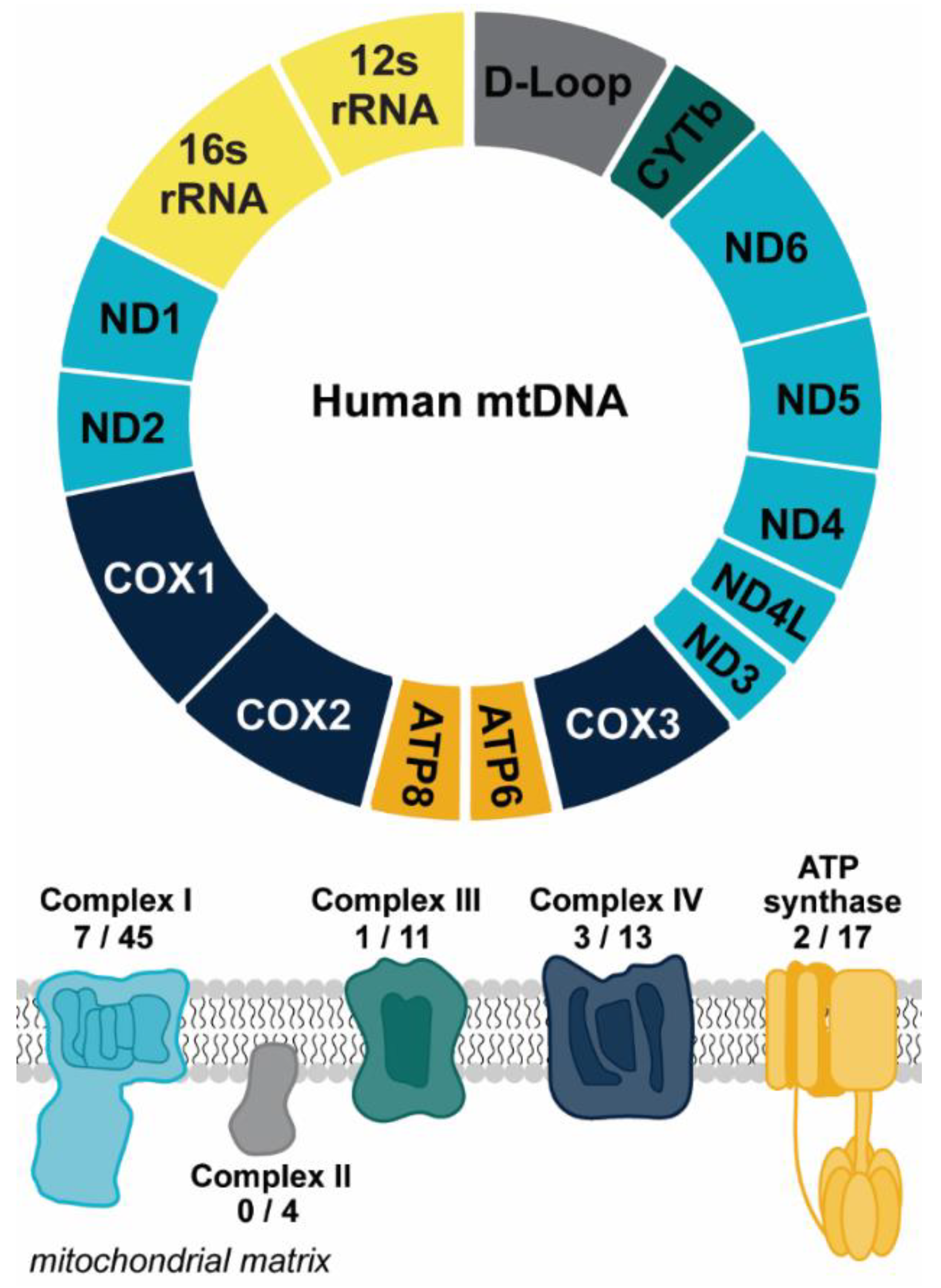
2. Mechanisms of Mitochondrial DNA Release
2.1. mtDNA Release in Response to Cellular Stress
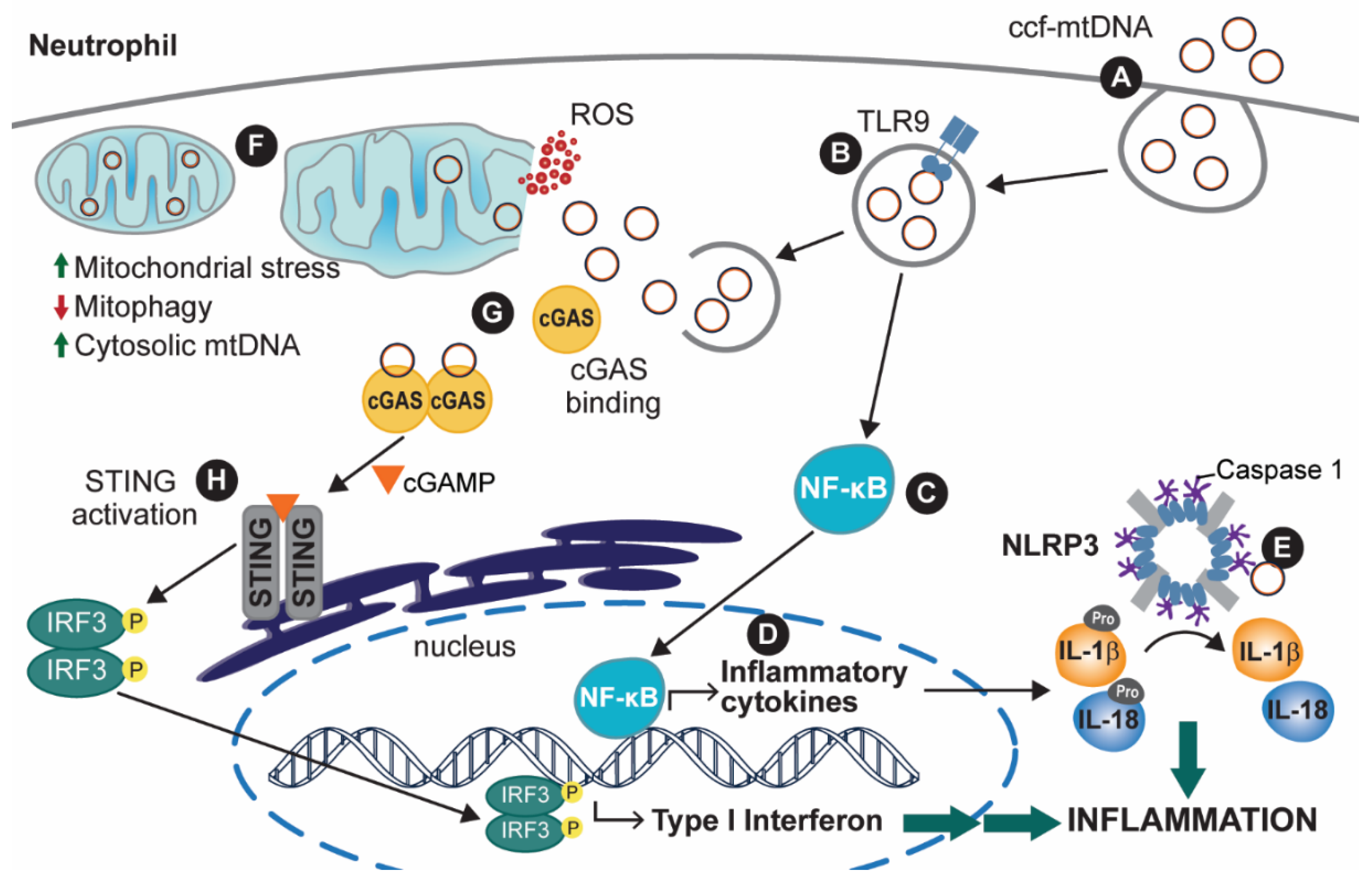
2.2. mtDNA Release as a Pillar for Immune and Inflammatory Processes
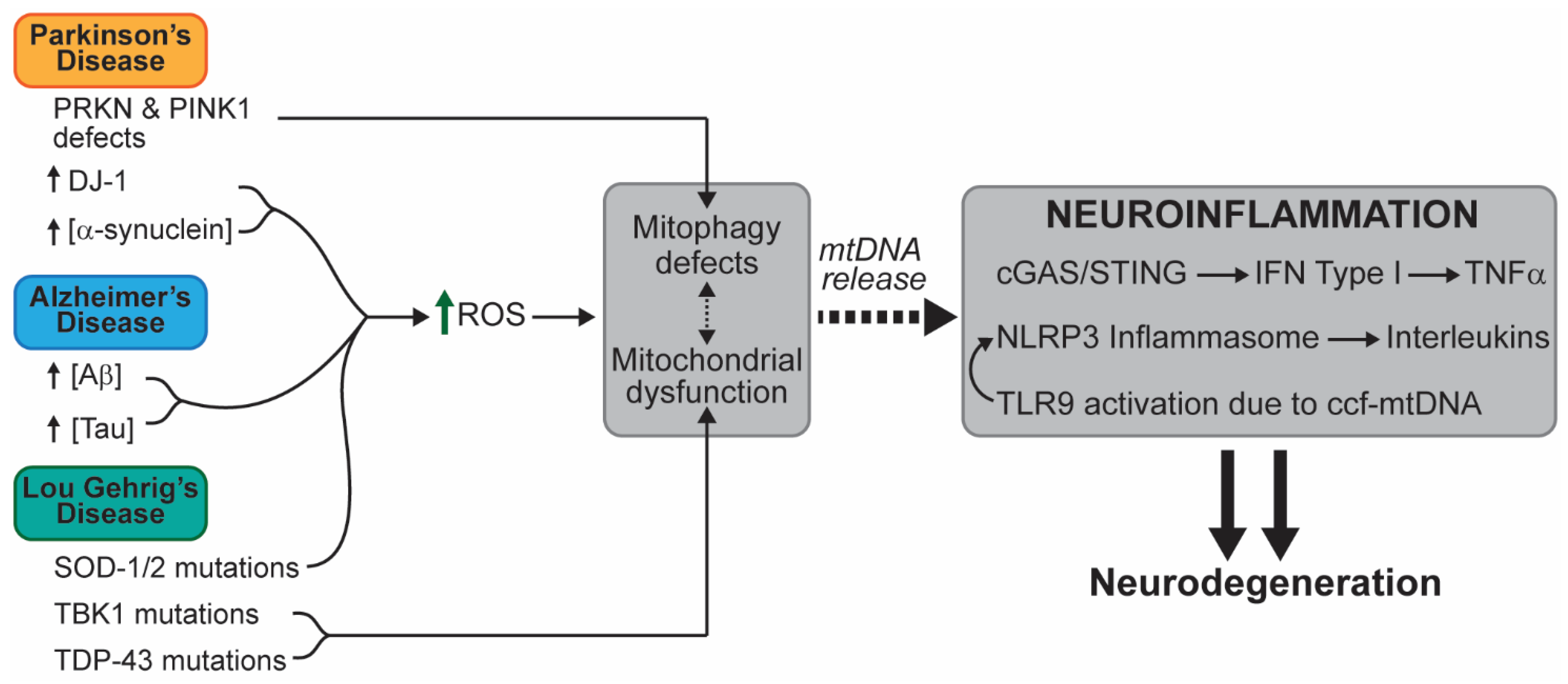
3. Analysis of a Potential Link between Mitochondrial Dysfunction and Progression of Neuronal Disease
3.1. Neurodegenerative Diseases
3.1.1. Amyotrophic Lateral Sclerosis (ALS)
mtDNA Release in ALS
Aberrant Mitochondrial Function in ALS
Inflammatory and Immune Responses in ALS
3.1.2. Parkinson’s Disease (PD)
mtDNA Release in PD
Aberrant Mitochondrial Function in PD
Inflammatory and Immune Responses in PD
3.1.3. Alzheimer’s Disease
mtDNA Release in AD
Aberrant Mitochondrial Function in AD
Inflammatory and Immune Responses in AD
3.2. Neuropsychiatric Disorders
3.2.1. Major Depressive Disorder (MDD)
mtDNA Release in MDD
Aberrant Mitochondrial Function in MDD
Inflammatory and Immune Responses in MDD
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Bestwick, M.L.; Shadel, G.S. Accessorizing the human mitochondrial transcription machinery. Trends Biochem. Sci. 2013, 38, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Kang, H. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, X.; Hu, Q.; Wu, J.; Wang, G.; Hong, Z.; Ren, J. Lab for Trauma and Surgical Infections Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019, 236, 116464. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Häcker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [Green Version]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- De Bont, R. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [Green Version]
- Cline, S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 979–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houten, B.V. Mitochondrial DNA damage induced autophagy cell death and disease. Front. Biosci. 2016, 21, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Garza-Lombó, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion 2020, 51, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Akira, S. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Amano, A. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Turco, D.D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Bampton, E.T.; Goemans, C.G.; Niranjan, D.; Mizushima, N.; Tolkovsky, A.M. The dynamics of autophagy visualised in live cells: From autophagosome formation to fusion with endo/lysosomes. Autophagy 2005, 1, 23–36. [Google Scholar] [CrossRef]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila Parkin requires PINK1 for mitochondrial translocation and ubiquitinates Mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Fitzgerald, K. Autophagy proteins regulate innate immune response by inhibiting NALP3 inflammasome-mediated mitochondrial DNA release. Nat. Immunol. 2011, 12, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Dai, Y.; Khaidakov, M.; Deng, X.; Mehta, J.L. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: Implications in atherogenesis. Cardiovasc. Res. 2014, 103, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 2041–2047. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Why F-ATP Synthase Remains a Strong Candidate as the Mitochondrial Permeability Transition Pore. Front. Physiol. 2018, 9, 1543. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Balka, K.R. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef]
- García, N.; Chávez, E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007, 81, 1160–1166. [Google Scholar] [CrossRef]
- Koulintchenko, M.; Temperley, R.J.; Mason, P.A.; Dietrich, A.; Lightowlers, R.N. Natural competence of mammalian mitochondria allows the molecular investigation of mitochondrial gene expression. Hum. Mol. Genet. 2006, 15, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Weber-Lotfi, F.; Ibrahim, N.; Boesch, P.; Cosset, A.; Konstantinov, Y.; Lightowlers, R.N.; Dietrich, A. Developing a genetic approach to investigate the mechanism of mitochondrial competence for DNA import. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Barbas, A.S.; Lin, L.; Scheuermann, U.; Bishawi, M.; Brennan, T.V. Mitochondria released by apoptotic cell death initiate innate immune responses. Immunohorizons 2018, 2, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.; Dzionek, A.; Pingel, J.T.; Colonna, M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004, 21, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoque, R.; Sohail, M.; Malik, A.; Sarwar, S.; Luo, Y.; Shah, A.; Mehal, W. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011, 141, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostjuk, S.; Loseva, P.; Chvartatskaya, O.; Ershova, E.; Smirnova, T.; Malinovskaya, E.; Ginter, E. Extracellular GC-rich DNA activates TLR9-and NF-kB-dependent signaling pathways in human adipose-derived mesenchymal stem cells (haMSCs). Expert Opin. Biol. Ther. 2012, 12, S99–S111. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Briard, B.; Burton, A.; Gingras, S.; Pelletier, S.; Kanneganti, T.D. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci. Rep. 2017, 7, 45126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollias, G.; Kontoyiannis, D. Role of TNF/TNFR in autoimmunity: Specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev. 2002, 13, 315–321. [Google Scholar] [CrossRef]
- Packard, A.E.; Leung, P.Y.; Vartanian, K.B.; Stevens, S.L.; Bahjat, F.R.; Stenzel-Poore, M.P. TLR9 bone marrow chimeric mice define a role for cerebral TNF in neuroprotection induced by CpG preconditioning. J. Cereb. Blood Flow Metab. 2012, 32, 2193–2200. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, J.T.; Chichester, K.L.; Bieneman, A.P. Toll-like receptor 9 suppression in plasmacytoid dendritic cells after IgE-dependent activation is mediated by autocrine TNF-α. J. Allergy Clin. Immunol. 2008, 121, 486–491. [Google Scholar] [CrossRef]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-α-induced apoptosis by NF-κB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Beg, A.A. Defining emerging roles for NF-κB in antivirus responses: Revisiting the interferon-β enhanceosome paradigm. PLoS Pathog 2011, 7, e1002165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasumoto, K.; Okamoto, S.I.; Mukaida, N.; Murakami, S.; Mai, M.; Matsushima, K. Tumor necrosis factor alpha and interferon gamma synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-kB-like binding sites of the interleukin 8 gene. J. Biol. Chem. 1992, 267, 22506–22511. [Google Scholar] [CrossRef]
- Sweiss, N.J.; Zhang, W.; Franek, B.S.; Kariuki, S.N.; Moller, D.R.; Patterson, K.C.; Niewold, T.B. Linkage of type I interferon activity and TNF-alpha levels in serum with sarcoidosis manifestations and ancestry. PLoS ONE 2011, 6, e29126. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Rentsendorj, A. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef]
- Ahn, J.; Barber, G.N. STING signaling and host defense against microbial infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J. Neurophysiol. 2019, 121, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Mathur, V.; Burai, R.; Vest, R.T.; Bonanno, L.N.; Lehallier, B.; Zardeneta, M.E.; Blurton-Jones, M. Activation of the STING-dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron 2017, 96, 1290–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, E.R.; Wang, B.; Wan, Y.W.; Chiu, G.; Cole, A.; Yin, Z.; Cao, W. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Investig. 2020, 130, 1912–1930. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.-X.; Sun, Y.; Roh, J.D.; Ng, R.P.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Lowes, H.; Pyle, A.; Duddy, M.; Hudson, G. Cell-free mitochondrial DNA in progressive multiple sclerosis. Mitochondrion 2019, 46, 307–312. [Google Scholar] [CrossRef]
- Nasi, M.; Bianchini, E.; De Biasi, S.; Gibellini, L.; Neroni, A.; Mattioli, M.; Pinti, M.; Iannone, A.; Mattioli, A.V.; Simone, A.M.; et al. Increased plasma levels of mitochondrial DNA and pro-inflammatory cytokines in patients with progressive multiple sclerosis. J. Neuroimmunol. 2020, 338, 577107. [Google Scholar] [CrossRef] [Green Version]
- Fissolo, N.; Cervera-Carles, L.; Villar Guimerans, L.M.; Lleó, A.; Clarimón, J.; Drulovic, J.; Dujmovic, I.; Voortman, M.; Khalil, M.; Gil, E.; et al. Cerebrospinal fluid mitochondrial DNA levels in patients with multiple sclerosis. Mult. Scler. J. 2019, 25, 1535–1538. [Google Scholar] [CrossRef]
- Leurs, C.E.; Podlesniy, P.; Trullas, R.; Balk, L.; Steenwijk, M.D.; Malekzadeh, A.; Teunissen, C.E. Cerebrospinal fluid mtDNA concentration is elevated in multiple sclerosis disease and responds to treatment. Mult. Scler. J. 2018, 24, 472–480. [Google Scholar] [CrossRef]
- Al-Kafaji, G.; Bakheit, H.F.; Alharbi, M.A.; Farahat, A.A.; Jailani, M.; Ebrahin, B.H.; Bakhiet, M. Mitochondrial DNA Copy Number in Peripheral Blood as a Potential Non-invasive Biomarker for Multiple Sclerosis. Neuromol. Med. 2020, 22, 304–313. [Google Scholar] [CrossRef]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.; Thouin, A.; Mollenhauer, B.; Burn, D.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef]
- Lowes, H.; Pyle, A.; Santibanez-Koref, M.; Hudson, G. Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol. Neurodegener. 2020, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- Podlesniy, P.; Puigròs, M.; Serra, N.; Fernández-Santiago, R.; Ezquerra, M.; Tolosa, E.; Trullas, R. Accumulation of mitochondrial 7S DNA in idiopathic and LRRK2 associated Parkinson’s disease. EBioMedicine 2019, 48, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeney, P.M.; Bennett, J.P. ALS spinal neurons show varied and reduced mtDNA gene copy numbers and increased mtDNA gene deletions. Mol. Neurodegener. 2010, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-W.; Ping, Y.-H.; Yen, J.-C.; Chang, C.-Y.; Wang, S.-F.; Yeh, C.-L.; Chi, C.-W.; Lee, H.-C. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol. Appl. Pharmacol. 2007, 220, 243–251. [Google Scholar] [CrossRef]
- Feng, Y.-M.; Jia, Y.-F.; Su, L.-Y.; Wang, D.; Lv, L.; Xu, L.; Yao, Y.-G. Decreased mitochondrial DNA copy number in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be salvaged by melatonin. Autophagy 2013, 9, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Trumpff, C.; Marsland, A.L.; Basualto-Alarcón, C.; Martin, J.L.; Carroll, J.E.; Sturm, G.; Vincent, A.E.; Mosharov, E.V.; Gu, Z.; Kaufman, B.A.; et al. Acute psychological stress increases serum circulating cell-free mitochondrial DNA. Psychoneuroendocrinology 2019, 106, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Fernström, J.; Grudet, C.; Ljunggren, L.; Träskman-Bendz, L.; Ohlsson, L.; Westrin, Å. Increased plasma levels of circulating cell-free mitochondrial DNA in suicide attempters: Associations with HPA-axis hyperactivity. Transl. Psychiatry 2016, 6, e971. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, D.; Wolkowitz, O.M.; Picard, M.; Ohlsson, L.; Bersani, F.S.; Fernström, J.; Westrin, Å.; Hough, C.M.; Lin, J.; Reus, V.I.; et al. Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology 2018, 43, 1557–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia de la Cruz, D.; Juárez-Rojop, I.E.; Tovilla-Zárate, C.A.; Martínez-Magaña, J.J.; Genis-Mendoza, A.D.; Nicolini, H.; González-Castro, T.B.; Guzmán-Priego, C.G.; López-Martínez, N.A.; Hernández-Cisneros, J.A.; et al. Association between mitochondrial DNA and cognitive impairment in schizophrenia: Study protocol for a Mexican population. Neuropsychiatr. Dis. Treat. 2019, 15, 1717–1722. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Efstathopoulos, P.; Millischer, V.; Olsson, E.; Wei, Y.B.; Brüstle, O.; Schalling, M.; Villaescusa, J.C.; Ösby, U.; Lavebratt, C. Mitochondrial DNA copy number is associated with psychosis severity and anti-psychotic treatment. Sci. Rep. 2018, 8, 12743. [Google Scholar] [CrossRef]
- Wang, D.; Li, Z.; Liu, W.; Zhou, J.; Ma, X.; Tang, J.; Chen, X. Differential mitochondrial DNA copy number in three mood states of bipolar disorder. BMC Psychiatry 2018, 18, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.; Dimick, M.K.; Sultan, A.; Duong, A.; Park, S.S.; El Sabbagh, D.E.S.; Goldstein, B.I.; Andreazza, A.C. Peripheral biomarkers of mitochondrial dysfunction in adolescents with bipolar disorder. J. Psychiatr. Res. 2020, 123, 187–193. [Google Scholar] [CrossRef]
- Kageyama, Y.; Kasahara, T.; Kato, M.; Sakai, S.; Deguchi, Y.; Tani, M.; Kinoshita, T. The relationship between circulating mitochondrial DNA and inflammatory cytokines in patients with major depression. J. Affect. Disord. 2018, 233, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Stertz, L.; Fries, G.R.; Rosa, A.R.; Kauer-Sant’anna, M.; Ferrari, P.; Paz, A.V.C.; Green, C.; Cunha, Â.B.M.; Dal-Pizzol, F.; Gottfried, C.; et al. Damage-associated molecular patterns and immune activation in bipolar disorder. Acta Psychiatr. Scand. 2015, 132, 211–217. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [Green Version]
- Weidberg, H.; Elazar, Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 2011, 4, pe39. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Zhang, Y.J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.; et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef] [PubMed]
- Sieverding, K.; Ulmer, J.; Bruno, C.; Satoh, T.; Tsao, W.; Freischmidt, A.; Akira, S.; Wong, P.C.; Ludolph, A.C.; Danzer, K.M.; et al. Hemizygous deletion of Tbk1 worsens neuromuscular junction pathology in TDP-43G298S transgenic mice. Exp. Neurol. 2021, 335, 113496. [Google Scholar] [CrossRef]
- Foster, A.D.; Downing, P.; Figredo, E.; Polain, N.; Stott, A.; Layfield, R.; Rea, S.L. ALS-associated TBK1 variant p.G175S is defective in phosphorylation of p62 and impacts TBK1-mediated signalling and TDP-43 autophagic degradation. Mol. Cell Neurosci. 2020, 108, 103539. [Google Scholar] [CrossRef]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; König, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Brüggemann, N.; Klein, C. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Release of mitochondrial Opa1 following oxidative stress in HT22 cells. Mol. Cell Neurosci. 2015, 64, 116–122. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Rahmani, Z. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [PubMed]
- Wang, L.; Xu, J.; Liu, H.; Li, J.; Hao, H.P. 5 inhibits SOD1 expression by up-regulating microRNA-206 and promotes ROS accumulation and disease progression in asthmatic mice. Int. Immunopharmacol. 2019, 76, 105871. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Klivenyi, P.; Klein, A.M.; Shinobu, L.A.; Epstein, C.J.; Flint Beal, M. Partial deficiency of manganese superoxide dismutase exacerbates a transgenic mouse model of amyotrophic lateral sclerosis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2000, 47, 447–455. [Google Scholar] [CrossRef]
- Squadrone, S.; Brizio, P.; Mancini, C.; Pozzi, E.; Cavalieri, S.; Abete, M.C.; Brusco, A. Blood metal levels and related antioxidant enzyme activities in patients with ataxia telangiectasia. Neurobiol. Dis. 2015, 81, 162–167. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; Bertolotti, A. Exposure of Hydrophobic Surfaces Initiates Aggregation of Diverse ALS-Causing Superoxide Dismutase-1 Mutants. J. Mol. Biol. 2010, 399, 512–525. [Google Scholar] [CrossRef]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis: Abnormal glutamate exocytotic release in ALS. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef]
- Liu, T.; Lu, B.; Lee, I.; Ondrovičová, G.; Kutejová, E.; Suzuki, C.K. DNA and RNA Binding by the Mitochondrial Lon Protease Is Regulated by Nucleotide and Protein Substrate. J. Biol. Chem. 2004, 279, 13902–13910. [Google Scholar] [CrossRef] [Green Version]
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174. [Google Scholar] [CrossRef]
- Carriedo, S.G.; Sensi, S.L.; Yin, H.Z.; Weiss, J.H. AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J. Neurosci. 2000, 20, 240–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G.; Ward, M.W. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 2000, 23, 166–174. [Google Scholar] [CrossRef]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Van Den Bosch, L. Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Hanger, D.P. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB–PTPIP 51 interaction and ER–mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef] [PubMed]
- Vielhaber, S.; Winkler, K.; Kirches, E.; Kunz, D.; Büchner, M.; Feistner, H.; Kunz, W.S. Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1999, 169, 133–139. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis. Model. Mech. 2019, 12, 4147. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Shaw, C.E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; McCluskey, L.F. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids 2016, 35, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Song, J.; Wang, G.; Cui, X.; Zheng, J.; Tang, Y.; Chen, X.; Li, J.; Cui, L.; Liu, C.-Y.; et al. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat. Commun. 2018, 9, 4468. [Google Scholar] [CrossRef] [Green Version]
- Puentes, F.; Malaspina, A.; Van Noort, J.M.; Amor, S. Non-neuronal cells in ALS: Role of glial, immune cells and blood-CNS barriers. Brain Pathol. 2016, 26, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Casula, M.; Iyer, A.M.; Spliet, W.G.M.; Anink, J.J.; Steentjes, K.; Sta, M.; Aronica, E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 2011, 179, 233–243. [Google Scholar] [CrossRef]
- West, X.Z.; Malinin, N.L.; Merkulova, A.A.; Tischenko, M.; Kerr, B.A.; Borden, E.C.; Byzova, T.V. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 2010, 467, 972–976. [Google Scholar] [CrossRef]
- Hartmann, G. Nucleic acid immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Taylor, R.W. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.-B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Autere, J.; Moilanen, J.S.; Finnilä, S.; Soininen, H.; Mannermaa, A.; Hartikainen, P.; Majamaa, K. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum. Genet. 2004, 115, 29–35. [Google Scholar]
- Campbell, G.R.; Reeve, A.; Ziabreva, I.; Polvikoski, T.M.; Taylor, R.W.; Reynolds, R.; Mahad, D.J. Mitochondrial DNA deletions and depletion within paraspinal muscles. Neuropathol. Appl. Neurobiol. 2013, 39, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [Green Version]
- Giannoccaro, M.P.; La Morgia, C.; Rizzo, G.; Carelli, V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. 2017, 32, 346–363. [Google Scholar] [CrossRef]
- Arthur, C.R.; Morton, S.L.; Dunham, L.D.; Keeney, P.M.; Bennett, J.P. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol. Neurodegener. 2009, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Cai, H. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Vilas, D.; Taylor, P.; Shaw, L.M.; Tolosa, E.; Trullas, R. Mitochondrial DNA in CSF distinguishes LRRK2 from idiopathic Parkinson’s disease. Neurobiol. Dis. 2016, 94, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [Green Version]
- She, H.; Yang, Q.; Shepherd, K.; Smith, Y.; Miller, G.; Testa, C.; Mao, Z. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Investig. 2011, 121, 930–940. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Keeney, P.M.; Quigley, C.K.; Dunham, L.D.; Papageorge, C.M.; Iyer, S.; Thomas, R.R.; Schwarz, K.M.; Trimmer, P.A.; Khan, S.M.; Portell, F.R.; et al. Mitochondrial gene therapy augments mitochondrial physiology in a Parkinson’s disease cell model. Hum. Gene Ther. 2009, 20, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y. DJ-1 as a Biomarker of Parkinson’s Disease. In DJ-1PARK7 Protein; Springer: Singapore, 2017; pp. 149–171. [Google Scholar]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Van Dongen, J.W. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Repici, M.; Hassanjani, M.; Maddison, D.C.; Garção, P.; Cimini, S.; Patel, B.; Outeiro, T.F. The Parkinson’s disease-linked protein DJ-1 associates with cytoplasmic mRNP granules during stress and neurodegeneration. Mol. Neurobiol. 2019, 56, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Mitsumoto, A.; Nakagawa, Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic. Res. 2001, 35, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, T.; Costa, C.; Bernardi, G. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Callaghan, S. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Riess, O. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterna, J.C.; Leng, A.; Weber, E.; Feldon, J.; Büeler, H. DJ-1 and Parkin modulate dopamine-dependent behavior and inhibit MPTP-induced nigral dopamine neuron loss in mice. Mol. Ther. 2007, 15, 698–704. [Google Scholar] [CrossRef]
- Bonello, F.; Hassoun, S.-M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef]
- Devine, M.J.; Lewis, P.A. Emerging pathways in genetic Parkinson’s disease: Tangles, Lewy bodies and LRRK2. FEBS J. 2008, 275, 5748–5757. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Palmer, T.D. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.H.; Laganière, J.; Cooper, O.; Mak, S.K.; Vu, B.J.; Huang, Y.A.; Langston, J.W. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: Reversal by gene correction. Neurobiol. Dis. 2014, 62, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Abarrategui, J.; Christian, H.; Lufino, M.M.P.; Mutihac, R.; Venda, L.L.; Ansorge, O.; Wade-Martins, R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 2009, 18, 4022–4034. [Google Scholar] [CrossRef] [Green Version]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.K.; Kang, H.Y.; Lu, B. Altered ER–mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Newman, L.E.; Shadel, G.S. Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J. Cell Biol. 2018, 217, 3327–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Liu, C.; Ji, S.; Yang, Q.; Ye, H.; Han, H.; Xue, Z. The NLRP3 inflammasome is involved in the pathogenesis of Parkinson’s disease in rats. Neurochem. Res. 2017, 42, 1104–1115. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, Y.H.; Chen, N.H.; Wang, H.B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2019, 67, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Bakota, L.; Brandt, R. Tau biology and tau-directed therapies for Alzheimer’s disease. Drugs 2016, 76, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossenkoppele, R.; Rabinovici, G.D.; Smith, R.; Cho, H.; Schöll, M.; Strandberg, O.; Ohlsson, T. Discriminative accuracy of [18F] flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 2018, 320, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [Green Version]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 1994, 36, 747–751. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Luong, T.; Neely, T.R.; Robinson, D.; Wands, J.R. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab. Investig. J. Tech. Methods Pathol. 2000, 80, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Nunomura, A.; Tamaoki, T.; Tanaka, K.; Motohashi, N.; Nakamura, M.; Hayashi, T.; Smith, M.A. Intraneuronal amyloid β accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol. Dis. 2010, 37, 731–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.-P.; Sarzi, E.; Aubert, S.; Chrétien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef]
- Zou, K.; Gong, J.S.; Yanagisawa, K.; Michikawa, M. A novel function of monomeric amyloid β-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 2002, 22, 4833–4841. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Frackowiak, J.; Mazur-Kolecka, B.; Imaki, H.; Deleon, M. Intraneuronal Aβ immunoreactivity is not a predictor of brain amyloidosis-β or neurofibrillary degeneration. Acta Neuropathol. 2007, 113, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Gallagher, J.J.; Finnegan, M.E.; Grehan, B.; Dobson, J.; Collingwood, J.F.; Lynch, M.A. Modest amyloid deposition is associated with iron dysregulation, microglial activation, and oxidative stress. J. Alzheimers Dis. 2012, 281, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Liang, X.; Wang, Q.; Hand, T.; Wu, L.; Breyer, R.M.; Montine, T.J.; Andreasson, K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 10180–10187. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Xu, I.M.-J.; Chiu, D.K.-C.; Lai, R.K.-H.; Tse, A.P.-W.; Lan Li, L.; Law, C.-T.; Tsang, F.H.-C.; Wei, L.L.; Chan, C.Y.-K.; et al. Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma. J. Clin. Investig. 2020, 127, 1856–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, K.; Giordano, T.; Brady, D.R.; Stoll, J.; Martin, L.J.; Rapoport, S.I. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Mol. Brain Res. 1994, 24, 336–340. [Google Scholar] [CrossRef]
- Parker, W.D.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090. [Google Scholar] [CrossRef] [PubMed]
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M.; et al. Protective Effects of Indian Spice Curcumin Against Amyloid-β in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2018, 61, 843–866. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Cervera-Carles, L.; Alcolea, D.; Estanga, A.; Ecay-Torres, M.; Izagirre, A.; Clerigué, M.; García-Sebastián, M.; Villanúa, J.; Escalas, C.; Blesa, R.; et al. Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. Neurobiol. Aging 2017, 53, 192.e1–192.e4. [Google Scholar] [CrossRef]
- Cottrell, D.A.; Blakely, E.L.; Johnson, M.A.; Ince, P.G.; Turnbull, D.M. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology 2001, 57, 260–264. [Google Scholar] [CrossRef]
- Maurer, I.; Zierz, S.; Möller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Naj, A.C.; Beecham, G.W.; Martin, E.R.; Gallins, P.J.; Powell, E.H.; Konidari, I.; Vance, J.M. Dementia revealed: Novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS Genet. 2010, 6, e1001130. [Google Scholar] [CrossRef] [Green Version]
- Pike, S.T.; Rajendra, R.; Artzt, K.; Appling, D.R. Mitochondrial C1-tetrahydrofolate synthase (MTHFD1L) supports the flow of mitochondrial one-carbon units into the methyl cycle in embryos. J. Biol. Chem. 2010, 285, 4612–4620. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.T.; Stover, P.J. Folate-mediated one-carbon metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [PubMed]
- Muntjewerff, J.W.; Kahn, R.S.; Blom, H.J.; Heijer, M. Homocysteine, methylenetetrahydrofolate reductase and risk of schizophrenia: A meta-analysis. Mol. Psychiatry 2006, 11, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osher, Y.; Sela, B.A.; Levine, J.; Belmaker, R.H. Elevated homocysteine levels in euthymic bipolar disorder patients showing functional deterioration. Bipolar Disord. 2004, 6, 82–86. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Obeid, R. Homocysteine and dementia: An international consensus statement. J. Alzheimers Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Wu, T.; Zhao, J.; Ji, L.; Song, A.; Zhang, M.; Huang, G. Plasma Homocysteine and Serum Folate and Vitamin B12 Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Case-Control Study. Nutrients 2017, 9, 725. [Google Scholar] [CrossRef] [Green Version]
- Salmon, E.; Sadzot, B.; Maquet, P.; Degueldre, C.; Lemaire, C.; Rigo, P.; Franck, G. Differential diagnosis of Alzheimer’s disease with PET. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1994, 35, 391–398. [Google Scholar]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.R.; Bussière, T.; Bouras, C.; Kövari, E.; Perl, D.P.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.Y. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat. Rev. Drug Discov. 2009, 8, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colangelo, V.; Schurr, J.; Ball, M.J.; Pelaez, R.P.; Bazan, N.G.; Lukiw, W.J. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: Transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 2002, 70, 462–473. [Google Scholar] [CrossRef]
- Hu, W.T.; Howell, J.C.; Ozturk, T.; Gangishetti, U.; Kollhoff, A.L.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF Cytokines in Aging, Multiple Sclerosis, and Dementia. Front. Immunol. 2019, 10, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Gelpi, E. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Iyer, S.; Thangavel, R.; Kempuraj, D.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, A. Co-localization of glia maturation factor with NLRP3 inflammasome and autophagosome markers in human Alzheimer’s disease brain. J. Alzheimers Dis. 2017, 60, 1143–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Wilton, D.K. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Rampe, D.; Wang, L.; Ringheim, G.E. P2X7 receptor modulation of β-amyloid-and LPS-induced cytokine secretion from human macrophages and microglia. J. Neuroimmunol. 2004, 147, 56–61. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef] [PubMed]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Di Virgilio, F. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef]
- Adinolfi, E.; Callegari, M.G.; Ferrari, D.; Bolognesi, C.; Minelli, M.; Wieckowski, M.R.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol. Biol. Cell 2005, 16, 3260–3272. [Google Scholar] [CrossRef] [Green Version]
- Massicot, F.; Hache, G.; David, L.; Chen, D.; Leuxe, C.; Garnier-Legrand, L.; Coudore, F. P2X7 cell death receptor activation and mitochondrial impairment in oxaliplatin-induced apoptosis and neuronal injury: Cellular mechanisms and in vivo approach. PLoS ONE 2013, 8, e66830. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef]
- Chung, I.C.; Chen, L.C.; Tsang, N.M.; Chuang, W.Y.; Liao, T.C.; Yuan, S.N.; Chang, Y.S. Mitochondrial oxidative phosphorylation complex regulates NLRP3 inflammasome activation and predicts patient survival in nasopharyngeal carcinoma. Mol. Cell Proteom. 2020, 19, 142–154. [Google Scholar] [CrossRef]
- Shida, M.; Kitajima, Y.; Nakamura, J.; Yanagihara, K.; Baba, K.; Wakiyama, K.; Noshiro, H. Impaired mitophagy activates mtROS/HIF-1α interplay and increases cancer aggressiveness in gastric cancer cells under hypoxia. Int. J. Oncol. 2016, 48, 1379–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel, E.M.; Hessas, E.; Müller, S.; Beiter, T.; Fisch, M.; Eibl, A.; Wolf, O.T.; Giebel, B.; Platen, P.; Kumsta, R.; et al. Cell-free DNA release under psychosocial and physical stress conditions. Transl. Psychiatry 2018, 8, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, N.; Li, Y.; Chang, S.; Liang, J.; Lin, C.; Zhang, X.; Liang, L.; Hu, J.; Chan, W.; Kendler, K.S.; et al. Genetic Control over mtDNA and Its Relationship to Major Depressive Disorder. Curr. Biol. CB 2015, 25, 3170–3177. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-C.; Jou, S.-H.; Lin, T.-T.; Lai, T.-J.; Liu, C.-S. Mitochondria DNA Change and Oxidative Damage in Clinically Stable Patients with Major Depressive Disorder. PLoS ONE 2015, 10, e0125855. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.K.; Lee, S.Y.; Park, M.; Joo, E.-J.; Kim, S.A. Investigation of mitochondrial DNA copy number in patients with major depressive disorder. Psychiatry Res. 2019, 282, 112616. [Google Scholar] [CrossRef]
- Andreazza, A.C.; Shao, L.; Wang, J.F.; Young, L.T. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch. Gen. Psychiatry 2010, 67, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shachar, D.; Karry, R. Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS ONE 2008, 3, e3676. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Onodera, Y.; Hamamoto, T.; Muraki, K.; Kato, N.; Kato, T. Hypofrontality and microvascular dysregulation in remitted late-onset depression assessed by functional near-infrared spectroscopy. Neuroimage 2005, 26, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Rasgon, N.L.; Kenna, H.A.; Geist, C.; Small, G.; Silverman, D. Cerebral metabolic patterns in untreated postmenopausal women with major depressive disorder. Psychiatry Res. Neuroimaging 2008, 164, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Tamura, M.; Chiba, Y.; Katsuse, O.; Suda, A.; Kamada, A.; Hirayasu, Y. Regional cerebral glucose metabolism in systemic lupus erythematosus patients with major depressive disorder. J. Neurol. Sci. 2017, 379, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, S.M.; Clark, A.G.; Stover, P.J.; Wells, M.T.; Litonjua, A.A.; Weiss, S.T.; Gaziano, J.M.; Tucker, K.L.; Baccarelli, A.; Schwartz, J.; et al. Folate network genetic variation, plasma homocysteine, and global genomic methylation content: A genetic association study. BMC Med. Genet. 2011, 12, 150. [Google Scholar] [CrossRef] [Green Version]
- Clarke-Harris, R.; Wilkin, T.J.; Hosking, J.; Pinkney, J.; Jeffery, A.N.; Metcalf, B.S.; Godfrey, K.M.; Voss, L.D.; Lillycrop, K.A.; Burdge, G.C. PGC1α Promoter Methylation in Blood at 5–7 Years Predicts Adiposity From 9 to 14 Years (EarlyBird 50). Diabetes 2014, 63, 2528–2537. [Google Scholar] [CrossRef] [Green Version]
- Jokinen, J.; Nordström, P. HPA axis hyperactivity and attempted suicide in young adult mood disorder inpatients. J. Affect. Disord. 2009, 116, 117–120. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W.J.H. Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The inflammatory & neurodegenerative (I & ND) hypothesis of depression: Leads for future research and new drug developments in depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar]
- Enache, D.; Pariante, C.M.; Mondelli, V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 2019, 81, 24–40. [Google Scholar] [CrossRef]
- Simon, N.M.; McNamara, K.; Chow, C.W.; Maser, R.S.; Papakostas, G.I.; Pollack, M.H.; Nierenberg, A.A.; Fava, M.; Wong, K.K. A detailed examination of cytokine abnormalities in Major Depressive Disorder. Eur. Neuropsychopharmacol. 2008, 18, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Basterzi, A.D.; Aydemir, Ç.; Kisa, C.; Aksaray, S.; Tuzer, V.; Yazici, K.; Göka, E. IL-6 levels decrease with SSRI treatment in patients with major depression. Hum. Psychopharmacol. Clin. Exp. 2005, 20, 473–476. [Google Scholar] [CrossRef]
- Tuglu, C.; Kara, S.H.; Caliyurt, O.; Vardar, E.; Abay, E. Increased serum tumor necrosis factor-alpha levels and treatment response in major depressive disorder. Psychopharmacology 2003, 170, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 Up-Regulates IL-18 Receptor Expression on T Cells, Th1 Cells, and B Cells: Synergism with IL-18 for IFN-γ Production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar]
- Alcocer-Gómez, E.; de Miguel, M.; Casas-Barquero, N.; Núñez-Vasco, J.; Sánchez-Alcazar, J.A.; Fernández-Rodríguez, A.; Cordero, M.D. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav. Immun. 2014, 36, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, X.-Y.; Zhang, Q.-Y.; Kong, L.-D. Microglial NLRP3 inflammasome activation mediates IL-1β-related inflammation in prefrontal cortex of depressive rats. Brain Behav. Immun. 2014, 41, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sheng, H.; Bao, Q.; Wang, Y.; Lu, J.; Ni, X. NLRP3 inflammasome activation mediates estrogen deficiency-induced depression- and anxiety-like behavior and hippocampal inflammation in mice. Brain Behav. Immun. 2016, 56, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Bremmer, M.A.; Beekman, A.T.F.; Deeg, D.J.H.; Penninx, B.W.J.H.; Dik, M.G.; Hack, C.E.; Hoogendijk, W.J.G. Inflammatory markers in late-life depression: Results from a population-based study. J. Affect. Disord. 2008, 106, 249–255. [Google Scholar] [CrossRef]
- Hung, Y.Y.; Huang, K.W.; Kang, H.Y.; Huang, G.Y.L.; Huang, T.L. Antidepressants normalize elevated Toll-like receptor profile in major depressive disorder. Psychopharmacology 2016, 233, 1707–1714. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Wang, T.; Wei, X.; Zhou, Y.; Li, J. CpG DNA-triggered upregulation of TLR9 expression affects apoptosis and immune responses in human plasmacytoid dendritic cells isolated from chronic hepatitis B patients. Arch. Physiol. Biochem. 2020, 1–8, Epub ahead of print. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | Species | Sample | Finding | Ref. |
|---|---|---|---|---|
| Neurodegenerative diseases | ||||
| MS | H. sapiens | CSF samples of MS patients | Association between lower ccf-mtDNA and MS and neurodegeneration | [57] |
| MS | H. sapiens | MS patients’ blood plasma samples | Increased ccf-mtDNA in MS patients compared to healthy patients | [58,59] |
| MS | H. sapiens | MS patients’ CSF samples | Increased ccf-mtDNA in CSF of MS patients compared to cognitively healthy patients | [60] |
| MS | H. sapiens | Relapsing-remitting MS patients’ peripheral blood samples | Decreased ccf-mtDNA levels in MS patients’ when compared to healthy patients | [61] |
| PD | H. sapiens | CSF samples from PD patients | Reduced ccf-mtDNA in CSF from PD patients compared to healthy subjects | [62] |
| PD | H. sapiens | CSF samples from PD patients | Significant reduction in ccf-mtDNA in PD patients Reduction in ccf-mtDNA associated with type and duration of treatment | [63] |
| AD | H. sapiens | CSF samples from preclinical AD patients | Lower ccf-mtDNA levels in CSF of preclinical AD patients (several studies failed to replicate results) | [64] |
| ALS | M. musculus | In vivo study of aberrant TDP-43 transgenic mice | TDP-43 Caused mtDNA release to the cytoplasm via the mPTP | [29] |
| ALS | H. Sapiens | ALS spinal cord neuron cell line | ALS spinal cord neurons showed significant reductions in mtDNA-cn and mitochondrial gene deletions | [65] |
| Neuropsychiatric diseases | ||||
| Addiction | H. sapiens | Methamphetamine-addiction model in human dopaminergic neuroblastoma SH-SY5Y cell line | Decrease in mtDNA-cn Apoptosis after 48 h of treatment | [66] |
| Addiction | M. musculus | Peripheral blood from murine model of morphine addiction Peripheral blood from heroin addiction patients | Decrease in mtDNA-cn in hippocampi; Melatonin-dosage restored mtDNA-cn | [67] |
| Anxiety | H. sapiens | Serum samples from a middle-aged cohort | Increased ccf-mtDNA levels with introduction of negative mood stimulus | [68] |
| MDD | H. sapiens | Plasma samples from suicide attempters | Increased levels of ccf-mtDNA | [69] |
| MDD | H. sapiens | Plasma samples from unmedicated MDD patients | Elevated ccf-mtDNA levels compared to healthy subjects; No significant difference in peripheral blood mononuclear cells’ mtDNA-cn | [70] |
| SZ | H. sapiens | Patient blood plasma samples | Increase in ccf-mtDNA in SZ patients; mtDNA release because of exacerbated apoptosis | [71] |
| SZ BD | H. sapiens | Whole-blood samples of screened patients | Increasing age and psychosis severity correlated with decreasing ccf-mtDNA levels; Risperidone treatments found to have a reducing effect on ccf-mtDNA at concentrations simulating clinical target level in plasma, but not at concentrations simulating CSF or brain interstitial target level | [72] |
| BD type I (BD-I) | H. sapiens | BD-I patients’ leukocyte samples | Negative association between number of manic-episode relapses and mtDNA-cn | [73] |
| BD | H. sapiens | Serum samples from an adolescent cohort | No significant difference in ccf-mtDNA levels between groups; Increase in serum lactate levels for adolescents diagnosed with BD | [74] |
| BD | H. sapiens | Plasma samples from MDD patients | MDD patients showed lower plasma mtDNA levels than healthy patients | [75] |
| BD | H. sapiens | Serum samples from BD patients | Higher levels of ccf-mtDNA in BD patients | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moya, G.E.; Rivera, P.D.; Dittenhafer-Reed, K.E. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 7030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137030
Moya GE, Rivera PD, Dittenhafer-Reed KE. Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders. International Journal of Molecular Sciences. 2021; 22(13):7030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137030
Chicago/Turabian StyleMoya, Gonzalo E., Phillip D. Rivera, and Kristin E. Dittenhafer-Reed. 2021. "Evidence for the Role of Mitochondrial DNA Release in the Inflammatory Response in Neurological Disorders" International Journal of Molecular Sciences 22, no. 13: 7030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137030